

Significance
Although error rates in protein translation, particularly under stress, are high, it is not known if these errors are inherent to the system, or if they may have an adaptive function. Here, we provide evidence that specific mistranslation of mycobacterial proteins is important for phenotypic resistance to the antibiotic rifampicin. This raises the possibility that errors in protein translation play a role in adapting to stress through increasing proteomic diversity.
Keywords: drug tolerance, persisters
Abstract
Errors are inherent in all biological systems. Errors in protein translation are particularly frequent giving rise to a collection of protein quasi-species, the diversity of which will vary according to the error rate. As mistranslation rates rise, these new proteins could produce new phenotypes, although none have been identified to date. Here, we find that mycobacteria substitute glutamate for glutamine and aspartate for asparagine at high rates under specific growth conditions. Increasing the substitution rate results in remarkable phenotypic resistance to rifampicin, whereas decreasing mistranslation produces increased susceptibility to the antibiotic. These phenotypic changes are reflected in differential susceptibility of RNA polymerase to the drug. We propose that altering translational fidelity represents a unique form of environmental adaptation.
Protein translation is critical for all cellular functions, and multiple mechanisms ensuring its fidelity have evolved (1, 2). Although the ribosome has recently been shown to have some degree of discrimination following peptide bond formation (2), the major determinant of fidelity of protein translation is at the level of aminoacyl tRNA formation (1). Typically, the measured error rate for aminoacyl tRNA synthesis using purified enzymes is 1 error per 10,000 reactions. Nonetheless, the very high discrimination of protein translation enzymes as measured in vitro does not explain the relatively high error rates witnessed in vivo (3–6). These high error rates of up to 10% per codon in both bacteria and eukaryotic cells were tolerated not only in genetically modified mutants (6) but also in WT cells (3, 4). The high error rate was due to lack of discrimination at aminoacyl tRNA formation (4, 7) and at the ribosome (3), suggesting that multiple different forms of translational error occur physiologically, especially under stress.
Error rates exceeding 1/100 amino acid substitutions per codon, such as under stressful conditions (3, 4, 7), result in a significant proportion of any given translated protein being represented by multiple variants (8), a phenomenon generally thought to be deleterious to the cell. However, these errors result in new variants that could result in new functions within the “statistical proteome” (9, 10), although none have been identified to date. We considered whether mycobacterial mistranslation may result in phenotypes associated with drug resistance or tolerance.
Phenotypic drug resistance, or drug tolerance, is distinct from genetic resistance to an antibiotic. Genetically resistant organisms grow relatively normally in the presence of the drug—due to either inactivation or removal of the antibiotic or mutation of its target. By contrast, drug tolerance usually results in a decrease in the degree of killing by a bactericidal antibiotic (11). Some drug-tolerant bacteria, nonreplicating persisters, do not grow in the presence of antibiotic, but recent studies have identified some drug-tolerant populations that can still grow in the drug (12). Importantly, even if drug-tolerant bacteria do grow in antibiotic, not all of their progeny will survive antibiotic action, which is in contradistinction to genetically resistant organisms.
Mycobacterial rifampicin resistance is almost entirely due to mutations in a small region of the β-subunit of RNA polymerase (RNAP)—the rifampicin resistance determining region (RRDR). We could therefore map “gain of function” changes in β that are likely to have functional consequences for rifampicin resistance more easily. Also, given that heterologous expression of rifampicin-resistant mutations is dominant (13), we decided to investigate the role of mistranslation in mycobacteria and found that mycobacterial mistranslation causes phenotypic resistance to the antibiotic rifampicin.
Results
High Mycobacterial Mistranslation Results in Rifampicin Phenotypic Resistance.
We chose to model excess mistranslation of glutamine and asparagine codons, because most bacteria, including mycobacteria (14) lack the specific tRNA synthetases for one or both of these two amino acids (15). By mutating the anticodons of glutamate and aspartate tRNAs to glutamine and asparagine, respectively, and expressing them in trans (Fig. 1A and Fig. S1), we created a mycobacterial strain that would increase substitution of glutamate for glutamine and aspartate for asparagine [Mycobacterium smegmatis strain “excess mistranslation of glutamate and aspartate” (EMED)]. Massive overexpression of the mutated tRNAs in EMED resulted in significant growth attenuation and death. To differentiate excess physiological (EMED) from nonphysiological mistranslation, we also created a strain that would misincorporate a different amino acid [strain “excess mistranslation of alanine for tryptophan” (EMAW)] that would insert an alanine instead of a coded tryptophan. Strain EMAW was constructed by expression of another mutated tRNA—the alanine tRNA alaT with the anticodon mutated to that of tryptophan (designated alaTCCA). Alanyl tRNAs tolerate mutation of their anticodons because recognition by their cognate synthetase is mediated by a highly conserved motif in the acceptor stem (16) and mutant tRNAs are still charged normally with alanine (16). In the absence of inducer, low-level expression of abnormal tRNAs in EMED from the tetracycline-inducible promoter (17) resulted in higher mistranslation rates (Fig. 1B) with limited growth inhibition (Fig. S2). Thus, increased rates of mistranslation are associated with modest fitness costs.
