


Abstract
Two modified 2′-deoxynucleoside 5′-triphosphates have been used for the in vitro selection of a modified deoxyribozyme (DNAzyme) capable of the sequence-specific cleavage of a 12 nt RNA target in the absence of divalent metal ions. The modified nucleotides, a C5-imidazolyl-modified dUTP and 3-(aminopropynyl)-7-deaza-dATP were used in place of TTP and dATP during the selection and incorporate two extra protein-like functionalities, namely, imidazolyl (histidine analogue) and primary amino (lysine analogue) into the DNAzyme. The functional groups are analogous to the catalytic Lys and His residues employed during the metal-independent cleavage of RNA by the protein enzyme RNaseA. The DNAzyme requires no divalent metal ions or other cofactors for catalysis, remains active at physiological pH and ionic strength and can recognize and cleave a 12 nt RNA substrate with sequence specificity. This is the first example of a functionalized, metal-independent DNAzyme that recognizes and cleaves an all-RNA target in a sequence-specific manner. The selected DNAzyme is two orders of magnitude more efficient in its cleavage of RNA than an unmodified DNAzyme in the absence of metal ions and represents a rate enhancement of 105 compared with the uncatalysed hydrolysis of RNA.
INTRODUCTION
It is now over 20 years since the initial discovery that RNA enzymes (ribozymes) could catalyse reactions in the absence of protein. Since this time, seven naturally occurring ribozymes have been studied in detail, several of these have been crystallized and most recently the catalytic function of RNA in the 30S ribosomal subunit has been proposed (1). All of these ribozymes normally utilize divalent metal ion cofactors such as magnesium for optimal activity. It has been shown that efficient catalysis may be achieved by the hairpin ribozyme when cobalt hexamine replaces magnesium ions (2–4), whilst high concentrations of monovalent cations can support catalysis by the hammerhead, hairpin and VS ribozymes (5,6). In contrast, the HDV ribozyme performs self-cleavage in the absence of divalent metals but at low pH (7,8). These results suggest the importance of electrostatic or structural roles for the metal ions. At least for the hammerhead, hairpin and VS ribozymes, the importance of electrostatic or structural roles for the metal ions is implied. Despite these results, the rate constants for these ribozymes at pH 7 and at cellular concentrations of sodium chloride are much reduced compared with those that are achieved in the presence of divalent metal ions.
Although catalytic DNAs (DNAzymes) have not been found in nature, they may be created using the combinatorial technique of SELEX (9–11). DNAzymes have been selected that are capable of accelerating a surprising variety of chemical reactions such as RNA cleavage, DNA cleavage, DNA ligation, DNA phosphorylation, porphyrin metallation, DNA capping, DNA depurination and the Diels-Alder reaction (12–15). In SELEX, nucleic acid molecules (either RNA or DNA) are selected from a library of typically 1012–1015 molecules of random sequence based on their ability to catalyse a given reaction. The technique exploits the wide range of structures that single-stranded nucleic acids can adopt and mimics the natural processes of evolution.
Ribozymes that are capable of the sequence-specific cleavage of RNA have potential uses in vitro as chemical nucleases and biosensors (16–19) and in vivo as therapeutic agents for the controlled destruction of mRNA for gene inactivation and the targeted cleavage of viral, oncogene or mutant mRNA (20–24). However, in vivo applications of ribozymes are limited by problems associated with their chemical instability, susceptibility toward cellular nucleases and the sub-optimal physiological concentrations of divalent metal ions, e.g. magnesium required for cleavage. Some progress has, however, been made by the incorporation of 2′-modified sugar or modified phosphate diester backbones into the RNA molecules to render them nuclease resistant (25). However, whilst these changes often produce the desired increased stability in vivo, they often severely decrease the catalytic efficiency of the ribozyme, thus reducing their efficacy. In contrast, DNAzymes designed to catalyse the sequence-specific cleavage of RNA for therapeutic applications in vivo offer advantages over analogous RNA-based enzymes due to their increased chemical stability. However, like RNA-based enzymes, most DNAzymes are divalent metal-ion dependent and require magnesium (26,27), calcium (28,29) or zinc (30) ions in order to produce catalytic activity. Furthermore, the concentrations of such divalent metal ions often exceed those present physiologically. Significantly, two metal-independent DNAzymes that were obtained following SELEX experiments in the absence of divalent metals displayed rate constants in the presence of monovalent cation concentrations of 0.25–1 M and at pH 7 of at best around 10–3 min–1 (28,31). Consequently, the development of DNAzymes that function effectively in the absence of divalent metal ions has begun to attract much interest.
In a divalent metal ion-independent DNAzyme the nucleobases may support catalysis using electrostatic or general acid–base mechanisms but the pKas of the functional groups are not well suited for such tasks. The poor catalytic rate enhancements achieved by nucleic acid-based enzymes relative to those attained by protein enzymes are probably in part due to the rather limited functional group repertoire of nucleic acids. In contrast, amino acids within proteins offer a wide variety of functional groups that are exploited for catalysis. Consequently, there has been growing interest in enhancing the catalytic activities of nucleic acid enzymes by equipping them with functionality that can support catalysis. Initial approaches to achieve this aim involved adding amino acid cofactors such as l-histidine (32) or by using oligonucleotide cofactors to regulate catalysis (33,34). However, a far more attractive approach is the direct modification of nucleic acids with suitable functional groups that are capable of catalysing the reaction of interest. The incorporation of extra functional groups into ribozymes may be conveniently achieved by employing suitably modified analogues of nucleoside 5′-triphosphates (35–44) in place of their natural counterparts during SELEX. The potential for this approach in developing catalytic nucleic acids with enhanced or novel activities was first demonstrated by Eaton and co-workers for an RNA-based Diels-Alderase enzyme. The Diels-Alderase was generated using SELEX in which a UTP bearing a pyridine moiety appended to C5 was used in place of natural UTP during the selection process (43).
The use of modified dNTPs for selecting more efficient DNAzymes was first exploited by Santoro et al. (45) who designed a zinc-dependent DNA-based ribonuclease functionalized with imidazolyl (histidine-like) groups. Thus, using the C5-imidazole functionalized dUTP analogue 1 (Fig. 1) in place of TTP during SELEX allowed the isolation of a DNAzyme that cleaves an RNA target with sequence specificity in the presence of zinc ions. The authors identified three essential imidazole moieties within the DNAzyme that they proposed could bind a zinc atom which in turn could activate a co-ordinated water molecule in order to achieve RNA cleavage, a mechanism that is comparable with that used by carboxypeptidase A.
Figure 1.

