

Introduction
Sleep apnea (SA), which is defined as intermittent respiratory arrest during sleep, is estimated to affect one in five adults in Western countries [1]. SA results in decreased oxygen saturation (hypoxia) and increased arterial CO2 levels (hypercapnia), and has been shown to initiate vascular changes that increase the patient’s risk of cardiovascular disease. SA is associated with increased incidence of hypertension, peripheral vascular disease, stroke, and sudden cardiac death [2; 3].
Our group has demonstrated that rats and mice exposed to intermittent hypoxia with CO2 supplementation (IH) during sleep have increased blood pressure [4; 5], increased circulating endothelin 1 (ET-1) levels [5], upregulation of pre-pro ET-1 in the lungs [4], and that the IH-induced increase in blood pressure is prevented by administration of ET receptor antagonists [5–7]. Our group has also reported that in rats, administration of the superoxide dismutase (SOD) mimetic tempol prevented the increase in blood pressure, oxidative stress, and plasma ET-1 levels in IH-exposed rats [8] suggesting that IH exposure increases reactive oxygen species (ROS) generation, which in turn stimulates ET-1 production. However, this has not been tested in mice.
ET-1 and ROS can activate the nuclear factor of activated T cells (NFAT) [9–14]. NFAT is a so-called master transcription factor that links Ca2+ signaling to other signaling pathways to induce specific genetic programs [15]. Our group has shown that the isoform NFATc3 is necessary for IH-induced systemic hypertension [4] and arterial remodeling [16].
We have previously reported that NFATc3 activity is increased in the aorta (AO) and mesenteric arteries (MA) in mice exposed to IH, that upregulation of ET-1 correlates with increased NFATc3 activation [4], and that blocking endothelin receptor type A (ETAR) during IH exposure prevents increases in NFATc3 activity, blood pressure and arterial remodeling [16]. However, the role of ROS in IH-induced NFATc3 activation is unknown.
Our group has also demonstrated that the arteries from IH-exposed rats express more ETAR than those of control animals, and that IH arteries show increased vasoconstrictor reactivity to ET-1 compared to those from control rats [17]. We have also shown that small rat mesenteric VSMC are depolarized in arteries IH exposed rats compared to arteries from air exposed rats [18] suggesting that K+ channels contribute to the enhanced vasoreactivity. However, it is unknown if IH exposure increases vasoreactivity in arteries from mice and if NFATc3 contributes to this response.
NFATc3 has been shown to regulate VSMC contractility. Specifically, NFATc3 activation by angiotensin II enhances cerebral artery contractility by decreasing voltage-dependent (isoform 2.1, KV 2.1) and large-conductance Ca2+-activated (BK) potassium channel expression [19; 20].
Therefore, we hypothesized that IH-induced increases in ROS activate NFATc3 via ET-1 to increase arterial smooth muscle vasoconstrictor reactivity by decreasing KV and/or BK channel expression. The aims of the present study were to determine whether: 1) IH exposure increases arterial ROS in mice; 2) IH-induced increases in ROS are downstream or upstream of ET-1; 3) IH-induced NFATc3 activation is mediated by ROS; 4) IH-induced increases in vasoreactivity are dependent on NFATc3; and 4) downregulation of KV 2.1 and/or BK channels is implicated in IH-induced NFATc3-mediated increased vasoreactivity.
In support of the hypothesis, our data demonstrate that: 1) IH exposure increases ROS in the arterial wall; 2) IH exposure increases protein carbonylation (a measure of oxidative stress) in the arterial wall upstream of ET-1; 3) administration of tempol to scavenge superoxide during IH exposure decreases NFATc3 activation, and 4) IH-induced increases in vasoconstrictor reactivity are dependent on NFATc3. Contrary to our expectations, KV 2.1 and BK channel mRNA expression were not affected by IH exposure. However, we found that transient receptor potential cation channel subtype C1 (TRPC1) is upregulated in an NFATc3-dependent manner suggesting that Ca2+ influx through TRPC1 might play a role in IH-induced increases in vasoconstrictor reactivity.
Materials and Methods
All protocols employed in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the University of New Mexico Health Science Center (Albuquerque, NM).
Animals
Adult male 9x-NFAT-luciferase reporter (NFAT-luc, FVBN background), adult male and female NFATc3 knockout (NFATc3 KO, BalB/C background) and BalB/C wild-type (WT) mice were used. NFAT-luc mice were provided by Dr. Jeffery D. Molkentin (Children’s Hospital Medical Center, Cincinnati, OH) [20; 21]. NFATc3 KO mice were kindly provided by Dr. Laurie Glimcher (Harvard University) [22].
Intermittent hypercarbic/hypoxia exposure
Animals were housed in regular cages with snug-fitting Plexiglas lids. During the normal sleep period, air in the cage was cycled between a low oxygen (5% O2), high CO2 (5% CO2) environment and room air [4; 5]. The mice were exposed to 20 IH episodes/h for 7 h/day for 2 or 7 days. Control (Air) animals were housed in similar cages with a constant flow of air, experiencing the same environment as the IH mice. All animals were maintained on a 12 h: 12h light-dark cycle.
On the morning of day 3 or day 8 of IH, before the initiation of the IH cycle, mice were euthanized with an overdose of pentobarbital (200 mg/kg ip). The mesentery and thoracic aorta (AO) were removed and placed in HEPES-buffered physiological saline solution (PSS; containing in mM 134 NaCl, 6 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 0.026 EDTA, and 10 glucose; pH adjusted to 7.4). Mesenteric arteries (MA) were dissected for isolated pressurized artery studies, placed in room temperature HEPES PSS to preserve viability. All other tissues were placed in ice-cold HEPES PSS. MA used for ROS measurement were dissected from the wall of the intestine but not from the surrounding connective tissue. AO used for ROS measurement and AO and MA used for luciferase assay were dissected from the surrounding connective tissue.
