

Significance
Despite the availability of a number of antibiotics and extensive use of the live vaccine bacillus Calmette–Guérin, tuberculosis remains a major global health problem. Antigen 85 is by far the most popular antigen in current clinical trials vaccines. However, a recent failure of a virus-based vaccine expressing antigen 85 emphasizes the continued need for identifying antigens, testing their protective efficacy, and learning from this process to develop a protective vaccine. The H65 vaccine demonstrates that it is possible to develop antigen 85-free vaccines that does not interfere with current immune-based diagnostic assays.
Keywords: T-cell immunology, gene expression, tuberculosis
Abstract
A central goal in vaccine research is the identification of relevant antigens. The Mycobacterium tuberculosis chromosome encodes 23 early secretory antigenic target (ESAT-6) family members that mostly are localized as gene pairs. In proximity to five of the gene pairs are ESX secretion systems involved in the secretion of the ESAT-6 family proteins. Here, we performed a detailed and systematic investigation of the vaccine potential of five possible Esx dimer substrates, one for each of the five ESX systems. On the basis of gene transcription during infection, immunogenicity, and protective capacity in a mouse aerosol challenge model, we identified the ESX dimer substrates EsxD-EsxC, ExsG-EsxH, and ExsW-EsxV as the most promising vaccine candidates and combined them in a fusion protein, H65. Vaccination with H65 gave protection at the level of bacillus Calmette–Guérin, and the fusion protein exhibited high predicted population coverage in high endemic regions. H65 thus constitutes a promising vaccine candidate devoid of antigen 85 and fully compatible with current ESAT-6 and culture filtrate protein 10-based diagnostics.
Despite the availability of a number of antibiotics and the extensive use of the live vaccine bacillus Calmette–Guérin (BCG), tuberculosis (TB) remains a major global health problem, with an estimated incidence rate of 9 million new cases per year (1). BCG vaccination protects against severe progressive TB in children but is not able to prevent reactivation of pulmonary disease in adult life (2). As a consequence, there is a need for a vaccine to supplement or replace BCG.
There are currently several TB vaccines in clinical trials, which include attenuated Mycobacterium tuberculosis (M.tb) strains, recombinant BCG strains, viral vectored vaccines, and protein-based subunit vaccines administered together with an adjuvant (3). Apart from the whole-cell vaccines, most are based on the same very limited number of M.tb antigens (Ags), with Ag85A and Ag85B in particular being present in many candidate vaccines (4). Recently, a TB efficacy study based on a recombinant strain of modified vaccinia Ankara virus expressing Ag85A (MVA85A) was unblinded. MVA85A was given to infants as a BCG booster but did not statistically improve the protective efficacy of the bacillus. In vivo gene expression data have further questioned the use of Ag85 proteins as vaccine targets, as the expression of all Ag85 genes after an initial peak during the first weeks of infection are dramatically reduced; in mice, this correlates with the onset of the adaptive immune response (5).
Adoptive transfer studies of Ag85B-specific T cells have shown that during the early phase of infection, ∼10% of the transferred T cells produced IFN-γ in vivo, whereas this percentage declined to <1% in the chronic phase (6). In TB granulomas, it was furthermore shown that the presentation of Ag85 epitopes on the surface of infected Ag-presenting cells was very low (7). Combined, these results suggest that an Ag85-specific adaptive immune response primed by vaccination should be effective in the early stage of infection but of relative low value during the later stages of infection. In support of this, a recent study showed that in patients with active pulmonary TB, less than 25% of the patients responded to Ag85A/B proteins, and that these responses in general were very modest (8). In contrast, more than 70% of the patients responded to the early secretory antigenic target-6 and culture filtrate protein 10 (ESAT-6-CFP10) fusion protein and the TB10.4 protein with overall strong Ag-specific responses. These three proteins are secreted T-cell Ags that are strongly recognized in mice, guinea pigs, cattle, and humans (9–12), and both ESAT-6 and TB10.4 have shown promising protective efficacy in subunit vaccines (13). The proteins are members of the ESAT-6 family that in M.tb includes 23 proteins, EsxA through EsxW, all localized as gene pairs except for EsxQ (14).