Selection strategy for the modified DNAzyme using modified nucleotides 1 and 2 (inset). DNA N50-template library 5′-d(GTGCCAAGCTTACCG-N50-GTCGCCATCTCTTCC)-3′ was copied using primer 5′-biotin-d(GGAAAAA)-r(GUAACUAGAGAU)-d(GGAAGAGATGGCGAC)-3′ and superscript II reverse transcriptase in the presence of dGTP, dCTP, modified dUTP (1) and modified dATP (2). The double-stranded products were added to streptavidin beads, followed by denaturation and washing to remove the non-biotinylated template. The pool of catalyst molecules was eluted following self-cleavage of captured biotinylated single-stranded catalyst-substrate molecules in the absence of divalent metal ions. Subsequent amplification by PCR using primers 5′-biotin-d(GGAAGAGATGGCGAC)-3′ and 5′-d(GTGCCAAGCTTACCG)-3′, followed streptavidin capture of the biotinylated strand, regenerated template molecules for the next cycle of selection. Incubation times (elution times) for the self-cleavage reaction were decreased from 1 h in cycles 1–6 to 1 min in cycle 13. See Materials and Methods for full experimental details.
In developing DNAzymes as sequence-specific ribonucleases the incorporation of modified nucleotides bearing side chains designed to mimic those of the amino acids His and Lys allows the delivery of catalysis through general acid–base mechanisms and electrostatic complementarity by analogy with RNaseA. The potential of this approach has been demonstrated by Perrin and co-workers (46,47) using a DNAzyme functionalized with both amino and imidazolyl groups to mimic the catalytic groups used by the corresponding protein. The modified DNAzyme was obtained using C5-aminoallyl-dUTP and a C8-imidazolyl-modified dATP (38) in place of TTP and dATP, respectively, during SELEX. The DNAzyme achieves the metal-independent catalytic cleavage of a single ribonucleotide unit within a DNA target molecule and may be acting as a mimic of RNaseA. We have also been actively pursuing this goal and have employed the imidazole-functionalized dUTP analogue 1 (39) together with the amino-modified dATP analogue 2 (36) during SELEX experiments. In these experiments, we used the 12 nt RNA target sequence employed previously by Santoro and Joyce (26) in their selection of a magnesium-dependent DNA-based ribonuclease. We describe here the first example of an amino- and imidazolyl-functionalized, divalent metal-independent DNAzyme that recognizes and achieves the sequence-specific cleavage of an all-RNA target and remains active in relatively low ionic strength buffer.
MATERIALS AND METHODS
Chemicals and reagents
All chemicals were molecular biology grade quality. All water solutions were made with MilliQ quality water purified by PurLite System and filtered through 0.22 µm filters. Natural 2′-deoxynucleoside 5′-triphosphates were from MBI Fermentas, whilst the two modified nucleotide triphosphates 1 and 2 (Fig. 1) were synthesized and purified as described previously (35,36).
Enzymes
SuperScript II Reverse Transcriptase was from Life Technologies. Taq DNA polymerase from Promega and BIO X ACT polymerase from Bioline were used for PCR. DNA ligase was from Qiagen as a part of pDrive cloning kit. EcoRI restriction endonuclease was from Promega.
DNA and oligonucleotides
Vector pDrive designed for cloning PCR products was from Qiagen. Oligodeoxyribonucleotides were synthesized on a 0.2 µmol scale with an ABI 392 DNA Synthesizer using conventional phosphoramidite reagents and standard synthesis and deprotection protocols. The syntheses of oligoribonucleotides and mixed DNA/RNA oligonucleotides were performed on a 1 µmol scale using 2′-O-tert-butyldimethylsilyl-3′-O-(2-cyanoethyl diisopropylphosphoramidite) monomers and solid supports with phenoxyacetyl protection for adenosine and guanosine and acetyl protection for cytidine (Glen Research), together with the same protecting groups for DNA monomers (Glen Research). Labelled oligonucleotides were prepared using 5′-biotin amidite (Glen Research) and 6-FAM fluorescein amidites (Transgenomic) in the final cycle of synthesis.
Oligodeoxyribonucleotides were purified by reversed-phase HPLC on a Jones C18 column (250 × 4.6 mm) using CH3CN in triethylammonium acetate. Oligoribonucleotides were deprotected and purified by denaturing preparative 20% PAGE as described (3).
Structures of all oligonucleotides were confirmed by MALDI-TOF mass spectra and displayed molecular weights within 4 Da of the expected masses.
In vitro selection
A DNA N50-template library 5′-d(GTGCCAAGCTTACCG-N50-GTCGCCATCTCTTCC)-3′ (20 pmol) was copied using 10 pmol of primer 1 [5′-biotin-d(GGAAAAA)-r(GUAACUAGAGAU)-d(GGAAGAGATGGCGAC)-3′] in a 50 µl mixture containing 500 U Superscript II Reverse Transcriptase (Life Technologies), 0.3 mM dGTP, 0.3 mM dCTP, 1.2 mM modified dUTP (1) (35,39), 1.2 mM modified dATP (2) (36), 50 mM Tris–HCl pH 8.3, 75 mM KCl, 3 mM MgCl2, 20 mM DTT incubated at 45°C for 2 h. One picomole of 5′-fluorescein-labelled primer 1 (5′-FAM label; Glen Research) was included to monitor the reaction using the denaturing HPLC (DHPLC) WAVE™ System equipped with a DNAsep™ column (Transgenomics) using a 5 µl aliquot of the reaction mixture in the conditions described below. The reaction mixture was purified by a Qiaquick Gel Extraction Kit (Qiagen), NaCl added to a final concentration of 0.2 M and the extension products immobilized on 100 µl of streptavidin agarose beads (Sigma) previously washed with 5 × 100 µl of wash buffer (0.2 M NaCl, 10 mM EDTA, 50 mM Tris–HCl, pH 7.5). The beads were incubated for 15 min at room temperature and then washed with 5 × 100 µl of wash buffer. The non-biotinylated DNA strand was removed by washing with 5 × 100 µl of ice-cold 0.1 N NaOH, 0.2 M NaCl and the beads then washed with 5 × 100 µl of 37°C wash buffer and self-cleaved catalysts eluted after incubation at 37°C for a given time with 3 × 100 µl of wash buffer. Elution times were: cycles 1–6, 1 h (i.e. 3 × 20 min) for the first six cycles; cycle 7, 45 min; cycle 8, 30 min; cycle 9, 15 min; cycle 10, 9 min; cycle 11, 6 min; cycle 12, 3 min; cycle 13 (final cycle), 1 min. Eluates were combined, concentrated four times on Microcon YM10 filters (Millipore) and made up to 30 µl with water. Five microlitres of this sample was diluted 105 times and 20 × 1 µl samples were amplified by PCR [30 cycles; 92°C (10 s), 50°C (30 s), 72°C (30 s)] using the primers (0.5 µM) 5′-biotin-d(GGAAGAGATGGCGAC)-3′ and 5′-d(GTGCCAAGCTTACCG)-3′, normal dNTPs (100 µM) and 5 U Taq polymerase (Promega) in 100 µl containing 1× Promega Taq buffer. PCR amplification was monitored by denaturing 14% PAGE with ethidium bromide staining or by using a DHPLC WAVE™ System (data not shown). The 20 PCR mixtures were combined and purified on a Qiaquick PCR Purification Kit (Qiagen), and immobilized on 0.5 ml streptavidin magnetic beads (Dynabeads M-280; Dynal) according to the manufacturer’s protocol and eluted with 3 × 100 µl of 0.1 N NaOH, 0.2 M NaCl to obtain the non-biotinylated strand. Eluates were neutralized with 30 µl of 7.5 M NH4OAc and desalted on Microcon YM10 filters. The isolated single-stranded DNA was used as a template in the next cycle of selection. After the 13th cycle of selection, enriched DNA molecules were amplified by PCR using non-biotinylated primers and cloned into pDrive Vector (Qiagen). Plasmids from selected colonies were purified by a QIAprep Spin Mini Prep Kit (Qiagen) and insertions were sequenced (DBS Genomics). Resulting sequences were analysed using RNA and DNA M-fold secondary structure prediction software (48).