Luciferase Activity
Isolated arteries (AO and second, third, and fourth order MA with outer diameter = 100 to 500 μm) from IH and Air NFAT-luc mice were lysed (Promega buffer). Luciferase activity was measured using Luciferase Assay System kit (Promega), and light was detected with a luminometer (TD20/20; Turner). Protein content determined by the Bradford method (Bio-Rad) was used to normalize luciferase activity [4; 16; 23; 24].
Dihydroethidium staining
The freely permeable superoxide-sensitive fluorescent dye dihydroethidium (DHE) was used to evaluate in situ production of superoxide as we have previously described [8; 25; 26]. In the presence of O2−, DHE is oxidized to the fluorescent products 2-hydroxyethidum and ethidium+ [27; 28]. DHE is a qualitative way to determine relative levels of oxidative stress [27]. Thoracic AO and MA from IH and Air NFATc3 WT and KO mice were harvested and rapidly frozen in optimal cutting temperature (OCT) embedding compound (TissueTek), stored at −20°C, and then cut into 10 μm sections. Sections were mounted on glass slides and kept at −20°C until stained. During staining, sections were thawed, incubated with 120U/mL polyethylene glycol conjugated SOD (PEG-SOD) in phosphate-buffered saline (PBS) or with PBS alone for 30 min at 37°C. The sections were then incubated with DHE in PBS (10 μM) plus or minus PEG-SOD for 30 min at 37°C. Slides were washed three times with PBS and coverslips were mounted using ProLong Gold (Life Technologies). Images were acquired with the 40x objective of a AMG EVOS fluorescence microscope. The sample was illuminated using the RFP LED light cube (531 nm) and emissions (593 nm) collected with a Sony ICX285AL CCD camera, a range that detects primarily DNA-bound ethidium+ [28]. Fluorescence intensity was analyzed using Image J software. Mean gray value in approximately 8 smooth muscle cells/section, four sections/artery were averaged from one AO per mouse for four IH and four Air mice, and approximately 8 smooth muscle cells/section, four sections/artery were averaged from one small MA per mouse for three IH and three Air mice. Elastin autofluorescence was excluded from the analysis.
ET-1 Assay
An ET-1 selective ELISA kit (QuantiGlo; R & D Systems, Minneapolis, MN) was used to measure the concentration of ET-1 in plasma samples collected from anesthetized IH on day 2 and 7, and from air mice. Blood was drawn into EGTA-coated vacutainer tubes and centrifuged at 3,000 g at 4°C for 10 min. Plasma was stored frozen at −70°C until the assay was performed.
Immunofluorescence detection of protein carbonyls
MA were fixed overnight in 4% paraformaldehyde in PBS and then mounted in paraffin. Sections (5-μm thick) were deparafinized with xylenes and a graded alcohol series, and then rinsed with PBS for 10 min. Carbonyl groups were converted into 2,4-dinitrophenyl (DNP) hydrazones by reaction with 1mg/mL 2,4-dinitrophenyl-hydrazine (Sigma) prepared in 2N HCl for 30min [29–31]. Sections were rinsed with PBS, blocked with 0.2% BSA (Fraction V) in PBS and 0.03% Tween 20 and incubated overnight with rabbit anti-DNP (1:1000) (Sigma). After removing the primary antibody, the sections were incubated with DyLight 549 donkey anti-rabbit (1:500, Jackson ImmunoResearch Laboratories). Sections were counter stained with Sytox Green to stain nuclei and Alexa Fluor 633 hydrazide in PBS to stain elastic lamina (Life Technologies). The positive and negative controls were treated for 30 min in 100 μmol/l FeSO4 + 10 mmol/l H2O2 or 80% methanol + 100 mmol/l NaBH4, respectively, before the derivatization step. Images were captured with a Leica TCS SP5 confocal microscope and analyzed using Image J (NIH). Integrated Fluorescence Intensity was measured in the media layer of mesenteric arteries, avoiding the perivascular staining. Images were thresholded to exclude background fluorescence, determined from negative control sections.
Isolated Pressurized Artery Studies
A small section of mesentery was removed and placed in a Silastic-coated Petri dish containing room-temperature HEPES-PSS. Third or fourth order artery segments were carefully dissected from surrounding connective tissue and placed in room temperature HEPES-PSS containing the cell-permeable ratiometric Ca2+-sensitive fluorescent dye fura 2-acetoxymethyl ester (fura 2-AM; 2 μM; Life Technologies) and 0.02% pluronic acid (Life Technologies) and incubated for 45 min. The artery segments were then placed in a vessel chamber (Living Systems) containing room temperature PSS (containing, in mM 129.8 NaCl, 5.4 KCl, 0.83 MgSO4, 19 NaHCO3, 1.8 CaCl2, and 5.5 glucose) oxygenated with normoxic gas (21% O2–6% CO2–73% N2). Arteries were cannulated onto glass micropipettes. The endothelium was disabled in all experiments by gently rubbing a piece of moose mane along the inner wall of the vessel and flushing the lumen with PSS. Integrity of the endothelium was assessed before and after denuding by measuring acetylcholine (ACh)-mediated dilation in arteries preconstricted to 50% of the initial diameter with phenylephrine (PE). ACh-mediated dilation was eradicated in vessels in which the endothelium was successfully disabled. The mounted vessels were then pressurized to 20 mm Hg with PSS using a servo-controlled peristaltic pump (Living Systems) and superfused with oxygenated 37°C PSS. After 10 min at 20 mm Hg, arteries were slowly pressurized to 60 mm Hg and allowed to equilibrate for an additional 15 min. Fura 2-AM-loaded vessels were alternately excited at 340 and 380 nm at a frequency of 10 Hz with an IonOptix Hyperswitch dual-excitation light source and the respective 510 nm emissions were collected with a photomultiplier tube (F340/F380) through a 20x objective. After subtracting background fluorescence, emissions ratios were calculated with Ion Wizard software (IonOptix) and recorded continuously throughout the experiment. Inner diameter was continuously and simultaneously measured from bright-field images as previously described [17; 32–34]. After determining baseline internal diameter and F340/F380, arteries were incubated with a single concentration (bolus administration) of ET-1 (10−9 M, each artery received only one bolus) or high K+ PSS (30 mM) in the superfusion media. After exposure, the artery was washed with Ca2+-free PSS three times, and allowed to sit in room temperature Ca2+-free PSS plus ionomycin (1 μM) for 15–20 minutes to cause complete dilation. This was performed to demonstrate that constriction was caused by reversible tone. Minimum (Ca2+ free) and maximum (1.8 mM Ca2+) fura 2 ratios were also recorded and compared to ensure that Ca2+ measurements were consistent in all experiments. Constrictions are expressed as percent of baseline diameter. Vessel wall [Ca2+] is expressed as Δ F340/F380 from baseline, because no difference in baseline fura 2-AM ratio was seen between groups (data not shown).