This protein family seems to be particularly interesting for vaccine purposes, as EsxV (Rv3619c) and EsxW (Rv3620c), or one of their paralogues, have been reported as T-cell targets in patients with TB (15), and both proteins have furthermore been included in a vaccine capable of boosting BCG (16). For five of the Esx gene pairs on the M.tb H37Rv chromosome, the flanking genes have been found to encode proteins involved in the secretion of the ESAT-6 family proteins. These clusters are known as the ESX or type VII secretion (T7S) systems, ESX-1 through ESX-5 (17). ESX-1 and ESX-5 are involved in the virulence of M.tb, whereas ESX-3 is essential for survival of M.tb and is required for the acquisition of metal ions. No function has been identified for ESX-2 or ESX-4. Most proteins secreted by T7S systems follow a pairwise dependency for secretion (18). Given the high frequency of antigenic and successful vaccine candidates among the secreted ESAT-6 family proteins, we decided to test the protective efficacy of a potential ESAT-6 family dimer-substrate for each of the five ESX systems. We show that four of the five dimer-substrates are protective against an aerosol challenge with virulent M.tb and that a fusion (H65) based on three of these has high predicted population coverage and is highly protective even without important Ags such as ESAT-6 or Ag85.
Results
Phylogeny and Selection of Esx Vaccine Candidates.
Our strategy was to test the vaccine potential of an Esx dimer substrate for each of the five ESX secretion systems and to combine the most promising ones into a fusion protein vaccine. To select relevant candidates among the 23 potential Esx substrates encoded on the M.tb H37Rv chromosome (Table S1), their amino acid sequences were aligned. Because the Esx proteins are secreted as heterodimers, the alignment was done for protein pairs organized in the order they are encoded on the chromosome (e.g., EsxB–EsxA). The esxQ gene is encoded as a singlet on the chromosome, and the protein has no obvious secretion partner and was therefore included in the alignment as a single protein. On the basis of sequence homology, the 11 possible ESX substrates cluster into 6 clades (Fig. S1A), consistent with at least one substrate for each of the five secretion systems, ESX-1 through ESX-5, plus a dimer pair (EsxF-EsxE) without an identifiable ESX secretion system (Table S1). None of the proteins within the dimer substrates for ESX-2 (EsxC and EsxD) or ESX-4 (EsxT and EsxU), nor the EsxQ singlet, contained the C-terminal YxxxD/E amino acid motif that has been linked with secretion via the ESX system (19). The genes encoding the dimer substrate for the ESX-5 secretion system have been duplicated several times during the evolution of M.tb, with the result that the chromosome encodes five dimer paralogues with at least 95% identity (Fig. S1B).
For this study we selected EsxW-EsxV, but a specific immune response against any of the five dimers will, in practice, target all of them. Sequence alignment shows that the ESX-3 system potentially has three ESAT-6 family-based substrates: two heterologous dimers plus the EsxQ single protein. The dimer substrates EsxG/EsxH and EsxS/EsxR are very similar in sequence (Fig. S1C). However, as EsxG and EsxH have both been identified in M.tb culture supernatants, and EsxH is recognized more frequently and strongly than EsxR by T cells isolated from TB patients (20), EsxG/EsxH were selected for the study. The singlet protein EsxQ is quite different in sequence from the two dimer substrates (43–44% identity) and could be a relevant vaccine Ag. However, EsxQ was excluded because it has no natural secretion partner, it lacks the ESX signature sequence found close to the C terminus in one of the ESX dimer substrates, secretion of the protein is not supported by proteome data, the physical characteristics are different from the other members (120 amino acids compared with 90–107 amino acids; isoelectric point, pI = 8.06 compared with 4.19–6.51), and the N-terminal 70 amino acids share 65% identity with EsxV, whereas the C-terminal 50 amino acids share 40% identity with EspG3 (ESX-3 secretion-associated protein, Rv0289). Most likely, esxQ is a pseudogene arising from a complex duplication event involving the genes encoding EsxV and EspG3.
T-Cell Recognition Pattern and Protective Efficacies.