Cloning of PCR products
PCR products were cloned by a pDrive Cloning Kit following the protocol recommended by Qiagen. Approximately 90 different clones were picked up and grown overnight in 3 ml of LB with 50 mg/ml Amp at 37°C. Plasmids were isolated by a QIAprep Spin Mini Prep Kit (Qiagen) and 100 bp insertions were checked by denaturing 14% PAGE after EcoRI digestion procedure (Supplementary fig. 3). Finally, 50 plasmids were found to have insertions of interest.
Preparation of single-stranded DNA
Plasmid DNA from an individual clone was amplified by PCR using primers 5′-d(GGAAGAGATGGCGAC)-3′ and 5′-biotin-d(GTGCCAAGCTTACCG)-3′ and single-stranded biotinylated DNA was obtained using conditions described (49) with some modifications as follows: captured double-stranded DNA was washed five times with 0.07 M ammonium phosphate, pH 7.5. The first strand was removed by 2 × 2 min treatment with 25% aqueous ammonia at room temperature. The second strand was recovered by 2 × 10 min treatment with 25% ammonia solution at 65°C, dialysed using a Microcon YM30 filter and used for cleavage studies. Cautionary note: we have found that the release of the biotinylated strand during this ammonolysis as published (49) appears to be batch specific. In particular, we have found that this procedure only works reproducibly with Dynabeads M-280 produced by Dynal Biotech sourced from Norway.
Analysis of self-cleavage reactions
The single-stranded DNA (20 pmol) was used as a template in the primer template extension reaction with Superscript II Reverse Transcriptase, modified dNTPs 1 and 2, dCTP and dGTP and 5′-fluorescein-labelled RNA/DNA mixed primer 1 as described above. The resulting DNA was immobilized on Dynabeads M-280 and single-stranded DNA was eluted with 10 µl of ice-cold 0.1 N NaOH, 0.1 M NaCl. The eluate was neutralized immediately by adding an equimolar quantity of ice-cold HCl to give final concentrations of Tris–HCl buffer at 50 mM, 10 mM EDTA and 200 mM NaCl in a final pH of 7.5 and a final volume of 13 µl. Where magnesium or zinc was added, MgCl2 or ZnCl2 was added immediately after neutralization to give the appropriate divalent metal ion concentration in a final volume of 13 µl. The mixtures were then incubated at 37°C for various times. The reactions were diluted with water to 100 µl and the whole sample analysed using DHPLC on the WAVE™ System as described below.
An RNA ladder was generated by limited alkaline hydrolysis of 5′-fluorescein-labelled DNA/RNA mixed primer 1 [conditions: 20 µl solution of 5′-fluorescein-labelled primer 1 (100 nM) heated at 99°C for 10 min in 50 mM NaHCO3]. Two microlitres of 1 M Tris–HCl, pH 7.5 was added to stop the reaction. One microlitre of this reaction was diluted to 100 µl with water and analysed using DHPLC on the WAVE™ System as described below.
Denaturing HPLC analysis
DHPLC used the Nucleic Acid Fragment Analysis WAVE™ System equipped with a DNAsep column (Transgenomics). For analysis of template extension during SELEX, the following gradient was used.
For analysis of primer extension reaction (see Supplementary fig. 4). Buffer A (0.1% acetonitrile, 0.025 mM tetrabutylammonium bromide, 0.01 mM EDTA) and buffer B (70% acetonitrile, 0.025 mM tetrabutylammonium bromide, 0.01 mM EDTA) at 50°C at a flow rate of 0.9 ml/min. The gradient used was: buffer B; 30% (at 0 min), 50% (1 min), 90% (13 min), 100% (at 13.1 min), 100% (at 13.6 min), 30% (at 13.7 min), with fluorescent detection with excitation at 494 nm and emission detected at 525 nm. Retention times: primer, 4.5 min; product (fully extended primer), 8.2 min.
For initial screening of clones (Fig. 3). The same gradient and conditions were used as for the primer extension reaction above. Retention times: DNAzyme/substrate, 11.2 min; products 1 and 2, 5.1 and 4.5 min, respectively.
Figure 3.

WAVE™ System DHPLC traces of self-cleavage reactions showing analysis of different clones or the selected pool. The reactions were performed for 1 h at 37°C in 50 mM Tris–HCl, pH 7.5; 10 mM EDTA; 0.2 M NaCl. ‘DNA pool’ means enriched pool of molecules after the last selection round; ‘Family 1’, one clone from family 1 sequences; ‘Family 2’, two different clones (clone 32 and clone 36) from family 2 sequences. Clone 32 (family 2) was the most active.
For analysis of self-cleavage reactions, 5′-fluorescein- labelled-modified and -unmodified clone 32 sequences (Figs 4, 6 and Supplementary fig. 6). Buffer A (0.1% acetonitrile, 0.025 mM tetrabutylammonium bromide, 0.01 mM EDTA) and buffer B (70% acetonitrile, 0.025 mM tetrabutylammonium bromide, 0.01 mM EDTA) at 50°C at a flow rate of 1 ml/min. The gradient used was: buffer B, 30% (at 0 min), 40% (1 min), 80% (23 min), 100% (at 23.1 min), 100% (at 23.6 min), 30% (at 23.7 min).
Figure 4.

WAVE™ System DHPLC traces of self-cleavage reactions using 5′-fluorescein-labelled modified and unmodified clone 32 sequences. ‘Natural’, unmodified sequence; ‘dA*’, sequence containing 2; ‘dU*’, sequence containing 1; ‘dA*dU*’ sequence containing 1 and 2. The RNA ladder was generated by limited alkaline hydrolysis of a 5′-fluorescein-labelled DNA/RNA mixed primer 1. See Materials and Methods for full experimental details.