Quantitative Real-time Polymerase Chain Reaction (qRT-PCR)
Small mesenteric arteries and aorta were isolated, cleaned of fat and connective tissue, and stored in RNAlater (Ambion). Total RNA was isolated using the RNeasy Mini Kit (Qiagen). Total RNA was reverse transcribed to cDNA using High Capacity Reverse Transcription kit (Life Technologies). For real time detection of Kv 1.5 and 2.1, BKα and β transcripts TaqMan Gene Expression Assays (Life Technologies) were used. β-Actin was used as endogenous control. The normalized gene expression method (2−ΔΔCT) for relative quantification of gene expression was used [35].
Results
IH exposure increases ROS in mouse MA and AO upstream of NFATc3 activation
Our group has shown that IH exposure increases arterial ROS in rats [8]. In this study, to determine if smooth muscle arterial ROS is increased in mice exposed to IH, both WT and NFATc3 KO mice were exposed to 1 week of IH or Air and their tissues collected and stained for ROS using DHE. AO (Fig. 1A and B) and MA (Fig. 1C and D) VSMC from mice exposed to IH showed increased ROS compared to arteries from air exposed mice. ROS was similarly elevated in both genotypes suggesting IH-induced increases in ROS were independent of NFATc3. Co-incubation with PEG-SOD significantly decreased ROS levels in both artery types to Air levels, validating that DHE staining detects superoxide (Fig. 1). These data indicate that IH increases superoxide generation upstream of NFATc3 activation.
Fig. 1.

IH increases superoxide in aorta (AO) and mesenteric arteries (MA) VSMC. Superoxide levels were measured in discrete regions of interest in the medial layer by DHE staining. Co-incubation with polyethylene glycol conjugated superoxide dismutase (PEG-SOD) reduced fluorescence intensity in IH arteries to or below that of control (Air) arteries. Summary data is shown for A) AO and C) MA of WT and NFATc3 KO mice after 1 week IH or Air exposure, in presence or absence of PEG-SOD. Representative images are shown in B) for AO and D) for MA.*p<0.05 vs. Air for AO; *p<0.01 vs. Air for MA; 2-way ANOVA and Bonferroni post hoc test; n = 4 animals for AO and n = 3 animals for MA. Scale= 100 μm.
IH exposure increases oxidative stress-mediated protein carbonylation in mouse MA upstream of ET receptor activation
We have previously shown that expression of pre-pro-ET-1 mRNA is increased in lungs of mice exposed to IH for 2 or 7 days compared to lungs from Air exposed mice [4]. Consistent with the notion that the lung is an important source of circulating ET-1, in the current study, ET-1 plasma levels were significantly elevated in the plasma of mice exposed to IH for 2 or 7 days (Fig. 2) compared to plasma from Air exposed mice.
Fig. 2.
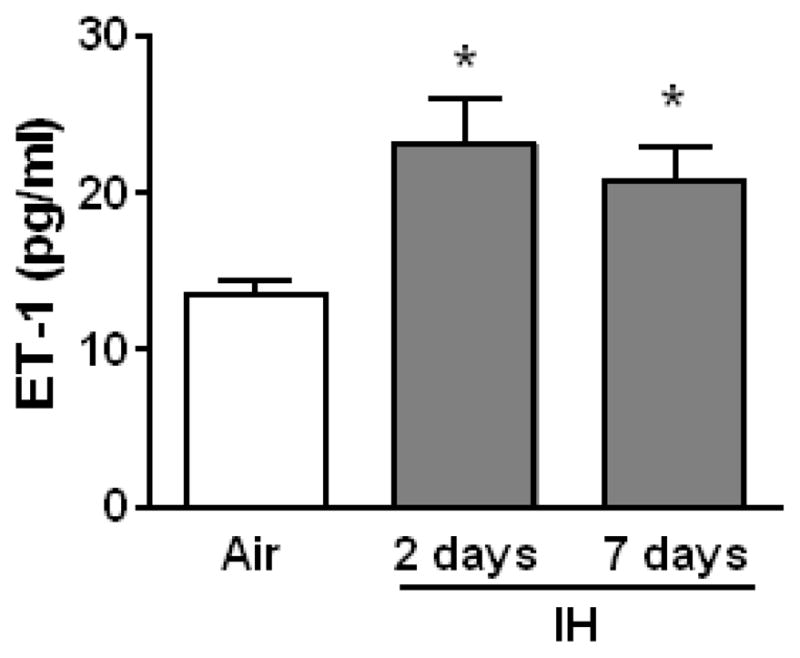
IH increases ET-1 plasma levels. Plasma levels of ET-1 were measured by ELISA in WT mice exposed to Air, and 2 days and 1 week of IH. *p<0.05 vs. Air, n=9.