The five recombinant dimer fusions were all constructed as (Gene1) – (Gene2), where Gene1 encodes the protein equivalent to EsxB and Gene2 encodes the protein equivalent to EsxA, mimicking how they are located and transcribed from the chromosome. For flexibility, the proteins are separated by a nine-amino acid linker. After successful expression and purification, the integrity and purity of the heterodimer proteins were confirmed by Coomassie blue-stained SDS/PAGE gels (Fig. S2A), and their identity was confirmed by mass spectrometry. The dimer proteins were formulated in a cationic adjuvant, and groups of CB6F1 or B6C3F1 mice were immunized with either one of the recombinant proteins. We used two hybrid mice strains to allow for a more diverse recognition of epitopes/Ags. Specific T-cell responses were measured in purified peripheral blood mononuclear cells 3 wk after vaccination. In CB6F1 vaccinated animals, we found vaccine-specific responses in all five vaccinated groups, but the immunogenicity of the proteins varied significantly, with EsxB-EsxA being the least and EsxW-EsxV the most immunogenic dimers (Fig. 1A). In B6C3F1 vaccinated animals, we found vaccine-specific responses against four of the five dimers, with EsxD-EsxC being the exception (Fig. 1B). No response was found in the two mice strains against the nine-amino acid linker inserted between the proteins.
Fig. 1.

No correlation between protective efficacy and immunogenicity. Groups of CB6F1 (A and C) or B6C3F1 (B and D) mice were immunized with either of the recombinant proteins. Peripheral blood mononuclear cells from CB6F1 (A) or B6C3F1 (B) mice (n = 3 per group) were purified 3 wk after immunization and stimulated with vaccine protein, after which the concentration of IFN-γ was measured in the culture medium. SEMs are indicated. Saline-injected mice gave no response against the proteins. Protective efficacies were measured in CB6F1 (C) and B6C3F1 (D) mice by enumerating the bacteria in the lung of individual animals 6 wk after challenge with M.tb Erdman (n = 6 per group). The values are shown as means of the log10 CFU in saline-injected mice minus means of the log10 CFU for each of the vaccination groups; SEMs are indicated. *P < 0.05, one-way ANOVA, Tukeys multiple comparison test.
Six weeks after vaccination, all mice were challenged with M.tb (Erdman) by the aerosol route, and bacteria were enumerated in the lungs 6 wk later. In CB6F1 vaccinated mice, two of the five dimer proteins (EsxB-EsxA and EsxG-EsxH) provided significant protection [0.46 ± 0.11 and 0.32 ± 0.14 log10 reduction of lung colony-forming units (CFUs)] compared with the adjuvant control group (Fig. 1C). However, this was a modest degree of protection compared with the high level of protection (1.39 ± 0.05 log10 reduction) obtained when vaccinating with BCG in this mouse strain. In B6C3F1 mice, four of the five dimers (EsxB-EsxA, EsxD-EsxC, EsxG-EsxH, and EsxW-EsxV) protected against M.tb Erdman infection (0.32 ± 0.12 to 0.58 ± 0.20 log10 reduction of lung CFUs) at a level more comparable to the reduction in the BCG-vaccinated group (0.87 ± 0.25 log10) (Fig. 1D). We did not observe a direct correlation between vaccine-specific T-cell secretion of IFN-γ and protective efficacy but cannot exclude the possibility of vaccine priming of an epitope that is not presented during infection.
Relative Gene Expression Pattern and Recognition During Infection.