Fig. 1.

High rates of mycobacterial mistranslation result in phenotypic drug resistance to rifampicin and isoniazid. (A) Schematic of tRNA anticodon swap construct. The anticodons of gluU, gluT, and aspT tRNAs were mutated to that of glnU, glnT, and asnT, respectively. The tRNAs were cloned into a mycobacterial expression plasmid under control of a tetracycline promoter and transformed into WT M. smegmatis mc2-155 to make strain EMED. Strain EMAW, which misincorporates alanine for tryptophan, was made by a similar strategy by mutation of the anticodon of tRNAAla. (B) Strain EMED has a basal mistranslation rate of ∼3% per codon for aspartate for asparagine, rising to ∼10% per codon upon induction of expression with anhydrotetracycline (ATc). Materials and Methods provides a description of the mistranslation reporter assay used to calculate mistranslation rates. WT M. smegmatis or mistranslating strains EMED and EMAW were plated on rifampicin media (100 µg/mL) (C and D) or isoniazid (30 µg/mL) or ciprofloxacin (0.5 µg/mL) media (E) and incubated at 37 °C for 5 d. (*P < 0.05, **P < 0.01, Student t test).
Mycobacterial infections are difficult to treat, as a subpopulation persists for extended periods even in the presence of effective antibiotics. We hypothesized that mistranslation might play a role. To test this, we selected for survivors in the presence of the drug rifampicin. On solid medium containing high concentrations (100 µg/mL) of rifampicin, WT M. smegmatis formed one-to-five colonies. However, after prolonged incubation, we found several thousand colonies using the EMED strain (Fig. 1 C and D).
One possible explanation for this could be mistranslation-induced error-prone replication leading to an increased mutation rate, a phenomenon described in Escherichia coli (18, 19). To test whether this was the case, we sequenced the RRDR of rpoB and found that 24/24 of the EMED colonies isolated from media containing rifampicin were WT in sequence. Moreover, strain EMED had the same minimum inhibitory concentration, MIC99, to rifampicin as WT M. smegmatis in both liquid culture (1.25 µg/mL) and on plate media (50 µg/mL). However, killing of the bulk population by rifampicin in liquid culture is considerably slowed in the mistranslating strain (Figs. S3 and S4), suggesting that resistance to rifampicin was phenotypic, not genetic in nature.
Does increased phenotypic resistance increase the likelihood of genetic resistance arising from the population? We plated both WT and EMED mycobacteria on very high (250 µg/mL) concentrations of rifampicin—on which only genetically resistant colonies can arise—and found no difference in the frequency of genetic rifampicin resistance between EMED and WT M. smegmatis (Fig. S5).
If the cells are not genetically resistant to the antibiotic, how are they surviving to form “colonies” on the agar plate? Up-regulation of chaperones in response to mistranslation-induced stress (6) might increase survivability (20). Such a response might be expected to increase survival to all antibiotics. In fact, there was a modest but reproducible survival benefit when EMED was plated on isoniazid (Fig. 1E), but the converse was true when bacteria were plated on ciprofloxacin (Fig. 1E). If a generalized up-regulated stress response was responsible for the survival phenotype, we reasoned that other types of translational error should produce a similar phenotype. We therefore tested strain EMAW for rifampicin tolerance. Expression of alaTCCA (EMAW) was associated with higher levels of some stress-related genes than EMED, including rbpA, which has been implicated in low-level rifampicin resistance (Fig. S6 and ref. 21). However, on rifampicin selection, EMED gave rise to far more colonies than EMAW (Fig. 1D). EMAW, in turn, gave rise to greater survival than WT M. smegmatis. Together, these data suggest that up-regulation of stress responses by mistranslation might make a modest contribution to survival in rifampicin but does not account for the bulk of phenotypic tolerance.
Phenotypic Resistance Is Mirrored by Increased Resistance at the Protein Level.