Since IH increases superoxide levels in mouse aortic (Fig. 1A and B) and mesenteric arterial smooth muscle cells (Fig. 1 C and D), and the rat [8] mesenteric arterial wall, this study evaluated IH effects on protein carbonylation, a marker of oxidative stress. Severe or prolonged oxidative stress leads to irreversible protein carbonylation [36] and we observed that protein carbonylation was indeed significantly greater in MA from IH exposed mice treated with vehicle compared to MA from Air exposed mice (Fig. 3). Our group has previously reported that in rats, IH (14 days) increases plasma ET-1 in a ROS-dependent manner [8]. In order to determine, whether the IH-induced increases in oxidative stress are upstream or downstream of ET receptor activation, bosentan (30 mg/kg/day, dual ET receptor antagonist) was administered in the drinking water for 7 days to mice exposed to Air or IH. (Bosentan kindly provided by Actelion Pharmaceuticals LTD, Switzerland). We have previously shown that this dose of bosentan prevents IH-induced NFATc3 activation, arterial remodeling and hypertension [16]. Consistent with our previous observations in rats demonstrating that ROS is upstream of ET-1 synthesis, we found that IH-induced increases in mesenteric arterial wall protein carbonylation (oxidative stress) is not affected by blocking ET receptors with bosentan (Fig. 3). These results suggest IH induces VSMC oxidative stress independent of ET receptor activation.
Fig. 3.

IH increases protein carbonylation in MA. Immunofluorescence detection of protein carbonyls in MA wall from WT mice exposed to 1 week of Air or IH and treated with vehicle or bosentan. A) Representative images. Green= nuclei stain; red= protein carbonylation; blue= elastin. The monochrome channel corresponds to protein carbonyls staining. B) Summary data of integrated fluorescence intensity (AU x 10−3).*p<0.05 vs. Air; 2-way ANOVA and Bonferroni post hoc test; n=4–6 mice. At least 5 arteries per mouse were analyzed. Scale= 75 μm.
IH-induced NFATc3 activation is mediated by superoxide
In this study, we found that IH increases superoxide and consequently protein carbonylation in the arterial wall (Fig. 1 and 3). ROS activate NFAT in T cells [12], mouse epidermal [13], human lung bronchoepithelial [13], cardiac cells [37], and pulmonary arterial smooth muscle cells [38]. Furthermore, we have previously reported that IH upregulates ET-1 in an ROS-dependent manner and that NFATc3 activation in AO and MA by IH is mediated by activation of ETAR [16]. These observations suggest increased superoxide may play a role in IH-induced increases in vascular NFATc3 activity.
To determine whether superoxide mediates IH-induced NFATc3 activation, NFAT-luc mice were given either regular water or water containing the SOD mimetic tempol (1 mmol/l; 0.17 g/kg) 1 day before and during 2 days of IH exposure, a time point when NFAT is maximally activated in MA [39]. This dose of tempol, previously shown to prevent increases in oxidative stress in IH-exposed rats [8], prevented increases in NFAT activity compared to vehicle-treated mice which had increased NFAT activity in both AO and MA (Fig. 4). These data indicate that superoxide mediates NFATc3 induction by IH.
Fig. 4.

The SOD mimetic, tempol, prevents IH-induced NFAT activation in aorta (AO) and mesenteric arteries (MA). Luciferase activity was measured in isolated AO and MA from NFAT-luc animals treated with tempol for 3 days and exposed to IH during the last 2 days of treatment. RLU, relative luciferase units. *p<0.05 vs. Air vehicle; #p<0.05 vs. IH Vehicle; 2-way ANOVA and Bonferroni post hoc test; n = 6 animals.
IH exposure increases NFATc3-dependent ET-1 constriction
Our group has published that IH increases reactivity to ET-1 in rat MA. Interestingly, it was found that this increased constriction was not accompanied by a concomitant increase in vessel wall [Ca2+] [17].
Therefore, to determine if IH increases ET-1 vasoreactivity in mice, and to assess the role of NFATc3 in any changes in reactivity, we incubated isolated pressurized small MA with a single concentration of ET-1 (10−9 mol/l). MA from IH WT mice constricted more to 10−9 mol/l ET-1 compared to constriction of MA from Air WT and IH NFATc3 KO mice (Fig. 5A and 6). The increased constriction was accompanied by a significantly greater increase in vessel wall [Ca2+], also dependent on NFATc3 (Fig. 5B and 6). A single concentration of ET-1 was used because mouse MA constricted less to 10−9 mol/l ET-1 when given in a cumulative manner than when administered in a bolus as a single concentration (Fig. 7). For example, in arteries from control mice (Air WT), 10−9 mol/l ET-1 (less than the pEC50 8.754 ± 0.122 mol/l) increased the fura ratio ~0.1 and caused a 40% vasoconstriction when applied as bolus concentration (Figure 5A and B, and 6). However, the same concentration as cumulative exposure increased the fura ratio ~0.075 and only caused a 25% vasoconstriction (Fig. 7). No significant differences were found in basal tone among groups (data not shown).
Fig. 5.

IH increases reactivity to ET-1. Representative traces of A) diameter and B) fura-2 ratios in isolated pressurized small MA from WT mice exposed to 7 days Air or IH and superfused with 10−9 mol/l ET-1.
Fig. 6.