Figure 6.

DHPLC WAVE™ System traces of self-cleavage of clone 32 sequence at different ionic strengths. Self-cleavage reactions of clone 32 sequence after 15 min at 37°C in 50 mM Tris–HCl, pH 7.5; 10 mM EDTA and indicated concentrations of sodium chloride. Arrows indicate positions of cleavage in comparison with RNA ladder (sequence inset).
Kinetic studies
DHPLC data of the cleavage reactions were fitted to first-order reactions with Kaleidagraph software (Synergy Software, Reading, PA, USA) using equation 1:
Pt = P∞ (1 – e–kt) 1
where Pt is the percentage product at time t, P∞ is the percentage product at the end point of the reaction and k is the first-order rate constant.
RESULTS AND DISCUSSION
In vitro selection of the DNAzyme
The objective of this work was to select a DNAzyme that cleaves a specific internucleotide bond in RNA and does not require the participation of divalent metal ions in either catalysis or formation of tertiary structure. For this purpose, we used a random 50 nt library and a 12 nt all-RNA target sequence that was utilized previously by Santoro and Joyce (26) to select a metal-dependant DNAzyme. The 12 nt RNA sequence corresponds to the start codon region of several HIV-1 mRNAs. Thus, our selection was designed to identify catalysts capable of RNA recognition and cleavage with sequence specificity and in the absence of divalent metal ions.
Previously, we described the syntheses of a set of imidazolyl- and amino-functionalized dUTP and 7-deaza-dATP derivatives and evaluated these as substrates for DNA polymerases (35,36). Based on this work, we chose to employ the most efficient substrates to achieve the incorporation of imidazolyl and amino functional groups, respectively, into DNAzymes during SELEX. Thus, a C5-urocanic acid modified allylamino dUTP analogue 1, a compound described earlier by Sakthivel and Barbas (39) and the 7-aminopropynyl modified 7-deaza-dATP analogue 2 (36) (Fig. 1) were chosen. Both of these analogues involve modifications within the major groove of the resulting DNA and do not affect the abilities of the modified bases to participate in Watson–Crick hydrogen bonding. Thus, nucleic acids containing these analogues are able to adopt tertiary structures that can support catalysis.
Using a 12 nt target RNA sequence and a randomized N50 library, the selection strategy corresponds to that employed previously by Santoro and Joyce (26) and is summarized in Figure 1 (Supplementary fig. 1 shows the full nucleic acid sequences). Two extra functional groups (imidazolyl and primary amino) were incorporated into the initial random 50 nt DNA library during DNA extension using template-directed synthesis. For this we employed SuperScript II Reverse Transcriptase (acting as DNA-dependant DNA polymerase), modified triphosphates 1 and 2 in place of TTP and dATP and a biotinylated DNA/RNA mixed primer containing the embedded target RNA sequence. The biotinylated products were then captured on streptavidin beads and the non-biotinylated template DNA strands removed following denaturation and washing. Upon incubation in pH 7.5 Tris buffer containing 0.2 M NaCl and EDTA, active catalysts within the pool were released from the beads following self-cleavage within the RNA target sequence and were then amplified by PCR using natural nucleotides prior to the next round of selection. The duration of the self-cleavage step of the immobilized single-stranded RNA/DNA pool performed in the absence of divalent metal ions (and in the presence of EDTA) was decreased from 1 h in cycles 1–6 to 1 min in the last round of selection (cycle 13).
Initially, during analysis of the PCRs using denaturing PAGE, the product of interest (comparable length to the initial random sequence) was accompanied by other slower migrating species. This problem was solved by dilution of the DNA template prior to PCR (Supplementary fig. 2), which produced pure PCR product with the correct length. In later cycles we used a proof-reading thermostable polymerase, which produced cleaner PCR products and was capable of a more accurate replication of the active catalysts than that achieved using Taq DNA polymerase. After the 13th cycle the enriched DNA pool was amplified by PCR using unmodified primers, the PCR products cloned into pDrive vector and 50 individual clones containing inserts of the correct length (Supplementary fig. 3) were sequenced.
Denaturing HPLC analysis of the fluorescein-labelled products
High-resolution, ion-pair, reversed-phase denaturing HPLC (DHPLC) in conjunction with fluorescent detection (Transgenomic WAVE™ machine) was used for both simultaneous separation and quantification of the products. This technique offers comparable resolution to PAGE of radiolabelled oligomers, whilst avoiding its disadvantages, such as the need for regular re-labelling of the DNA using radioactive ATP and the time-consuming processes of gel preparation, electrophoresis, gel drying and quantification using phosphoimaging. In previous studies we have found good correlation between kinetic data obtained using fluorescently labelled substrates and that obtained using radioactively labelled substrates in a variety of enzyme assays (50–52). The same technique was also used for analysis of the RNA cleavage reaction (Figs 3–6) and monitoring the DNA extension steps during selection rounds (see Supplementary fig. 4). In the latter case a trace amount of 5′-fluorescein-labelled primer was included in order to monitor the extension reaction.
Analysis of the enriched DNA pool of sequences
Fifty sequences from different clones were analysed using alignment and folding, or secondary structure prediction (48), and revealed two main families (family 1 and family 2), each of four sequences with more than 95% primary structure homology (see Supplementary fig. 5). Family 1 contained ∼55% of the modified dU analogue. In family 2, the content of the dU analogue was slightly less (40%), but in contrast to the members of family 1, these sequences appeared to have more promising secondary structures in terms of Watson–Crick base pairing between the DNAzyme and RNA target sequence after virtual folding. In all, five sequences, one from family 1, two from family 2, together with two more based on their folded secondary structures, were chosen for further study. In addition, the total enriched pool of DNA after the last 13th round of selection was used as a control in cleavage analysis.
A 5′-fluorescently labelled mixed RNA/DNA primer containing the embedded 12 nt target RNA sequence (identical in sequence to the biotinylated primer in Fig. 1) was used to prepare samples of the five selected clones for studying their intramolecular cleavage. Thus, PCR amplification of the cloned sequences using a biotinylated ‘template’ strand, produced double-stranded PCR products that were captured on streptavidin beads. Following removal of the non-biotinylated strand, the biotinylated strand was eluted from the beads following heating in ammonia (49). The isolated biotinylated strands then served as templates for preparing the fluorescently tagged DNAzyme–substrate sequences (Fig. 2) using superscript II according to the scheme shown in Figure 1. In addition, a fluorescently tagged library of all the sequences from the enriched DNA pool after the last round of selection was prepared analogously. When the same primer (i.e. lacking the 3′-catalytic sequence) was incubated under conditions employed during the cleavage assay, there was no evidence of any reaction after 3 days of incubation (data not shown).