Ninety-six percent of rifampicin resistance maps to the RRDR (22). EMED will mistranslate glutamate for glutamine and aspartate for asparagine with relatively high frequency, including two critical residues in the RRDR of β that confer a rifampicin resistance phenotype when altered (Fig. 2A). By contrast, the mistranslation variants in strain EMAW do not map to the RRDR of β in RNAP. Although the EMED strain could produce amino acid substitutions that would mimic genetic resistance, the EMAW strain should not as this region does not contain any tryptophans. These results suggested that the mechanism underlying phenotypic resistance is synthesis of protein variants that are relatively resistant to rifampicin. To test this, we purified mycobacterial RNAP from the three strains (Fig. S7) and assayed activity in vitro in the presence and absence of rifampicin. We found that protein isolated from EMED was relatively resistant to rifampicin compared with RNAP from both WT mycobacteria and strain EMAW (Fig. 2 B and C and Fig. S8). At concentrations of rifampicin sufficient to suppress polymerase activity in the other two strains, RNAP from EMED retained significant residual activity. In fact, because a variety of quasi-species are produced even in WT cells, we see a range of inhibition at various rifampicin concentrations.
Fig. 2.
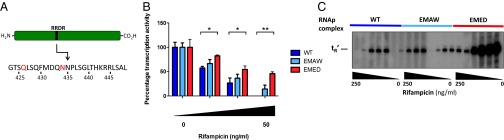
Rifampicin phenotypic resistance from mistranslation is due to variation in the protein target. (A) The RRDR of the β-subunit of RNAP of M. smegmatis. The highlighted residues refer to a glutamine or asparagine residue, that if substituted for glutamate or aspartate, respectively, results in rifampicin resistance of the protein. The relative activity of RNAP isolated from WT or mistranslating strains of M. smegmatis in the presence of rifampicin was measured by rolling circle assay (B) or radioactively labeled multiround in vitro transcription. The gel is representative of multiple experiments, and results are quantified and analyzed in Fig. S7 (C) (*P < 0.05, **P < 0.01, Student t test).
WT Mycobacteria Exhibit High and Variable Mistranslation Rates.
Although increasing the rate of mistranslation can produce phenotypic antibiotic resistance, does this play a role in WT bacteria? To investigate this, we measured mistranslation rates in WT mycobacteria grown under different environmental conditions. We used gain of function reporters (3, 6, 23) that can sensitively detect small increases in the mistranslation rate. The kanamycin kinase (Aph) protein contains a critical aspartate that is necessary to produce resistance to kanamycin (24). We mutated that residue to asparagine (D214N), rendering the enzyme inactive. Mistranslation of aspartate for asparagine would result in some small amount of active enzyme, which would be measured by survival of otherwise susceptible cells to kanamycin. We found that under several growth conditions, including low pH and stationary phase, there was a significant increase in survival in the presence of kanamycin (Fig. S9).
Although the increase in kanamycin resistance demonstrates an increased substitution rate, this rate is difficult to quantify. To measure rates accurately we used a dual luciferase fusion protein (23). This reporter consists of two different luciferase proteins, one containing a mutation that substitutes either a glutamine for a glutamate or an asparagine for an aspartate, in each case producing inactive enzyme (Fig. 3A). The ratio of activity of the WT and mutant proteins allows us to precisely control for protein expression to provide accurate estimations of the error rate. We found that during log-phase growth, rates of substitution of aspartate for asparagine were 0.8% per codon and of glutamate for glutamine were 0.2% per codon (Fig. 3B). Error rates increased substantially in stationary phase, up to 1.8% per codon, and also with low pH (Fig. 3B), and were at least one order of magnitude higher than error rates measured in E. coli (23). Mistranslation rates for other near-synonymous substitutions not likely to be encountered physiologically such as aspartate or glutamate for valine were significantly lower (Fig. 3C and Fig. S9).
Fig. 3.

Mycobacteria have high rates of misincorporation of glutamate and aspartate. (A) Schematic of the dual-luciferase mistranslation reporter based on ref. 23. The reporter is a fusion protein of luciferase enzymes from Renilla reniformis (blue, Renilla) and Photinus pyralis (yellow, Firefly). The WT enzyme exhibits luciferase activity from both enzymes (i). If an active site residue of the Renilla enzyme is mutated to render it inactive (e.g., D120N), the firefly enzyme retains activity, but the Renilla enzyme now has no activity (ii). However, if the mutated enzyme from ii is expressed under conditions of low translational fidelity, a proportion of the protein will be translated with a reconstituted active site, which results in some measurable activity (iii). The firefly activity controls for total protein expression and thus allows accurate measurement of mistranslation rates. (B) Amino acid misincorporation rates under different physiological conditions. Misincorporation of aspartate for asparagine in stationary phase (DN stationary) is higher than in log phase (DN log) and similarly for glutamate misincorporation for glutamine (EQ). Amino acid misincorporation is higher at pH 6.1 compared with pH 7.0. (C) Mistranslation rates of aspartate or glutamate for valine are lower than aspartate for asparagine and glutamate for glutamine (*P < 0.05, **P < 0.01, Student t test).
Mistranslation Is Necessary for Mycobacterial Rifampicin Phenotypic Resistance.