For a vaccine-primed immune response to be effective, the target Ag has to be expressed during infection. We therefore followed the expression pattern of the Esx genes from before infection (day 0, the in vitro culture used for inoculation) to 140 d after infection. For comparison, the combined expression of the genes encoding the fibronectin-binding proteins Ag85A, Ag85B, and Ag85C were included (Fig. 2A; Table S2). In this model, the host cell adaptive immunity is measurable at day 12–14 after infection, and the expression analysis covers the period during which the bacteria adapt to the inflicted stress. Consistent with the lack of protection, we found no expression of esxT and esxU at any of the times. The relative expression of esxC + D and esxV + W was constant at the four points (2–3% and 5–9%, respectively). In contrast, the relative esxA + B and esxH + G expression changed quite significantly during the infection. The esxG + H expression increased steadily from ∼5% of the investigated transcripts at day 0 to ∼49% after 140 d of infection. In contrast, the relative esxA + B expression dropped from ∼81% to ∼37% during this period. To confirm that the high gene expression at least partly correlated with Ag presentation and priming of T cells, the specific T-cell release of IFN-γ was measured by stimulating isolated lung cells with dimer proteins in CB6F1 (Fig. 2B) and B6C3F1 (Fig. 2C). The highly expressed EsxB-EsxA and EsxG-EsxH dimer proteins were both strongly recognized by T cells in CB61F1 and B6C3F1 mice after 21 d of infection. The intermediately expressed EsxW-EsxV was recognized in B6C3F1 but not CB6F1 mice, and we did not detect any specific T-cell response against the modestly expressed EsxD-EsxC dimer or the nonexpressed EsxU-EsxT dimer. Despite the fact that EsxB-EsxA and EsxG-EsxH were the most highly expressed and best recognized Esx dimers after infection, they were not significantly more protective than the immunogenic EsxW-EsxV dimer (Fig. 1 C and D) in either mice strains nor EsxD-EsxC in B6C3F1 mice.
Fig. 2.

Expression dynamic and Ag recognition during infection. (A) The relative expression of the genes encoding the five secretion dimers and Ag85A, Ag85B, and Ag85C was measured in M.tb (strain Erdman) in vitro cultures (0 d) and in lungs of infected CB6F1 mice (n = 4 per time). The relative expression for each of the 5 dimer operons was calculated by [(RGCNgene1+ RGCNgene2)/∑(RGCNtotal)]*100, and for the fbp genes by [(RGCNfbpA + RGCNfbpB + RGCNfbpC)/ ∑(RGCNtotal)]*100. RGCN = RNA gene copy number. RGCNtotal is the sum of the 13 genes investigated (10 esx and 3 fbp genes). For more information, see Table S2. We found no detectable expression of esxU-T under the conditions tested. The Esx and Ag85B specificity of T cells recruited to the lung was measured in nonvaccinated CB6F1 (B) or B6C3F1 (C) mice 21 d after infection with M.tb (strain Erdman) (n = 3 per mice strain).
The ESX Substrate-Based Fusion Protein H65 as a TB Vaccine.
On the basis of the immunogenicity, expression pattern, and protective efficacy data shown earlier, the ESX dimer substrates EsxD-EsxC, EsxG-EsxH, and EsxW-EsxV were combined into a fusion protein termed H65. To allow some freedom for the subunits to fold, the three dimer substrates were separated by two 20-amino acid-long linker sequences from the M.tb protein Rv1986 (Fig. S2B). Both sequences have previously been shown to hold human T-cell epitopes (21). Despite being immunogenic, highly expressed, and protective, EsxB-EsxA was not included in the fusion because of the widespread use of ESAT-6 and CFP10 as TB diagnostic Ags. The H65 fusion protein was expressed in Escherichia coli and recovered with a very high purity, although with minor degrees of degraded product visible in both the Coomassie-stained gels and Western blot developed with anti-His-tag antibodies (Fig. S2C).
Groups of CB6F1 mice were either vaccinated with H65 or BCG Danish or injected with the adjuvant alone. Three weeks after the third vaccination, Ag-specific T-cell responses were analyzed in purified splenocytes isolated from H65-vaccinated and adjuvant control animals (Fig. 3A). In H65-vaccinated CB6F1 mice, two of the vaccine dimers were recognized by T cells, with the order of immunogenicity being EsxG-EsxH > EsxW-EsxV. No IFN-γ release could be detected after restimulation with either EsxD-EsxC or a mixture of the linker sequences. In the adjuvant control group, there was no detectable response toward any of the proteins/peptides. The vaccinated groups of animals were challenged with M.tb, and after 6 wk of infection, their lungs were analyzed for vaccine-induced protection. All vaccinated animals had reduced bacterial burdens compared with the adjuvant control group (Fig. 3B). H65 vaccination resulted in a CFU reduction of ∼0.85 log10 (P < 0.01) relative to the adjuvant control, whereas BCG vaccination gave a 1.39 log10 (P < 0.001) reduction. The level of protection obtained with H65 was identical to the protection obtained after vaccination with the H56 fusion protein (Ag85B-ESAT6-Rv2660c). As a snapshot of the in vivo immune activity at the site of infection, the IFN-γ levels were measured in lung homogenates from individual animals (Fig. S3). The highest IFN-γ level was found in the adjuvant control group, and only the H65-vaccinated group had a statistically lower level (P < 0.05).