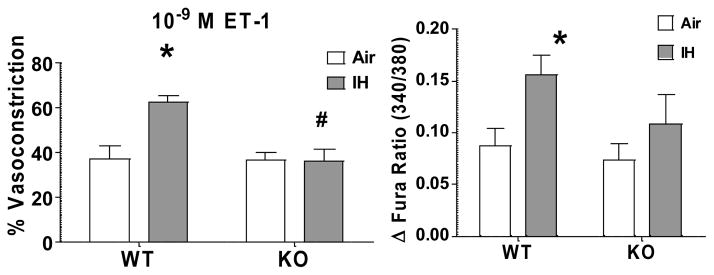
IH increases reactivity to ET-1 in an NFATc3-dependent manner. Summary results of % Vasoconstrictor reactivity and vessel wall [Ca2+] response to 10−9 mol/l ET-1 in MA from WT and NFATc3 KO mice exposed to 7 days Air or IH. *p<0.05 vs. Air WT; #p<0.01 vs. IH WT; 2-way ANOVA and Bonferroni post hoc test; n = 6 animals.
Fig. 7.

Concentration response curve to ET-1 in isolated pressurized mouse mesenteric arteries. Reactivity to increasing concentrations of ET-1 was assessed in isolated pressurized small MA from WT mice exposed to 7 days of Air. No significant increase in vessel wall [Ca2+] was observed in response to ET-1 from baseline although there was a significant vasoconstriction. One-way repeated measures ANOVA and Bonferroni post hoc test; *p<0.05 vs. 10−10 mol/l; n = 7 animals. pEC50 (−8.754 ± 0.122 mol/l) was calculated by fitting the data to the following equation Y=Bottom + (Top-Bottom)/(1+10^((LogEC50-X)))
IH exposure does not alter KCl constriction
Our group has previously reported that IH exposure does not increase vasoconstrictor reactivity to KCl in rat MA (2). In the present study, we assessed reactivity of mouse MA to 30 mmol/l KCl, and similarly saw no differences between groups (Fig. 8).
Fig. 8.
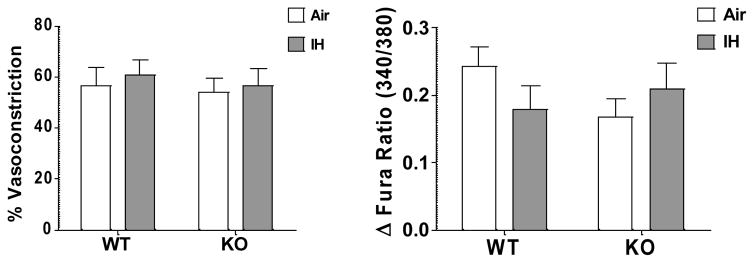
IH does not increase reactivity to KCl. Reactivity to high K+ PSS (30 mmol/l KCl) was assessed in isolated pressurized small MA from WT and NFATc3 KO animals exposed to 7 days IH or Air. 2-way ANOVA and Bonferroni post hoc test, n = 7 animals.
IH Exposure does not Change KV 1.5 and 2.1, BKα and β or TRPC6 mRNA Levels but Increases TRPC1
NFATc3 has been implicated in the regulation of VSMC contractility by regulating the expression of KV and BK channels [19; 20] and transient receptor potential channels (TRPC) isoforms c1 and c6 channels [40; 41]. No significant differences were found between groups for KV 1.5 and 2.1, TRPC6 or BKα and β mRNA levels (Fig. 9). However, IH id induced increases in TRPC1 mRNA levels in MA from WT but not KO mice (Fig. 9).
Discussion
The main findings of this study are that superoxide is an important upstream factor in IH-induced NFATc3 activation, and that NFATc3 mediates increased vasoconstrictor reactivity to ET-1 that may be partly responsible for IH-induced hypertension.
Our group has demonstrated that IH increases ROS which causes an increase in ET-1 plasma levels in rats [8]. This study is consistent with this previous finding since IH increased mesenteric arterial wall protein carbonylation, a marker of oxidative stress, upstream of ET receptor activation. Our laboratory has also shown that IH-induced upregulation of lung pre-pro ET-1 transcript levels in mice and that IH-induced VSMC NFATc3 activation requires ET-1 [16]. The current results extend these observations to show that IH-induced increases in superoxide elevate circulating ET-1 levels to cause NFATc3 activation in VSMC. This conclusion is supported by other reports that oxidative stress, particularly elevated superoxide, increases ET-1 synthesis in both endothelial and VSMC [42; 43].
We have previously demonstrated that NFATc3 is required for IH-induced hypertension [4] in part through NFATc3-dependent increases in arterial wall thickness due to VSMC hypertrophy [16]. This new study suggests that another important role for NFATc3 in IH-induced hypertension is to mediate increases in ET-1 vasoreactivity.
Unlike previous studies in rats which observed IH-induced increases in ET-1 reactivity with no concomitant increase in vessel wall [Ca2+] due to activation of Ca2+-sensitizing pathways [17], this study found that a bolus administration of ET-1 (10−9 mol/l) increased vessel wall [Ca2+] and constriction in arteries from IH mice compared to arteries from Air exposed mice. These results suggest that increased VSMC [Ca2+] contributes to IH-induced increases in vasoconstrictor reactivity to ET-1 in small mouse MA and that bolus versus cumulative exposures to ET-1 may elicit different Ca2+ responses.