Do these increased rates of mistranslation play a role in phenotypic resistance in WT cells? To measure this, we took advantage of a ribosomal mutation that results in increased fidelity of translation (Figs. S10 and S11). This (K43N) streptomycin-resistant mutation maps to the rpsL gene (25). Other (RpsL–K43R) streptomycin-resistant mutants do not have increased translational fidelity (Fig. S11). Indeed, in the EMED strain, the K43N mutation resulted in decreased survival upon exposure to rifampicin (Fig. 4A). In WT cells, the K43N mutation similarly resulted specifically in a decreased number of survivors upon plating in the presence of rifampicin (Fig. 4B) but not ciprofloxacin (Fig. 4B). Exposure of stationary-phase cells to rifampicin resulted in more rapid killing of RpsL–K43N (Fig. 4C). RNAP isolated from RpsL–K43N mycobacteria is also more sensitive than WT RNAP to rifampicin (Fig. 4D), verifying that amino acid substitutions resulting from translational error are necessary for rifampicin tolerance. To model how, and how much, mistranslation may be necessary for the difference in phenotypic rifampicin resistance between the WT and K43N strains, we performed an in vitro transcription assay with mixtures of RNAP from strain K43N and a rifampicin-resistant strain (Fig. S12). With a moderate rifampicin concentration, by adding >1% and <5% of rifampicin-resistant RNAP to K43N, we could “phenocopy” RNAP isolated from WT mycobacteria. Thus, mistranslation is an important contributor to mycobacterial phenotypic resistance.
Fig. 4.

Mistranslation is necessary for mycobacterial rifampicin phenotypic resistance. (A) Survival of strain EMED on rifampicin plates is significantly attenuated in the context of a mycobacterial strain with high-fidelity ribosomes (RpsL–K43N). WT mycobacteria (109 cfu per plate) expressing high fidelity (RpsL–K43N) but not other streptomycin-resistant (RpsL–K43R) mutations are also less likely to survive on rifampicin but not ciprofloxacin plates (B). High protein translation fidelity (K43N) mycobacteria are rapidly killed in stationary phase by rifampicin (C). The RNA polymerase from high protein translation fidelity (RpsL–K43N) mycobacteria is more susceptible to in vitro inhibition by rifampicin (D). (*P < 0.05, **P < 0.01, ***P < 0.001, Student t test).
Discussion
All biological systems are constrained by the sometimes opposing needs for efficiency and accuracy. Multiple, redundant mechanisms to ensure protein translation fidelity exist in all cells, and the relatively high error rate for protein translation compared with DNA replication was thought to be acceptable given the nonheritability of proteins. Given the existence of disease associated with mistranslation (26, 27) and the toxicity of mistranslation-inducing drugs such as aminoglycosides, minimizing errors in translation to an acceptable rate would appear to be a universal evolutionary constraint. It was therefore surprising that both eukaryotic and bacterial cells appear to tolerate extremely high rates of translational error. Because error rates increased under stress, it was unclear whether this was a maladaptive response to environmental deterioration or a physiological response. In our study, we show that mycobacterial mistranslation—specifically aspartate for asparagine and glutamate for glutamine—has an unexpected role in tolerance to the important antibiotic rifampicin due to the generation of gain of function protein variants.
Error rates for protein translation—even when measured under optimal conditions in model organisms—at one error per 104 codons (23) were orders of magnitude higher than for DNA replication (8). Given that bacterial strains with hyperaccurate ribosomes have a growth defect (25), it was thought that the relatively high error rate for protein translation compared with DNA replication was a necessary tradeoff to enable sufficiently rapid growth. Coupled with the observations that aminoacyl tRNA synthetases have either inherently low error rates or proofreading, editing functions (28) and that the bactericidal activity of aminoglycoside antibiotics is due to their inducing mistranslation (29), it was assumed that elevated error rates would be universally detrimental. However, increasing evidence has emerged recently that not only can cells tolerate elevated translational error rates remarkably well (ref. 6 and Fig. 1), but physiological stress can induce extremely high mistranslation rates (refs. 3, 4, and 7 and Fig. 3), at least two orders of magnitude higher than what had been considered tolerable.
The extremely high protein translation error rates witnessed under stress in both bacterial and eukaryotic systems raised the possibility that mistranslation may play a physiological role (3, 4, 7)—but no direct evidence for such a role had been identified. Furthermore, it was not clear whether any adaptive phenotypes would be due to up-regulation of stress-associated chaperones (20) or due to a direct gain of function in the protein variants arising from mistranslation. We chose to investigate potentially adaptive roles for mistranslation in mycobacterial phenotypic resistance to the antibiotic rifampicin, because expression of a mixture of WT and resistant forms of the target protein (β) have been shown to be sufficient for drug tolerance (13).