Fig. 3.

H65-vaccine-specific responses reduce bacteria load. (A) Splenocytes were isolated from H65-vaccinated animals (n = 3) 3 wk after vaccination and stimulated in vitro with single proteins, after which secreted IFN-γ was measured in the supernatants. Linkers were a 1:1:1 mix of the three sequences included in the fusion (Fig. S2B). Bacterial load in groups of vaccinated CB6F1 mice (B) were measured in the lungs of individual mice 6 wk after challenge with M.tb (strain Erdman); means ± SEM are indicated (n = 6 per group). **P < 0.01, ***P < 0.001, one-way ANOVA, Tukeys multiple comparison test. The frequency of H65-specific CD4+ T cells producing IFN-γ, TNF-α, or IL-2 was measured in splenocytes by flow cytometry 3 wk after immunization with CAF01 (gray bars) or H65 (black bars) in B6C3F1 mice after in vitro stimulation with single proteins (C). The bacterial burden in vaccinated B6C3F1 mice (D) was measured by enumerating the bacteria in the lung of individual animals 6 wk after challenge with M.tb (strain Erdman) (n = 6 per group). *P < 0.05, one-way ANOVA, Tukeys multiple comparison test. For both mice strains, the tests have been done twice with similar results.
Because the recognition of the Esx dimer proteins during infection is broader (Fig. 2C) and their protective efficacy closer to the BCG vaccine in B6C3F1 mice (Fig. 1D), the H65 fusion was also evaluated in this mouse strain. Three weeks after vaccination, purified splenocytes were analyzed for vaccine-induced responses by multicolor flow cytometry. The frequencies of Esx dimer-specific CD4+ T cells, producing IL-2, TNF-α, or IFN-γ, were measured after in vitro stimulation with each of the three Esx vaccine dimers or EsxB-EsxA. In H65-vaccinated animals, the three Esx vaccine dimers were recognized by T cells with frequencies ranging from 0.19% to 0.64%; none of these were recognized in the adjuvant control group (Fig. 3C). The broad vaccine-induced immune response resulted in a statistically significant (P < 0.01) reduction of mycobacteria (0.66 log10 ± 0.15) 6 wk after aerosol infection with virulent M.tb compared with the adjuvant control group, a level equivalent to the reduction obtained with BCG (0.70 log10 ± 0.09) (Fig. 3D).
Polyfunctionality of T Cells.
Week 6 postinfection, cells were isolated from perfused lungs and stimulated in vitro with either of the three vaccine-containing ESX dimers or EsxB-EsxA to determine the frequency of cytokine-producing Esx dimer-specific CD4+ T cells (Fig. 4A). In both the H65-vaccinated and adjuvant control groups, there was a strong recruitment of cytokine-producing Esx dimer-specific CD4+ T cells to the lungs. The strong recruitment supports the theory that all four dimers were expressed during infection and confirmed their immunogenicity. In addition to responses against the vaccine proteins EsxD-EsxC, EsxG-EsxH, and EsxW-EsxV, the infection also induced a strong response against EsxB-EsxA, particularly in the CAF01 vaccinated group (Fig. 4A). In a more detailed analysis, we compared the differentiation status of the Esx-specific CD4+ T cells not only between but also within the vaccination groups. In the infected adjuvant control animals, we found primarily IFN-γ-producing CD4+ T cells that were of an effector phenotype (IFN-γ+, TNF-α+, or IFN-γ+) or, to a minor degree, memory T cells (IL-2+, TNF+, IFN-γ+), regardless of their Esx protein specificity (Fig. 4B). However, in H65-vaccinated/infected animals, the T-cell functionality depended on whether the cells were vaccine-specific or not. The functionality of the nonvaccine-specific T cells (recognizing EsxB-EsxA) were, despite the lower CFU counts in these animals, practically identical to the effector and memory T-cell populations found in the infected adjuvant control animals. In contrast, the vaccine-specific CD4+ T-cell population was more diverse in terms of functionality. It consisted preferentially of memory cells (IL-2+, TNF+, IFN-γ+), a solid frequency of effector cells (IFN-γ+, TNF-α+, or IFN-γ+), and a smaller but distinct fraction of less-differentiated CD4+ T cells that were IL-2+, TNF+ double-positive or IL-2+/TNF+ single-positive.