Figure 2.

Self-cleavage scheme. Plasmid DNA from individual clones or the enriched DNA pool after the last selection round were amplified by PCR and immobilized on streptavidin beads. The two individual DNA strands were recovered by treatment with concentrated ammonia under different conditions as described (49) (see also Materials and Methods). The biotinylated strand was used for a primer extension reaction with modified triphosphates 1 and 2 and a 5′-fluorescein (Fl)-labelled primer 1. The resulting double-stranded product was immobilized on streptavidin magnetic beads and single-stranded molecules were eluted by treatment with ice-cold sodium hydroxide. Eluates were neutralized immediately and incubated at 37°C. The RNA sequence following cleavage that was initially unknown is indicated as ‘??????’.
For all five sequences, the 12 nt RNA target sequence was cleaved specifically at one of two positions; after the second and sixth ribonucleotides (Figs 3 and 4), the major products being formed following reaction at the former site. In each case the scissile phosphate diester linkage was between rU and rA residues. The selectivity of the cleavage reaction is different to that reported by Joyce and co-workers for the zinc-dependent DNAzyme (45) and presumably reflects differences in the tertiary folding and mechanism of cleavage between the respective DNAzyme–substrate complexes. The specificity also differs from that observed with the magnesium-dependent, unmodified DNAzyme described earlier by the same group (26). The preference for selected cleavage at U-A sites determined for the DNAzyme described here is consistent with findings of others and based on such sites being more favourable toward cleavage due to decreased base stacking and/or altered hydrogen bonding networks around the scissile linkage (53,54).
In the earlier studies by Joyce and co-workers (26,45), early rounds of selection were characterized by multiple cleavage sites within the RNA substrate, whilst in later rounds a DNAzyme cleaving at a single one of these sites was evolved. We anticipate that for the DNAzyme described here, after several further rounds of SELEX, a single cleavage site would also be selected.
Of the sequences that we investigated, clone 32, a member of family 2, was more active than any of the other catalysts tested (Fig. 3 and Supplementary fig. 5) including the total DNA pool after cycle 13 and was therefore studied further.
Characterization of the DNAzyme
In order to investigate the influence of the base modifications on catalysis, we prepared 5′-fluorescein-labelled clone 32 sequences containing no modifications, the modified dU only (using analogue 1) and the modified dA only (using analogue 2) and compared the efficiency of their self-cleavage reactions with that of the doubly modified sequences. Analysis of the reactions using DHPLC revealed that both of the base modifications were required for optimal cleavage (Fig. 4), but the influence of the imidazolyl-modified dU analogues was more crucial. After 1 h, only a small amount of reaction was observed in the case of the natural, unmodified sequence (7%) that was comparable with the amount of reaction achieved by the dA-modified sequence (10% cleavage). For the dU-modified sequence, more than 30% cleavage had been achieved in a comparable time, whilst the amount of product formation was further increased upon incorporation of both of the dU- and dA-modified nucleotides into the sequence. These results clearly demonstrate that both imidazolyl and primary amino functionalities are necessary for achieving the optimum rate of RNA cleavage by the DNAzyme.
The determined first-order rate constants for cleavage at the two sites within the RNA sequence using the doubly modified DNAzyme were quite similar, with values of 0.06 ± 0.01 and 0.07 ± 0.01 min–1 at the major and minor sites, respectively (Fig. 5). In previous work, Geyer and Sen selected unmodified DNAzymes cabable of RNA hydrolysis and then reselected DNAzymes with increased activity from the initial pool of catalysts (31). In 0.2 M NaCl and in the absence of divalent metal ions, DNAzymes with kcat values of 0.0015 min–1 were reported. The cleavage by DNAzymes at an adenosine within a DNA target, in the absence of metal ions, reported by the Famulok group, proceeds with a similar efficiency (28). Thus, the DNAzyme selected herein is ∼50-fold more efficient than DNAzyme-catalysed cleavage of RNA in the absence of metal ions (31) and represents a rate enhancement of ∼105 compared with the uncatalysed reaction (55).
Figure 5.

Kinetic analysis of self-cleavage. Plots of percentage product versus time for self-cleavage of clone 32 sequence between nucleotides 2 and 3 (circles) and 6 and 17 (squares) indicated in the inset structure. For cleavage between nucleotides 2 and 3 and between 6 and 7, the rate constants were 0.07 ± 0.01 and 0.06 ± 0.01 min–1, with end points of 44 and 13%, respectively.
We also investigated the effect of ionic strength on the cleavage reaction. Thus, decreasing the NaCl concentration from 200 to 100 mM had little effect on the reaction rates. However, cleavage was substantially inhibited at concentrations below 50 mM (Fig. 6), presumably conditions under which correct folding of the DNAzyme and substrate does not take place. Nevertheless, the reaction is supported under physiological salt concentration with the DNAzyme displaying comparable activity in the presence of 1 mM magnesium and 10 µM zinc (Supplementary fig. 6). No significant loss of activity was observed up to 5 mM concentrations of Mg2+.
The predicted secondary structure of the clone 32 catalytic core derived from simulation folding (48) is presented in Figure 7 (two possible structures are shown in Supplementary fig. 7). It should be pointed out that this structure is necessarily based on the folding of unmodified sequences. Whilst Watson–Crick base pairing for the modified nucleotides in this study can occur as normal, folding programs such as M-fold do not take into account possible interactions that might involve imidazolyl or amino groups within the 3D structure. A 6–8 nt triplet AAG near the 5′ end was forced to interact with a 3′-TTC-5′ triplet at the 3′ end to form double helix prior to the folding calculation. As with other DNAzymes, recognition appears to be via Watson–Crick base pairing between the catalytic sequence and its substrate. Residues within the DNAzyme which involve imidazolyl-modified dU as well as primary amino-modified 7-deaza-dA are contained within looped regions opposite both sites of the reaction. In addition, both sites have an upstream rU-dG ‘wobble’ pair. This site has been described by others (45) as being crucial for catalytic activity and might be involved in facilitating the separation of the DNAzyme and substrate strands in the vicinity of the cleavage sites. Whilst it is possible that the imidazolyl and amino groups are exploited in an analogous fashion to those in RNaseA, it cannot be ruled out that these appended functional groups facilitate the folding of the DNAzyme rather than acting directly as catalytic groups. Confirmation of the role of the imidazolyl and amino groups within the DNAzyme will require a more detailed investigation of the roles of individual modifications and reaction conditions on the cleavage reaction.
Figure 7.