What is the source of mistranslation? Most bacteria with the exception of some proteobacteria such as E. coli lack one or more synthetases for aminoacylation of tRNA. Mycobacteria lack the synthetases for both glutamine and asparagine aminoacyl tRNA synthesis (14). It is possible that the two-step pathway for synthesis of these two tRNAs may be associated with increased error. Mistranslation rates of aspartate for asparagine and glutamate for glutamine were higher than for other near-cognate codons (Fig. 3C and Fig. S9). However, the partial rescue of both high mistranslation rates and the rifampicin survival phenotype by high-fidelity ribosomal mutants suggests that errors in ribosomal decoding are at least partially responsible (Fig. 4).
Tolerance to antibiotics, like other stresses encountered by bacteria, is an important part of the survival of a population. Phenotypic variation among genetically identical individuals can be considerable, resulting in clones that are not killed and can even grow in the presence of antibiotics (12, 30). The resulting subpopulations can survive until stresses pass or serve as founder populations from which resistant mutants arise. Our data show that errors in translation might be an important component of individual variation with significant consequences for stress survival. By use of two strains with different forms of mistranslation—one of which would not cause protein variants in β that would result in classical drug resistance—we were able to show that mistranslation-mediated phenotypic resistance to rifampicin was largely, although not exclusively, due to protein variants that resulted in drug resistance (Figs. 2 and 4).
How can mistranslation in RNAP cause rifampicin tolerance? There are several possibilities. The simplest model is that mistranslation of residues critical for rifampicin resistance in β (Fig. 2A) results in a subpopulation of protein that is now rifampicin-resistant. We modeled this by mixing RNAP from a high-fidelity (K43N) strain with small amounts of RNAP from a rifampicin-resistant strain. Small (< 5%) amounts of rifampicin-resistant protein was sufficient to phenocopy the degree of resistance of WT RNAP at moderate rifampicin concentrations (Fig. S12). Another distinct possibility that is more difficult to model biochemically is that there are multiple protein variants in RNAP (perhaps in β′ – RpoC, as well as β) that may cause a degree of phenotypic resistance but are insufficient for robust, genetic resistance to the drug and thus have not been identified by selection of rifampicin-resistant mutations. Each of these variants may contribute individually a small degree to rifampicin phenotypic resistance with the entire, heterogeneous population having a significant phenotype. When we compared the rifampicin resistance of RNAP from our different strains, we noted that at 50 ng/mL rifampicin, where we observe almost complete suppression of transcription from RNAP from WT cells, there is residual transcription from RNAP from EMAW (Fig. 2B). Although this difference does not reach statistical significance, it might suggest that there may be other residues outside of the RRDR that may contribute in a minor way to rifampicin resistance—and recent genetic studies support a role for mutations in rpoC in rifampicin resistance (31).
We observed a stark contrast in the degree of rifampicin tolerance due to mistranslation when measured on plates (Fig. 1 C and D) and in liquid culture (Figs. S3 and S4). The difference may in part be due to the fact that at concentrations of rifampicin well above the MIC for liquid culture (10–50 µg/mL), a large number of M. smegmatis colonies still arise on plate culture, which are not genetically resistant to the drug. Furthermore, a very small percentage of mycobacteria in a population may have the required mistranslation threshold for survival under drug selection, with the probability of any one bacterium being above this threshold higher in EMED than WT mycobacteria, but the absolute numbers still relatively low. In axenic culture, this will be represented by a slower kill kinetic to rifampicin; but over the 20 generations required to form a colony on a plate, even such small differences may be greatly amplified.
Our data also show that the mistranslating strain EMED has increased tolerance to isoniazid but not ciprofloxacin (Fig. 1E). Resistance to isoniazid is mediated either by mutations inactivating the mycobacterial catalase, katG, which is required to activate the prodrug isoniazid to its active form, or overexpression of the drug target inhA (32). Some naturally occurring isoniazid-resistant mutants of Mycobacterium tuberculosis have mutations in katG that might occur with greater frequency in EMED, such as KatG–N138D (32), potentially explaining the result. Purifying KatG from WT and EMED mycobacteria and performing an enzyme assay similar to that which we performed for RNAP may help resolve whether this is a possible mechanism for the observation. Resistance to the quinolone ciprofloxacin is usually mediated by mutations in the DNA gyrase gene gyrA, and less commonly gyrB (33). Although there is one isolated mutant in gyrB, N510D, that mediates quinolone resistance via an asparagine to aspartate mutation (33), rather than increased resistance of EMED to ciprofloxacin, we saw decreased resistance (Fig. 1E). It is not clear why this may be, although higher mistranslation would also result in alteration of an asparagine critical for gyrase function (34), which may offset any advantages from resistant variants of the protein.