Fig. 4.
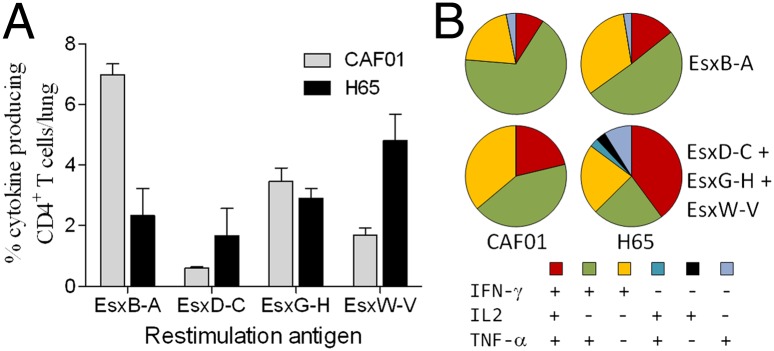
Vaccine-primed T cells maintains a memory phenotype. (A) The frequency of dimer-specific CD4+ T cells producing IFN-γ, TNF-α, or IL-2 after in vitro stimulation with dimer substrates. Cells were isolated from perfused lungs 6 wk after M.tb (strain Erdman) infection from B6C3F1 mice immunized with CAF01 adjuvant alone or in combination with H65 (n = 3 per group). (B) The polyfunctionality of H65 vaccine epitopes (EsxD-EsxC + EsxG-EsxH + EsxW-EsxV) was compared with nonvaccine epitopes (EsxB-EsxA) in infected (labeled CAF01) and H65 vaccinated/infected groups of animals (labeled H65) by multicolor flow cytometry. Only cytokine expression combinations with frequencies above 0.05% were included.
The H65 Fusion Protein Has Broad Human Coverage.
Despite proven success in animal studies, the large host genetic variability on a global scale could compromise the potential efficacy of the vaccine in humans. We therefore used in silico epitope-binding predictions to estimate the population coverage of the H65 vaccine and the individual protein components in countries with a high burden of TB. Because CD4+ T-cell-mediated immunity is essential to combat M.tb infection and class 2 HLA proteins are responsible for stimulating CD4+ T cells, we examined the class 2 HLA-DRB1 diversity (22).The DRB1 allele was chosen because DR alleles bind the vast majority of known M.tb epitopes (23), and among the DR alleles, the DRB1 surface expression is five times greater than for other DR alleles (24). Finally, epitope prediction programs for DRB1 alleles are more frequently available than for other class 2 HLA alleles. Binding predictions for epitopes in the H65 fusion protein were generated for 34 HLA-DBR1 alleles, representing the three most common HLA-DRB1 alleles in the 22 countries that, according to the World Health Organization, have the highest burden of TB (25).
The predictions were generated using four methods: CombLib, SMM-align, NN-align, and/or NetMHCIIpan. Wherever possible, we used a consensus method approach based on two or three of the methods, but for many of the alleles, only the NetMHCIIpan method was available (Table S3). The median number of vaccine epitopes predicted to bind any of DRB1 allele ranged from 7 to 67 for the H65 fusion protein and from 0 to 14 for the individual proteins, clearly supporting the importance of including multiple proteins in vaccines. Among the individual proteins, the highly expressed, ESX-3-secreted EsxG holds the largest total number of predicted human CD4 T-cell epitopes for the 34 alleles, followed by its secretion partner EsxH and the much lower expressed EsxD from the ESX-2 system. The other substrate for the ESX-2 system, EsxC, holds the lowest number of predicted epitopes among the single proteins, even less than what was predicted for the two 20-amino acid long linkers combined. The combination of low expression and relatively few human epitopes may suggest that in a potential future improvement of the H65 vaccine, EsxC could be replaced with another Ag.