The mechanism by which NFATc3 contributes to the IH-induced increases in vasoconstriction and [Ca2+]i in response to ET-1 is not yet clear. Because previous reports found that NFATc3 augments VSMC contractility by downregulating KV 2.1 and BKβ-subunit [19; 20] and that VSMC in small mesenteric arteries from IH treated rats are depolarized compared to VSMC in arteries from Air exposed rats [18], we hypothesized that IH activation of NFATc3 might increase ET-1 vasoreactivity by decreasing the expression of KV and/or BK channels. However, transcripts levels of these channels were not different between Air and IH, and reactivity to KCl was not different between groups suggesting changes in these channels do not contribute to the augmented reactivity. Since both TRPC1 and 6 are also regulated by NFAT [44; 45] and play a role in ET-1-induced extracellular Ca2+ entry in VSMC [46; 47], we further explored IH regulation of these channels in an NFATc3-dependent manner. We found that IH increases TRPC1 transcript but not TRPC6 mRNA in MA and that the increase in TRPC1 was NFATc3-dependent. This finding is consistent with a report showing that NFAT inhibition reduces the expression of TRPC1 but not TRPC6 in VSMC [48]. The role of TRPC1 upregulation in the enhanced vasoreactivity present in arteries from IH exposed animals remains to be addressed.
Conclusions
In summary, this study demonstrates the novel finding that IH increases superoxide in mouse arteries, independent of NFATc3 and ET receptor activation. Rather, IH-induced increases in superoxide activate NFATc3, likely by increasing ET-1 synthesis [8]. In addition, NFATc3 activation mediates increased vasoconstrictor sensitivity to ET-1 which is accompanied by greater increases in [Ca2+]i compared to arteries from control mice. These studies define a novel regulation of ET-1 vasoconstriction by NFATc3, highlighting the potential clinical relevance of NFAT inhibition to ameliorate hypertension in SA patients.
Fig. 9.

IH does not affect K channel or TRPC6 expression in small MA but upregulates TRPC1 in an NFATc3-dependent manner. KV1.5, KV2.1, BKα, BKβ, TRPC1 and TRPC6 mRNA levels were measured in MA from WT and NFATc3 KO mice exposed to 7 days IH or Air. 2-way ANOVA and Bonferroni post hoc test; *p<0.05 vs. air WT. n = 5–10 mice.
Acknowledgments
Funding support: NIH Grants R01-HL-088151 (and a supplement from the American Recovery and Reinvestment Act of 2009 to R01-HL-088151), R01 HL-82799 and 0535347N Scientist Development Award, American Heart Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Tishler PV, Larkin EK, Schluchter MD, Redline S. Incidence of sleep-disordered breathing in an urban adult population: the relative importance of risk factors in the development of sleep-disordered breathing. JAMA. 2003;289:2230–2237. doi: 10.1001/jama.289.17.2230. [DOI] [PubMed] [Google Scholar]
- 2.Fletcher EC. Invited review: Physiological consequences of intermittent hypoxia: systemic blood pressure. J Appl Physiol. 2001;90:1600–1605. doi: 10.1152/jappl.2001.90.4.1600. [DOI] [PubMed] [Google Scholar]
- 3.Neubauer JA. Physiological and Genomic Consequences of Intermittent Hypoxia: Invited Review: Physiological and pathophysiological responses to intermittent hypoxia. J Appl Physiol. 2001;90:1593–1599. doi: 10.1152/jappl.2001.90.4.1593. [DOI] [PubMed] [Google Scholar]
- 4.De Frutos S, Duling L, Alo D, Berry T, Jackson-Weaver O, Walker M, Kanagy N, Gonzalez BL. NFATc3 is required for intermittent hypoxia-induced hypertension. Am J Physiol Heart Circ Physiol. 2008;294:H2382–H2390. doi: 10.1152/ajpheart.00132.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanagy NL, Walker BR, Nelin LD. Role of endothelin in intermittent hypoxia-induced hypertension. Hypertension. 2001;37:511–515. doi: 10.1161/01.hyp.37.2.511. [DOI] [PubMed] [Google Scholar]
- 6.Allahdadi KJ, Cherng TW, Pai H, Silva AQ, Walker BR, Nelin LD, Kanagy NL. Endothelin type A receptor antagonist normalizes blood pressure in rats exposed to eucapnic intermittent hypoxia. Am J Physiol Heart Circ Physiol. 2008;295:H434–H440. doi: 10.1152/ajpheart.91477.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Frutos S, Spangler R, Alo D, Gonzalez Bosc LV. NFATc3 mediates chronic hypoxia-induced pulmonary arterial remodeling with alpha -actin up-regulation. J Biol Chem. 2007;282:15081–15089. doi: 10.1074/jbc.M702679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Troncoso Brindeiro CM, da Silva AQ, Allahdadi KJ, Youngblood V, Kanagy NL. Reactive oxygen species contribute to sleep apnea-induced hypertension in rats. Am J Physiol Heart Circ Physiol. 2007;293:H2971–H2976. doi: 10.1152/ajpheart.00219.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abbott KL, Loss JR, Robida AM, Murphy TJ. Evidence that Galpha(q)-coupled receptor-induced interleukin-6 mRNA in vascular smooth muscle cells involves the nuclear factor of activated T cells. Mol Pharmacol. 2000;58:946–953. doi: 10.1124/mol.58.5.946. [DOI] [PubMed] [Google Scholar]
- 10.Gomez MF, Stevenson AS, Bonev AD, Hill-Eubanks DC, Nelson MT. Opposing Actions of Inositol 1,4,5-Trisphosphate and Ryanodine Receptors on Nuclear Factor of Activated T-cells Regulation in Smooth Muscle. J Biol Chem. 2002;277:37756–37764. doi: 10.1074/jbc.M203596200. [DOI] [PubMed] [Google Scholar]