Slow or nongrowing “persisters” are a key type of drug-tolerant bacteria (11). Given that EMED grows slightly more slowly than WT bacteria (Fig. S2), could that be the reason for increased phenotypic resistance? If slow growth was responsible, we would expect increased phenotypic resistance to all conventional antibiotics (11), which is in contrast to the observed decreased resistance to quinolones (Fig. 1E). Furthermore, the high-fidelity mutant RpsL–K43N, which has lower rifampicin tolerance, also grows more slowly than WT mycobacteria (Fig. S10), together suggesting that our observed phenotype is specific to translational fidelity and not bacterial growth rates.
Distinct from stress-induced error-prone DNA replication (35), protein sequence variation arising from mistranslation is a potentially enormous and reversible resource of diversity for cells undergoing stress. We propose that protein sequence variation, whether by RNA editing (36), yeast prion protein variants (37), or tuning of translational fidelity represent unique forms of environmental adaptation.
Materials and Methods
Bacterial Culture and Strains.
WT M. smegmatis, strain mc2-155 (38), was cultured in Middlebrook 7H9 medium supplemented with albumin, dextrose, catalase (ADC), glycerol, and 0.05% Tween-20 unless otherwise specified. Plate culture was performed on LB–agar plates supplemented with antibiotics as indicated unless otherwise specified. For experiments performed in different pH, cells were grown in M9 minimal medium, supplemented with 0.4% glucose and 0.025% Tyloxapol (Sigma). The medium pH was buffered using HCl or NaOH. All chemicals were from Sigma unless otherwise specified.
The strains M. smegmatis–EMED and M. smegmatis–EMAW were made by transforming competent WT M. smegmatis with plasmids EMED and EMAW, respectively, and plating on selective media. Reporter strains were also constructed by transformation of WT M. smegmatis with the appropriate reporter constructs. The strains RpsL–K43N and RpsL–K43R were generated by plating M. smegmatis mc2-155 onto LB–Streptomycin (10 µg/mL). Streptomycin mutants were selected and the rpsL gene sequenced. The strains were routinely grown on 5 µg/mL Streptomycin, except for the rifampicin survival experiments, when streptomycin was omitted.
Antibiotic Survival Assays.
Approximately 108 (rifampicin) or 107 (isoniazid and ciprofloxacin) bacteria were plated onto LB–agar plates containing the appropriate antibiotic incubated at 37 °C for 7 d. For liquid culture antibiotic killing/survival, cells were grown in supplemented 7H9 medium until stationary phase. Rifampicin was added. Aliquots of cells were taken at various time points, the antibiotic diluted out, and survivors plated onto nonselective medium.
His-Tagged rpoC Strain Construction.
A recombination cassette was synthesized by GenScript in a pUC57 plasmid containing a 200-bp region homologous to rpoC 3′ end followed by a 6× His-tag, kanamycin resistance marker and a 200-bp region homologous to rpoC 3′ end-flanking region (Supporting Information). A linear dsDNA fragment was amplified by PCR. A M. smegmatis strain expressing an isovalonitrile-inducible Che9c recombinase [mc2 pNITRILE Che9C::zeoR (39)] was transformed with 2 μg of linear His-tagged RpoC:: kanR.
Isolation of Mycobacterial RNAP.
Mycobacterial RNAP was isolated from strains engineered with a His6-tag to the C terminus of rpoC (β′). Cells were harvested at late exponential growth phase and lysed by French Press. The cleared lysate was bound to a preequilibrated TALON cobalt resin (Clontech). The final concentration of the enzymes was determined by Bradford assay (Bio-Rad) and the identity of the RNAP subunits verified by SDS/PAGE and mass spectrometry.
In Vitro Transcription Assay.
The activity-isolated RNAP was measured by two methods, radio-labeled in vitro transcription and via rolling circle transcription assay using the Kool NC-45 RNAP Activity & Inhibitor Screening kit (EpiCentre) in the absence and presence of rifampicin.
Radio-labeled in vitro transcription assay is described in detail in the SI Appendix. Briefly, holoenzymes (formed by preincubating 50 nM RNAPs with 100 nM σA at 37 °C for 10 min) were incubated in the absence or in the presence of rifampicin for 5 min. Reactions were stopped after 15 min by adding formamide-loading buffer and heating to 95 °C, and RNA transcripts with similar activity were loaded and electrophoresed on 6% (wt/vol) denaturing polyacrylamide sequencing gels. Bands were visualized by PhosphorImager and the data analyzed using ImageQuant software.
Dual-Luciferase Assay.