Discussion
The mycosyltransferases Ag85A and Ag85B are frequently used Ags in TB vaccines currently in clinical trials (3). Both are primarily expressed during the initial stage of infection, where M.tb divides rapidly and thus has a need for synthesis and assembly of cell wall components. As the growth rate decreases, there is less need for mycolyl synthesis and the expression of Ag85 proteins is reduced to a low level. Thus, this study is a step toward identifying vaccine targets that have a more constitutive expression profile and then testing the protective efficacy of a TB vaccine on the basis of a multiple of these targets.
The reported density of B- and T-cell epitopes in mycobacterial proteins is at least five to six times higher for extracellular proteins than for cytoplasmic, membrane, or cell wall proteins (23). It is therefore logical to look for new vaccine targets among the secreted proteins. Protein secretion is essential for all bacteria to interact with their environment, and mycobacteria have, during coevolution with their host cell, acquired specialized protein secretory pathways that deliver effector proteins to the host cell. These include the ESAT-6 secretion system (ESX) that secretes proteins lacking a classical signal sequence (26). The genome of M.tb encodes five ESX regions (ESX-1 to ESX-5) arranged in conserved clusters. The ESX systems secrete high quantities of proteins with different biological functions, and as we know several of the ESX substrates are strong vaccine Ags, we speculate that a combination of substrates from different ESX systems could generate an efficient vaccine.
On the basis of phylogenetic relationships, we identified the most likely Esx dimer substrate for each of the five ESX systems. From our protection and recognition studies using the Esx dimer substrates, it is clear that the substrates for four of the ESX systems are protective. Only the ESX-4 substrate EsxU-EsxT had no protective efficacy against a M.tb challenge, nor was it recognized during infection. This could be because of the use of inbred mice strains and lack of T-cell epitopes, but vaccination with EsxU-EsxT did induce strong T-cell responses, suggesting that there are indeed potential T-cell epitopes represented in this protein. Gene expression studies up until 20 wk of infection showed no expression of the esxT or esxU genes, suggesting that the absence of protection 6 wk after infection is a result of the lack of expression of EsxT and EsxU. The ESX-2 substrate EsxD-EsxC is constitutively expressed at a low but seemingly sufficient level for protection against M.tb, even though there was no detectable T-cell response in the lungs after 6 wk of infection. EsxD and EsxC have neither been identified in the culture supernatants of in vitro cultures nor do they contain the ESX signature tag, and it is therefore questionable whether the proteins are actually being secreted by the ESX-2 system.
Within the first 3 wk of infection, esxA/B and esxG/H were the strongest expressed esx genes. However, as a consequence of the encoded proteins’ biological functions, the relative expression of the esxA/B and esxG/H genes changes radically during this period. The ESX-1 substrates EsxB and EsxA are virulence factors that are used for escaping the phagosome and possibly facilitating cell-to-cell spread during the stages of infection in which M.tb is multiplying (e.g., during the early infection or reactivation from latency). Both EsxB and EsxA are strongly recognized in infected individuals and are therefore exploited in TB diagnostic tests. The ESX-3 substrates EsxG and EsxH are described to be involved in the acquisition of divalent metal ions such as iron and zinc (27). When grown in vitro in rich medium, there are plenty of metal ions available in an accessible form. Thus, there is little need for these proteins, and the gene expression is relatively low. Under normal physiological conditions, the concentration of free iron in body fluids is very low (10−18 M). As a consequence, intracellular mycobacteria are competing with the host for iron and zinc and to sequester metals for survival M.tb strongly up-regulates the expression of EsxG and EsxH. In M.tb-infected guinea pigs, iron has been shown to accumulate within the primary lesions, but most of it accumulates as extracellular ferric iron, and it is unknown whether M.tb can exploit this source (28). ESX-5 is the most recently evolved ESX cluster and is only present in the group of slow-growing pathogenic species of mycobacteria (29). ESX-5 is responsible for the secretion of several proteins involved in virulence/pathogenicity (26) and is a major modulator of the host immune response and a key virulence determinant of M.tb. The inactivation of ESX-5 results in severe attenuation of the mutant strains, which are unable to replicate even in immune-deficient mice (30).