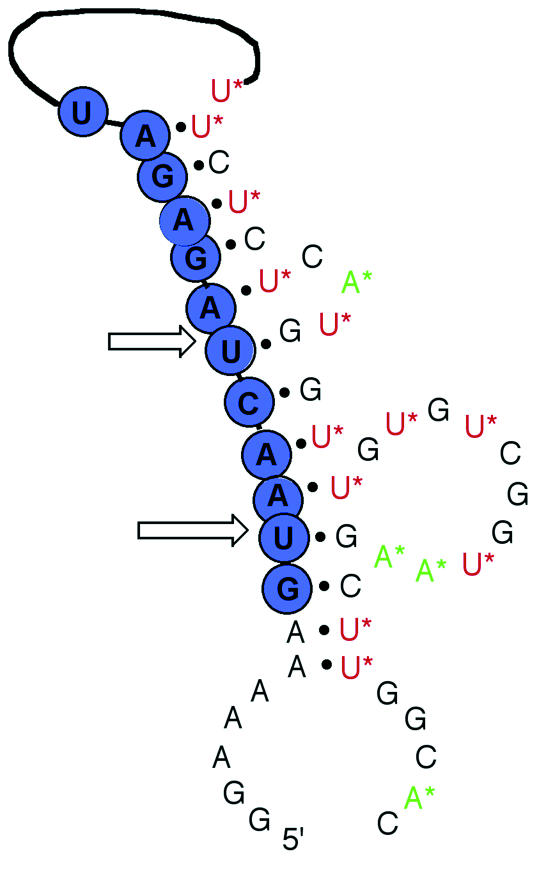
Secondary structure prediction for enzyme catalytic core of clone 32 following analysis using RNA and DNA secondary structure prediction using M-fold software (48). Circled letters correspond to ribonucleotides, uncircled letters correspond to deoxynucleotides and starred letters correspond to modified nucleotides 1 (U*) and 2 (A*). Arrows indicate the sites of RNA cleavage. A 6–8 nt triplet AAG near the 5′ end was forced to interact with a 3′-TTC-5′ triplet at the 3′ end to form a double helix prior to the folding calculation.
In work described by the Perrin group (46,47) the selection of a metal-independent DNAzyme was from an initial library containing a 20 nt random sequence expected to achieve self-cleavage 3′ to a single cytidine within a 29mer DNA target sequence derived from the HIV-LTR promoter. The selected DNAzyme recognizes the target sequence through Watson–Crick base pairing and presents a looped region containing both imidazolyl and amino groups opposite the site of cleavage within the substrate. A rate constant of 0.045 min–1 for the self-cleaving reaction was observed (46), whilst in a later report the corresponding reaction was achieved using a separate DNAzyme and substrate component (47). In contrast, we used a random 50 nt library and a 12 nt all-RNA target sequence together with a different set of imidazolyl- and amino-functionalized dNTPs. The recognition and cleavage of the target sequences by both the Perrin DNAzyme and that described here involves Watson–Crick base pairing between the DNAzyme and the target with catalytic looped regions within the DNAzyme placed opposite the respective cleavage sites.
The 10–23 metal-dependent DNAzyme reported by Joyce and coworkers is capable of cleaving its RNA target sequence with rates up to 10 min–1 by judicious choice of metal ion cofactor, its concentration or pH (56,57). In addition, a recent report for the cleavage of a chimeric RNA/DNA target by a different metal-dependent DNAzyme reported rates of 7 min–1 (17). These rates represent the most efficient DNAzymes that have thus far been identified. Under physiological conditions the 10–23 DNAzyme achieves RNA cleavage with a rate of ∼0.1 min–1 (26,56). The current study demonstrates that sequence-specific cleavage of an RNA target may be achieved in the absence of metal ions with a comparable rate using imidazolyl and amino functional groups to achieve catalysis. The goal of isolating more efficient metal-independent DNAzymes than these using the technique of SELEX is unlikely to be achieved by current protocols due to the time required for the washing step, prior to any self-cleaving selection reaction: The very efficient DNAzymes are likely to be lost during the washing step, limiting maximal rates for RNA cleavage to this order of magnitude [see Perrin et al. (46) for useful discussion]. It is anticipated that improvements to SELEX protocols will allow the development of catalysts that offer rate enhancements approaching that obtained by RNaseA.
CONCLUSIONS
We have selected and described a novel, modified DNAzyme that may act as a mimic of RNaseA by virtue of two protein-like functionalities, namely imidazolyl (histidine analogue) and primary amine (lysine analogue). The DNAzyme has optimal activity under physiological conditions, requires no divalent metal ions or other cofactors for catalysis and achieves the sequence-specific cleavage of a 12 nt RNA substrate. Whilst the rate enhancement achieved by this DNAzyme does not match that displayed by RNaseA, it is comparable with that of other functionalized and metal-dependent DNAzymes that cleave RNA under physiological conditions and represents a substantial rate enhancement over the uncatalysed reaction (55). This work demonstrates the potential of SELEX for the designing of catalytically active nucleic acids enriched by extra functional groups that are employed by proteins during catalysis. Since these nucleic acids also display substrate selectivity, improvements in the practical aspects of the SELEX protocol should allow more efficient catalysts to be selected. To this end, we are currently engaged in developing efficient trans-acting DNAzymes that exploit these catalytic functional groups.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Ms E. Frary for her expert technical assistance and Drs T. Shibata and M. Tock for helpful discussions. This work was supported by the Wellcome Trust grant ref. no. 065789. J. A. Grasby is a BBSRC Advanced Research Fellow.