This assay was based on that developed by Kramer and Farabaugh (23). The sequence of the protein fusion of Renilla and firefly luciferase genes was codon-optimized for expression in M. smegmatis and synthesized synthetically (Genscript–GenBank JQ606807). Using structural data suggesting the critical residues for Renilla luciferase activity (40), mutations in the Renilla gene were made by site-directed mutagenesis. The resulting sequences were subcloned into a tetracycline-inducible expression shuttle plasmid, pTetG (41) using gateway cloning (Invitrogen) and transformed in competent M. smegmatis cells. Approximately 5 × 108 cells were harvested, pelleted, and lysed by the passive lysis buffer supplied with the dual-luciferase assay kit (Promega). Luciferase activity was measured by the dual luciferase assay kit according to the manufacturer’s instructions using a Fluoroskan Ascent FL luminometer using integration times of 1 s for measurements. Mistranslation rates were calculated as per Kramer and Farabaugh (23).
Supplementary Material
Acknowledgments
We thank Sarah Fortune and Chris Sassetti for critical reading of the manuscript and Paul Lehner for helpful discussion. We thank Dr. Jun-Rong Wei (Harvard School of Public Health) for the kind gift of plasmid pJW3 and Rong-Jun Cai (Tsinghua University School of Medicine) for generation of strains RpsL–K43N and RpsL–K43R. This work was in part funded by a Medical Research Council (United Kingdom) Clinician Scientist Fellowship, Bill and Melinda Gates Foundation Grand Challenges phase I grant, and start-up funds from Tsinghua University School of Medicine (to B.J.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1317580111/-/DCSupplemental.
References
- 1.Ibba M, Söll D. Quality control mechanisms during translation. Science. 1999;286(5446):1893–1897. doi: 10.1126/science.286.5446.1893. [DOI] [PubMed] [Google Scholar]
- 2.Zaher HS, Green R. Quality control by the ribosome following peptide bond formation. Nature. 2009;457(7226):161–166. doi: 10.1038/nature07582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyerovich M, Mamou G, Ben-Yehuda S. Visualizing high error levels during gene expression in living bacterial cells. Proc Natl Acad Sci USA. 2010;107(25):11543–11548. doi: 10.1073/pnas.0912989107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Netzer N, et al. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature. 2009;462(7272):522–526. doi: 10.1038/nature08576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reynolds NM, et al. Cell-specific differences in the requirements for translation quality control. Proc Natl Acad Sci USA. 2010;107(9):4063–4068. doi: 10.1073/pnas.0909640107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruan B, et al. Quality control despite mistranslation caused by an ambiguous genetic code. Proc Natl Acad Sci USA. 2008;105(43):16502–16507. doi: 10.1073/pnas.0809179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ling J, Söll D. Severe oxidative stress induces protein mistranslation through impairment of an aminoacyl-tRNA synthetase editing site. Proc Natl Acad Sci USA. 2010;107(9):4028–4033. doi: 10.1073/pnas.1000315107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drummond DA, Wilke CO. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet. 2009;10(10):715–724. doi: 10.1038/nrg2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pezo V, et al. Artificially ambiguous genetic code confers growth yield advantage. Proc Natl Acad Sci USA. 2004;101(23):8593–8597. doi: 10.1073/pnas.0402893101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moura GR, Carreto LC, Santos MA. Genetic code ambiguity: An unexpected source of proteome innovation and phenotypic diversity. Curr Opin Microbiol. 2009;12(6):631–637. doi: 10.1016/j.mib.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 11.Lewis K. Persister cells. Annu Rev Microbiol. 2010;64:357–372. doi: 10.1146/annurev.micro.112408.134306. [DOI] [PubMed] [Google Scholar]
- 12.Wakamoto Y, et al. Dynamic persistence of antibiotic-stressed mycobacteria. Science. 2013;339(6115):91–95. doi: 10.1126/science.1229858. [DOI] [PubMed] [Google Scholar]
- 13.Miller LP, Crawford JT, Shinnick TM. The rpoB gene of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1994;38(4):805–811. doi: 10.1128/aac.38.4.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cole ST, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393(6685):537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 15.Curnow AW, et al. Glu-tRNAGln amidotransferase: A novel heterotrimeric enzyme required for correct decoding of glutamine codons during translation. Proc Natl Acad Sci USA. 1997;94(22):11819–11826. doi: 10.1073/pnas.94.22.11819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swairjo MA, et al. Alanyl-tRNA synthetase crystal structure and design for acceptor-stem recognition. Mol Cell. 2004;13(6):829–841. doi: 10.1016/s1097-2765(04)00126-1. [DOI] [PubMed] [Google Scholar]