- 11.Stevenson AS, Gomez MF, Hill-Eubanks DC, Nelson MT. NFAT4 movement in native smooth muscle. A role for differential Ca(2+) signaling. J Biol Chem. 2001;276:15018–15024. doi: 10.1074/jbc.M011684200. [DOI] [PubMed] [Google Scholar]
- 12.Kalivendi SV, Konorev EA, Cunningham S, Vanamala SK, Kaji EH, Joseph J, Kalyanaraman B. Doxorubicin activates nuclear factor of activated T-lymphocytes and Fas ligand transcription: role of mitochondrial reactive oxygen species and calcium. Biochemical Journal. 2005;389:527–539. doi: 10.1042/BJ20050285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ke Q, Li J, Ding J, Ding M, Wang L, Liu B, Costa M, Huang C. Essential role of ROS-mediated NFAT activation in TNF-{alpha} induction by crystalline silica exposure. Am J Physiol Lung Cell Mol Physiol. 2006;291:L257–L264. doi: 10.1152/ajplung.00007.2006. [DOI] [PubMed] [Google Scholar]
- 14.Ramiro-Diaz JM, Nitta CH, Maston LD, Codianni SJ, Giermakowska W, Resta TC, Gonzalez Bosc LV. NFAT is Required for Spontaneous Pulmonary Hypertension in Superoxide Dismutase 1 Knockout Mice. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2013 doi: 10.1152/ajplung.00408.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 16.De Frutos S, Caldwell E, Nitta CH, Kanagy NL, Wang J, Wang W, Walker MK, Gonzalez Bosc LV. NFATc3 contributes to intermittent hypoxia-induced arterial remodeling in mice. Am J Physiol Heart Circ Physiol. 2010;299:H356–H363. doi: 10.1152/ajpheart.00341.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allahdadi KJ, Walker BR, Kanagy NL. Augmented Endothelin Vasoconstriction in Intermittent Hypoxia-Induced Hypertension. Hypertension. 2005;45:705–709. doi: 10.1161/01.HYP.0000153794.52852.04. [DOI] [PubMed] [Google Scholar]
- 18.Jackson-Weaver O, Paredes DA, Bosc LVG, Walker BR, Kanagy NL. Intermittent Hypoxia in Rats Increases Myogenic Tone Through Loss of Hydrogen Sulfide Activation of Large-Conductance Ca2+-Activated Potassium Channels / Novelty and Significance. Circ Res. 2011;108:1439–1447. doi: 10.1161/CIRCRESAHA.110.228999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amberg GC, Rossow CF, Navedo MF, Santana LF. NFATc3 regulates Kv2.1 expression in arterial smooth muscle. J Biol Chem. 2004;279:47326–47334. doi: 10.1074/jbc.M408789200. [DOI] [PubMed] [Google Scholar]
- 20.Nieves-Cintron M, Amberg GC, Nichols CB, Molkentin JD, Santana LF. Activation of NFATC3 down-regulates the beta 1 subunit of large conductance, calcium-activated K+ channels in arterial smooth muscle and contributes to hypertension. J Biol Chem. 2006;282:3231–3240. doi: 10.1074/jbc.M608822200. [DOI] [PubMed] [Google Scholar]
- 21.Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, Braunwart J, Glascock BJ, Klevitsky R, Kimball TF, Hewett TE, Molkentin JD. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest. 2003;111:1475–1486. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oukka M, Ho IC, de la Brousse FC, Hoey T, Grusby MJ, Glimcher LH. The transcription factor NFAT4 is involved in the generation and survival of T cells. Immunity. 1998;9:295–304. doi: 10.1016/s1074-7613(00)80612-3. [DOI] [PubMed] [Google Scholar]
- 23.De Frutos S, Spangler R, Alo D, Gonzalez Bosc LV. NFATc3 mediates chronic hypoxia-induced pulmonary arterial remodeling with alpha -actin up-regulation. J Biol Chem. 2007;282:15081–15089. doi: 10.1074/jbc.M702679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Frutos S, Ramiro-Diaz JM, Nitta CH, Sherpa ML, Gonzalez Bosc LV. Endothelin-1 contributes to increased NFATc3 activation by chronic hypoxia in pulmonary arteries. American Journal of Physiology - Cell Physiology. 2011;301:C441–C450. doi: 10.1152/ajpcell.00029.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cherng TW, Paffett ML, Jackson-Weaver O, Campen MJ, Walker BR, Kanagy NL. Mechanisms of Diesel-Induced Endothelial Nitric Oxide Synthase Dysfunction in Coronary Arterioles. Environ Health Perspect. 2010 doi: 10.1289/ehp.1002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Norton CE, Jernigan NL, Kanagy NL, Walker BR, Resta TC. Intermittent hypoxia augments pulmonary vascular smooth muscle reactivity to NO: regulation by reactive oxygen species. J Appl Physiol. 2011;111:980–988. doi: 10.1152/japplphysiol.01286.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao H, Kalivendi S, Zhang H, Joseph J, Nithipatikom K, Vasquez-Vivar J, Kalyanaraman B. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radic Biol Med. 2003;34:1359–1368. doi: 10.1016/s0891-5849(03)00142-4. [DOI] [PubMed] [Google Scholar]
- 28.Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vasquez-Vivar J, Kalyanaraman B. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc Natl Acad Sci U S A. 2005;%19;102:5727–5732. doi: 10.1073/pnas.0501719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smerjac SM, Bizzozero OA. Cytoskeletal protein carbonylation and degradation in experimental autoimmune encephalomyelitis. J Neurochem. 2008;105:763–772. doi: 10.1111/j.1471-4159.2007.05178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dasgupta A, Zheng J, Bizzozero OA. Protein carbonylation and aggregation precede neuronal apoptosis induced by partial glutathione depletion. ASN NEURO. 2012;4 doi: 10.1042/AN20110064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Z, Dmitrieva NI, Park JH, Levine RL, Burg MB. High urea and NaCl carbonylate proteins in renal cells in culture and in vivo, and high urea causes 8-oxoguanine lesions in their DNA. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9491–9496. doi: 10.1073/pnas.0402961101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naik JS, Earley S, Resta TC, Walker BR. Pressure-induced smooth muscle cell depolarization in pulmonary arteries from control and chronically hypoxic rats does not cause myogenic vasoconstriction. J Appl Physiol. 2005;98:1119–1124. doi: 10.1152/japplphysiol.00819.2004. [DOI] [PubMed] [Google Scholar]