REFERENCES
- 1.Fedor M.J. and Westhof,E. (2002) Ribozymes: the first 20 years. Mol. Cell, 10, 703–704. [DOI] [PubMed] [Google Scholar]
- 2.Nesbitt S., Hegg,L.A. and Fedor,M.J. (1997) An unusual pH-independent and metal-ion-independent mechanism for hairpin ribozyme catalysis. Chem. Biol., 4, 619–630. [DOI] [PubMed] [Google Scholar]
- 3.Young K.J., Gill,F. and Grasby,J.A. (1997) Metal ions play a passive role in the hairpin ribozyme catalysed reaction. Nucleic Acids Res., 25, 3760–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hampel A. and Cowan,J.A. (1997) A unique mechanism for RNA catalysis: the role of metal cofactors in hairpin ribozyme cleavage. Chem. Biol., 4, 513–517. [DOI] [PubMed] [Google Scholar]
- 5.Murray J.B., Seyhan,A.A., Walter,N.G., Burke,J.M. and Scott,W.G. (1998) The hammerhead, hairpin and VS ribozymes are catalytically proficient in monovalent cations alone. Chem. Biol., 5, 587–595. [DOI] [PubMed] [Google Scholar]
- 6.O'Rear J.L., Wang,S.L., Feig,A.L., Beigelman,L., Uhlenbeck,O.C. and Herschlag,D. (2001) Comparison of the hammerhead cleavage reactions stimulated by monovalent and divalent cations. RNA, 7, 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakano S., Chadalavada,D.M. and Bevilacqua,P.C. (2000) General acid-base catalysis in the mechanism of a hepatitis delta virus ribozyme. Science, 287, 1493–1497. [DOI] [PubMed] [Google Scholar]
- 8.Wadkins T.S., Shih,I.H., Perrotta,A.T. and Been,M.D. (2001) A pH-sensitive RNA tertiary interaction affects self-cleavage activity of the HDV ribozymes in the absence of added divalent metal ion. J. Mol. Biol., 305, 1045–1055. [DOI] [PubMed] [Google Scholar]
- 9.Ellington A.D. and Szostak,J.W. (1990) In vitro selection of RNA molecules that bind specific ligands. Nature, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- 10.Tuerk C. and Gold,L. (1990) Systematic evolution of ligands by exponential enrichment—RNA ligands to bacteriophage-T4 DNA-polymerase. Science, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 11.Beaudry A.A. and Joyce,G.F. (1992) Directed evolution of an RNA enzyme. Science, 257, 635–641. [DOI] [PubMed] [Google Scholar]
- 12.Li Y.F. and Breaker,R.R. (1999) Deoxyribozymes: new players in the ancient game of biocatalysis. Curr. Opin. Struct. Biol., 9, 315–323. [DOI] [PubMed] [Google Scholar]
- 13.Jaschke A. and Seelig,B. (2000) Evolution of DNA and RNA as catalysts for chemical reactions. Curr. Opin. Chem. Biol., 4, 257–262. [DOI] [PubMed] [Google Scholar]
- 14.Jaschke A. (2001) Artificial ribozymes and deoxyribozymes. Curr. Opin. Struct. Biol., 11, 321–326. [DOI] [PubMed] [Google Scholar]
- 15.Wilson D.S. and Szostak,J.W. (1999) In vitro selection of functional nucleic acids. Annu. Rev. Biochem., 68, 611–647. [DOI] [PubMed] [Google Scholar]
- 16.Liu Z.J., Mei,S.H.J., Brennan,J.D. and Li,Y.F. (2003) Assemblage of signaling DNA enzymes with intriguing metal-ion specificities and pH dependences. J. Am. Chem. Soc., 125, 7539–7545. [DOI] [PubMed] [Google Scholar]
- 17.Mei S.H.J., Liu,Z.J., Brennan,J.D. and Li,Y.F. (2003) An efficient RNA-cleaving DNA enzyme that synchronizes catalysis with fluorescence signaling. J. Am. Chem. Soc., 125, 412–420. [DOI] [PubMed] [Google Scholar]
- 18.Liu J.W. and Lu,Y. (2003) A colorimetric lead biosensor using DNAzyme-directed assembly of gold nanoparticles. J. Am. Chem. Soc., 125, 6642–6643. [DOI] [PubMed] [Google Scholar]
- 19.Fahlman R.P. and Sen,D. (2002) DNA conformational switches as sensitive electronic sensors of analytes. J. Am. Chem. Soc., 124, 4610–4616. [DOI] [PubMed] [Google Scholar]
- 20.Lewin A.S. and Hauswirth,W.W. (2001) Ribozyme gene therapy: applications for molecular medicine. Trends Mol. Med., 7, 221–228. [DOI] [PubMed] [Google Scholar]
- 21.Toyoda T., Imamura,Y., Takaku,H., Kashiwagi,T., Hara,K., Iwahashi,J., Ohtsu,Y., Tsumura,N., Kato,H. and Hamada,N. (2000) Inhibition of influenza virus replication in cultured cells by RNA-cleaving DNA enzyme. FEBS Lett., 481, 113–116. [DOI] [PubMed] [Google Scholar]
- 22.Unwalla H. and Banerjea,A.C. (2001) Inhibition of HIV-1 gene expression by novel macrophage-tropic DNA enzymes targeted to cleave HIV-1 TAT/Rev RNA. Biochem. J., 357, 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cieslak M., Niewiarowska,J., Nawrot,M., Koziolkiewicz,M., Stec,W.J. and Cierniewski,C.S. (2002) DNAzymes to beta(1) and beta(3) mRNA down-regulate expression of the targeted integrins and inhibit endothelial cell capillary tube formation in fibrin and matrigel. J. Biol. Chem., 277, 6779–6787. [DOI] [PubMed] [Google Scholar]
- 24.Kurreck J., Bieber,B., Jahnel,R. and Erdmann,V.A. (2002) Comparative study of DNA enzymes and ribozymes against the same full-length messenger RNA of the vanilloid receptor subtype I. J. Biol. Chem., 277, 7099–7107. [DOI] [PubMed] [Google Scholar]
- 25.Heidenreich O., Kang,S.H., Brown,D.A., Xu,X., Swiderski,P., Rossi,J.J., Eckstein,F. and Nerenberg,M. (1995) Ribozyme-mediated RNA degradation in nuclei suspension. Nucleic Acids Res., 23, 2223–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santoro S.W. and Joyce,G.F. (1997) A general purpose RNA-cleaving DNA enzyme. Proc. Natl Acad. Sci. USA, 94, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feldman A.R. and Sen,D. (2001) A new and efficient DNA enzyme for the sequence-specific cleavage of RNA. J. Mol. Biol., 313, 283–294. [DOI] [PubMed] [Google Scholar]
- 28.Faulhammer D. and Famulok,M. (1997) Characterization and divalent metal-ion dependence of in vitro selected deoxyribozymes which cleave DNA/RNA chimeric oligonucleotides. J. Mol. Biol., 269, 188–202. [DOI] [PubMed] [Google Scholar]
- 29.Peracchi A. (2000) Preferential activation of the 8–17 deoxyribozyme by Ca2+ ions—evidence for the identity of 8–17 with the catalytic domain of the MG5 deoxyribozyme. J. Biol. Chem., 275, 11693–11697. [DOI] [PubMed] [Google Scholar]
- 30.Li J., Zheng,W.C., Kwon,A.H. and Lu,Y. (2000) In vitro selection and characterization of a highly efficient Zn(II)-dependent RNA-cleaving deoxyribozyme. Nucleic Acids Res., 28, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geyer C.R. and Sen,D. (1997) Evidence for the metal-cofactor independence of an RNA phosphodiester-cleaving DNA enzyme. Chem. Biol., 4, 579–593. [DOI] [PubMed] [Google Scholar]