- 17.Carroll P, Muttucumaru DG, Parish T. Use of a tetracycline-inducible system for conditional expression in Mycobacterium tuberculosis and Mycobacterium smegmatis. Appl Environ Microbiol. 2005;71(6):3077–3084. doi: 10.1128/AEM.71.6.3077-3084.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bacher JM, Schimmel P. An editing-defective aminoacyl-tRNA synthetase is mutagenic in aging bacteria via the SOS response. Proc Natl Acad Sci USA. 2007;104(6):1907–1912. doi: 10.1073/pnas.0610835104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murphy HS, Humayun MZ. Escherichia coli cells expressing a mutant glyV (glycine tRNA) gene have a UVM-constitutive phenotype: implications for mechanisms underlying the mutA or mutC mutator effect. J Bacteriol. 1997;179(23):7507–7514. doi: 10.1128/jb.179.23.7507-7514.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feder ME, Cartaño NV, Milos L, Krebs RA, Lindquist SL. Effect of engineering Hsp70 copy number on Hsp70 expression and tolerance of ecologically relevant heat shock in larvae and pupae of Drosophila melanogaster. J Exp Biol. 1996;199(Pt 8):1837–1844. doi: 10.1242/jeb.199.8.1837. [DOI] [PubMed] [Google Scholar]
- 21.Dey A, Verma AK, Chatterji D. Role of an RNA polymerase interacting protein, MsRbpA, from Mycobacterium smegmatis in phenotypic tolerance to rifampicin. Microbiology. 2010;156(Pt 3):873–883. doi: 10.1099/mic.0.033670-0. [DOI] [PubMed] [Google Scholar]
- 22.Gagneux S, et al. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science. 2006;312(5782):1944–1946. doi: 10.1126/science.1124410. [DOI] [PubMed] [Google Scholar]
- 23.Kramer EB, Farabaugh PJ. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA. 2007;13(1):87–96. doi: 10.1261/rna.294907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boehr DD, Thompson PR, Wright GD. Molecular mechanism of aminoglycoside antibiotic kinase APH(3′)-IIIa: Roles of conserved active site residues. J Biol Chem. 2001;276(26):23929–23936. doi: 10.1074/jbc.M100540200. [DOI] [PubMed] [Google Scholar]
- 25.Zaher HS, Green R. Hyperaccurate and error-prone ribosomes exploit distinct mechanisms during tRNA selection. Mol Cell. 2010;39(1):110–120. doi: 10.1016/j.molcel.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JW, et al. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443(7107):50–55. doi: 10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
- 27.Schimmel P, Guo M. A tipping point for mistranslation and disease. Nat Struct Mol Biol. 2009;16(4):348–349. doi: 10.1038/nsmb0409-348. [DOI] [PubMed] [Google Scholar]
- 28.Reynolds NM, Lazazzera BA, Ibba M. Cellular mechanisms that control mistranslation. Nat Rev Microbiol. 2010;8(12):849–856. doi: 10.1038/nrmicro2472. [DOI] [PubMed] [Google Scholar]
- 29.Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell. 2008;135(4):679–690. doi: 10.1016/j.cell.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levin-Reisman I, et al. Automated imaging with ScanLag reveals previously undetectable bacterial growth phenotypes. Nat Methods. 2010;7(9):737–739. doi: 10.1038/nmeth.1485. [DOI] [PubMed] [Google Scholar]
- 31.Farhat MR, et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat Genet. 2013;45(10):1183–1189. doi: 10.1038/ng.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hazbón MH, et al. Population genetics study of isoniazid resistance mutations and evolution of multidrug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2006;50(8):2640–2649. doi: 10.1128/AAC.00112-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aubry A, et al. Novel gyrase mutations in quinolone-resistant and -hypersusceptible clinical isolates of Mycobacterium tuberculosis: Functional analysis of mutant enzymes. Antimicrob Agents Chemother. 2006;50(1):104–112. doi: 10.1128/AAC.50.1.104-112.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gross CH, et al. Active-site residues of Escherichia coli DNA gyrase required in coupling ATP hydrolysis to DNA supercoiling and amino acid substitutions leading to novobiocin resistance. Antimicrob Agents Chemother. 2003;47(3):1037–1046. doi: 10.1128/AAC.47.3.1037-1046.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galhardo RS, Hastings PJ, Rosenberg SM. Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol Biol. 2007;42(5):399–435. doi: 10.1080/10409230701648502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li JB, et al. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324(5931):1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- 37.Halfmann R, et al. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature. 2012;482(7385):363–368. doi: 10.1038/nature10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR., Jr Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol. 1990;4(11):1911–1919. doi: 10.1111/j.1365-2958.1990.tb02040.x. [DOI] [PubMed] [Google Scholar]
- 39.Wei JR, et al. Depletion of antibiotic targets has widely varying effects on growth. Proc Natl Acad Sci USA. 2011;108(10):4176–4181. doi: 10.1073/pnas.1018301108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loening AM, Fenn TD, Gambhir SS. Crystal structures of the luciferase and green fluorescent protein from Renilla reniformis. J Mol Biol. 2007;374(4):1017–1028. doi: 10.1016/j.jmb.2007.09.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siegrist MS, et al. Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc Natl Acad Sci USA. 2009;106(44):18792–18797. doi: 10.1073/pnas.0900589106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.