- 33.Allahdadi KJ, Walker BR, Kanagy NL. ROK contribution to endothelin-mediated contraction in aorta and mesenteric arteries following intermittent hypoxia/hypercapnia in rats. Am J Physiol Heart Circ Physiol. 2007;293:H2911–H2918. doi: 10.1152/ajpheart.00217.2007. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez Bosc LV, Wilkerson MK, Bradley KN, Eckman DM, Hill-Eubanks DC, Nelson MT. Intraluminal pressure is a stimulus for NFATc3 nuclear accumulation - Role of calcium, endothelium-derived nitric oxide, and cGMP-dependent protein kinase. J Biol Chem. 2004;279:10702–10709. doi: 10.1074/jbc.M312920200. [DOI] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-[Delta][Delta]CT Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36.le-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A. Protein carbonylation in human diseases. Trends in Molecular Medicine. 2003;9:169–176. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 37.Fujii T, Onohara N, Maruyama Y, Tanabe S, Kobayashi H, Fukutomi M, Nagamatsu Y, Nishihara N, Inoue R, Sumimoto H, Shibasaki F, Nagao T, Nishida M, Kurose H. G{alpha}12/13-mediated Production of Reactive Oxygen Species Is Critical for Angiotensin Receptor-induced NFAT Activation in Cardiac Fibroblasts. J Biol Chem. 2005;280:23041–23047. doi: 10.1074/jbc.M409397200. [DOI] [PubMed] [Google Scholar]
- 38.Ramiro-Diaz JM, Nitta CH, Maston LD, Codianni S, Giermakowska W, Resta TC, Gonzalez BL. NFAT is required for spontaneous pulmonary hypertension in superoxide dismutase 1 knockout mice. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2013;304:L613–L625. doi: 10.1152/ajplung.00408.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Frutos S, Duling L, Alo D, Berry T, Jackson-Weaver O, Walker M, Kanagy N, Gonzalez BL. NFATc3 is required for intermittent hypoxia-induced hypertension. Am J Physiol Heart Circ Physiol. 2008;294:H2382–H2390. doi: 10.1152/ajpheart.00132.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koitabashi N, Aiba T, Hesketh GG, Rowell J, Zhang M, Takimoto E, Tomaselli GF, Kass DA. Cyclic GMP/PKG-dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation Novel mechanism of cardiac stress modulation by PDE5 inhibition. J Mol Cell Cardiol. 2010;48:713–724. doi: 10.1016/j.yjmcc.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang C, Li JF, Zhao L, Liu J, Wan J, Wang YX, Wang J, Wang C. Inhibition of SOC/Ca2+/NFAT pathway is involved in the anti-proliferative effect of sildenafil on pulmonary artery smooth muscle cells. Respir Res. 2009;10(123):123. doi: 10.1186/1465-9921-10-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kahler J, Mendel S, Weckmuller J, Orzechowski HD, Mittmann C, Koster R, Paul M, Meinertz T, Munzel T. Oxidative stress increases synthesis of big endothelin-1 by activation of the endothelin-1 promoter. J Mol Cell Cardiol. 2000;32:1429–1437. doi: 10.1006/jmcc.2000.1178. [DOI] [PubMed] [Google Scholar]
- 43.Kahler J, Ewert A, Weckmuller J, Stobbe S, Mittmann C, Koster R, Paul M, Meinertz T, Munzel T. Oxidative stress increases endothelin-1 synthesis in human coronary artery smooth muscle cells. J Cardiovasc Pharmacol. 2001;38:49–57. doi: 10.1097/00005344-200107000-00006. [DOI] [PubMed] [Google Scholar]
- 44.Koitabashi N, Aiba T, Hesketh GG, Rowell J, Zhang M, Takimoto E, Tomaselli GF, Kass DA. Cyclic GMP/PKG-dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation Novel mechanism of cardiac stress modulation by PDE5 inhibition. J Mol Cell Cardiol. 2010;48:713–724. doi: 10.1016/j.yjmcc.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang C, Li JF, Zhao L, Liu J, Wan J, Wang YX, Wang J, Wang C. Inhibition of SOC/Ca2+/NFAT pathway is involved in the anti-proliferative effect of sildenafil on pulmonary artery smooth muscle cells. Respir Res. 2009;10(123):123. doi: 10.1186/1465-9921-10-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tai K, Hamaide MC, Debaix H, Gailly P, Wibo M, Morel N. Agonist-evoked calcium entry in vascular smooth muscle cells requires IP3 receptor-mediated activation of TRPC1. European Journal of Pharmacology. 2008;583:135–147. doi: 10.1016/j.ejphar.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 47.Lee HA, Baek EB, Park KS, Jung HJ, Kim JI, Kim SJ, Earm YE. Mechanosensitive nonselective cation channel facilitation by endothelin-1 is regulated by protein kinase C in arterial myocytes. Cardiovascular Research. 2007;76:224–235. doi: 10.1016/j.cardiores.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 48.Nilsson LM, Nyberg M, Zetterqvist AV, Bengtsson JME, Sward K, Gomez MF, Hellstrand P. Inhibition of Ca2+-calcineurin/Nuclear Factor of Activated T-cells (NFAT) signaling reduces the expression of TRPC1 but not TRPC6 in vascular smooth muscle. The FASEB Journal. 2007;21:A1243–A124a. [Google Scholar]