- 32.Roth A. and Breaker,R.R. (1998) An amino acid as a cofactor for a catalytic polynucleotide. Proc. Natl Acad. Sci. USA, 95, 6027–6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horn S. and Schwenzer,B. (1999) Oligonucleotide facilitators enhance the catalytic activity of RNA-cleaving DNA enzymes. Antisense Nucleic Acid Drug Dev., 9, 465–472. [DOI] [PubMed] [Google Scholar]
- 34.Wang D.Y., Lai,B.H.Y., Feldman,A.R. and Sen,D. (2002) A general approach for the use of oligonucleotide effectors to regulate the catalysis of RNA-cleaving ribozymes and DNAzymes. Nucleic Acids Res., 30, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S.E., Sidorov,A., Gourlain,T., Mignet,N., Thorpe,S.J., Brazier,J.A., Dickman,M.J., Hornby,D.P., Grasby,J.A. and Williams,D.M. (2001) Enhancing the catalytic repertoire of nucleic acids: a systematic study of linker length and rigidity. Nucleic Acids Res., 29, 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gourlain T., Sidorov,A., Mignet,N., Thorpe,S.J., Lee,S.E., Grasby,J.A. and Williams,D.M. (2001) Enhancing the catalytic repertoire of nucleic acids. II. Simultaneous incorporation of amino and imidazolyl functionalities by two modified triphosphates during PCR. Nucleic Acids Res., 29, 1898–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matulic-Adamic J., Daniher,A.T., Karpeisky,A., Haeberli,P., Sweedler,D. and Beigelman,L. (2000) Functionalized nucleoside 5′-triphosphates for in vitro selection of new catalytic ribonucleic acids. Bioorg. Med. Chem. Lett., 10, 1299–1302. [DOI] [PubMed] [Google Scholar]
- 38.Perrin D.M., Garestier,T. and Helene,C. (1999) Expanding the catalytic repertoire of nucleic acid catalysts: simultaneous incorporation of two modified deoxyribonucleoside triphosphates bearing ammonium and imidazolyl functionalities. Nucl. Nucl., 18, 377–391. [DOI] [PubMed] [Google Scholar]
- 39.Sakthivel K. and Barbas,C.F. (1998) Expanding the potential of DNA for binding and catalysis: highly functionalized dUTP derivatives that are substrates for thermostable DNA polymerases. Angew. Chem. Int. Ed., 37, 2872–2875. [DOI] [PubMed] [Google Scholar]
- 40.Vaish N.K., Fraley,A.W., Szostak,J.W. and McLaughlin,L.W. (2000) Expanding the structural and functional diversity of RNA: analog uridine triphosphates as candidates for in vitro selection of nucleic acids. Nucleic Acids Res., 28, 3316–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sawai H., Ozaki,A.N., Satoh,F., Ohbayashi,T., Masud,M.M. and Ozaki,H. (2001) Expansion of structural and functional diversities of DNA using new 5-substituted deoxyuridine derivatives by PCR with superthermophilic KOD Dash DNA polymerase. Chem. Commun., 2604–2605. [Google Scholar]
- 42.Thum O., Jager,S. and Famulok,M. (2001) Functionalized DNA: a new replicable biopolymer. Angew. Chem. Int. Ed., 40, 3990–3993. [DOI] [PubMed] [Google Scholar]
- 43.Tarasow T.M., Tarasow,S.L. and Eaton,B.E. (1997) RNA-catalysed carbon–carbon bond formation. Nature, 389, 54–57. [DOI] [PubMed] [Google Scholar]
- 44.Wiegand T.W., Janssen,R.C. and Eaton,B.E. (1997) Selection of RNA amide syntheses. Chem. Biol., 4, 675–683. [DOI] [PubMed] [Google Scholar]
- 45.Santoro S.W., Joyce,G.F., Sakthivel,K., Gramatikova,S. and Barbas,C.F. (2000) RNA cleavage by a DNA enzyme with extended chemical functionality. J. Am. Chem. Soc., 122, 2433–2439. [DOI] [PubMed] [Google Scholar]
- 46.Perrin D.M., Garestier,T. and Helene,C. (2001) Bridging the gap between proteins and nucleic acids: a metal-independent RNAseA mimic with two protein-like functionalities. J. Am. Chem. Soc., 123, 1556–1563. [DOI] [PubMed] [Google Scholar]
- 47.Lermer L., Roupioz,Y., Ting,R. and Perrin,D.M. (2002) Toward an RNaseA mimic: a DNAzyme with imidazoles and cationic amines. J. Am. Chem. Soc., 124, 9960–9961. [DOI] [PubMed] [Google Scholar]
- 48.Zuker M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res., 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jurinke C., vandenBoom,D., Collazo,V., Luchow,A., Jacob,A. and Koster,H. (1997) Recovery of nucleic acids from immobilized biotin-streptavidin complexes using ammonium hydroxide and applications in MALDI-TOF mass spectrometry. Anal. Chem., 69, 904–910. [DOI] [PubMed] [Google Scholar]
- 50.Harris V.H., Smith,C.L., Cummins,W.J., Hamilton,A.L., Adams,H., Dickman,M., Hornby,D.P. and Williams,D.M. (2003) The effect of tautomeric constant on the specificity of nucleotide incorporation during DNA replication: support for the rare tautomer hypothesis of substitution mutagenesis. J. Mol. Biol., 326, 1389–1401. [DOI] [PubMed] [Google Scholar]
- 51.Tock M.R., Frary,E., Sayers,J.R. and Grasby,J.A. (2003) Dynamic evidence for metal ion catalysis in the reaction mediated by a flap endonuclease. EMBO J., 22, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patel D., Tock,M.R., Frary,E., Feng,M., Pickering,T.J., Grasby,J.A. and Sayers,J.R. (2002) A conserved tyrosine residue aids ternary complex formation, but not catalysis, in phage T5 flap endonuclease. J. Mol. Biol., 320, 1025–1035. [DOI] [PubMed] [Google Scholar]
- 53.Kaukinen U., Lyytikainen,S., Mikkola,S. and Lonnberg,H. (2002) The reactivity of phosphodiester bonds within linear single-stranded oligoribonucleotides is strongly dependent on the base sequence. Nucleic Acids Res., 30, 468–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bibillo A., Figlerowicz,M., Ziomek,K. and Kierzek,R. (2000) The nonenzymatic hydrolysis of oligoribonucleotides VII. Structural elements affecting hydrolysis. Nucleosides Nucleotides Nucleic Acids, 19, 977–994. [DOI] [PubMed] [Google Scholar]
- 55.Williams N.H., Takasaki,B., Wall,M. and Chin,J. (1999) Structure and nuclease activity of simple dinuclear metal complexes: Quantitative dissection of the role of metal ions. Acc. Chem. Res., 32, 485–493. [Google Scholar]
- 56.Santoro S.W. and Joyce,G.F. (1998) Mechanism and utility of an RNA-cleaving DNA enzyme. Biochemistry, 37, 13330–13342. [DOI] [PubMed] [Google Scholar]
- 57.Breaker R.R. and Joyce,G.F. (1994) A DNA enzyme that cleaves RNA. Chem. Biol., 1, 223–229. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.