To target a broad range of relevant epitopes and maximize the likelihood of one or more of the vaccine targets being expressed at any time during an infection, we included the ESX-2, ESX-3, and ESX-5 substrates (EsxC, EsxD, EsxG, EsxH, EsxV, and EsxW) in a six-protein fusion (H65) and evaluated its protective efficacy in two different inbred mice strains. Although fulfilling all criteria, EsxA and EsxB were excluded because of their common use as Ags in diagnostic tests. In CB6F1 mice, H65 vaccination induced an EsxG-EsxH- and EsxW-EsxV-specific immune response but did not protect to the same degree as Mycobacterium bovis BCG 6 wk after infection. Reflecting the reduced bacteria load, the overall in vivo IFN-γ level was significantly reduced in lungs of H65-vaccinated animals. Reduction of T-cell activation is likely to extend the time to exhaustion, a problem associated with chronic pathogens such as M.tb (31). In B6C3F1 mice, there is a broader vaccine specific immune response, and the protective efficacy of H65 vaccination was comparable to the protection observed with BCG. The H65 vaccinated and adjuvant control animals from this study were used for multiparameter flow-cytometry analysis to compare the quality of the T-cell response at the site of infection.
This analysis demonstrates that the CD4 T cells maintained at the site of infection in H65-vaccinated animals has a larger fraction of polyfunctional CD4 T cells compared with the T cells that infiltrate the lungs of nonvaccinated animals. It has previously been shown that in mice vaccinated with BCG, the T cells are primarily effector or effector-memory T cells 6 mo after vaccination (32). In contrast we have shown that 1 y after vaccination with a fusion protein (H1) formulated in the same adjuvant used in this study, the T cells are primarily central memory T cells (33). In HIV-infected individuals, polyfunctional CD4 T cells are a characteristic feature observed in HIV controllers, and an inverse correlation has been shown with viral load, whereas noncontrollers elicit responses dominated by IFN-γ single-positive CD4 T cells (34). However, in this study, it was not clear whether the high degree of polyfunctional CD4 T cells caused improved control of HIV or whether the improved quality of the T-cell response was an effect of a lower viral load. We have the possibility to address this question by simultaneously monitoring the quality of the T-cell responses to Ags that are present in the H65 vaccine and comparing these to responses to Ags that are absent from the vaccine but promoted by the infection. EsxB-EsxA serves as a marker of a purely infection-promoted response, whereas the Esx molecules in the H65 vaccine are markers for a vaccine-primed and infection-expanded response. In H65-vaccinated animals, there is a striking difference in the quality of the EsxB-EsxA- and vaccine-specific T cells at the site of infection that is not seen in nonvaccinated animals. Because the quality of the EsxB-EsxA-specific response in the H65-vaccinated animals is identical to the EsxA-EsxB response in the adjuvant control group, it demonstrates that the polyfunctional quality of the response is maintained selectively for the vaccine-promoted part of the mycobacteria-specific response. Thus, T-cell polyfunctionality is not a consequence of efficient bacterial containment but, rather, a vaccine-related phenomenon. In mice, vaccine-induced multifunctional CD4 T cells have been shown to be superior to their single-positive counterparts in terms of protection against HIV, M.tb, and Leishmania major infections (34–36). The improved effector/memory CD4 T-cell balance in H65-vaccinated animals may have implications for their long-term ability to suppress the infection; this is currently under investigation (37).
Although successful in mice, we did find variations in the protective efficacy of H65 in the two mice strains tested. This underlines the importance of the genetic background and that there is no guarantee that the protective efficacy for any vaccine tested in animals is transferable to humans, given the large genetic variability worldwide. From the literature, it is clear that the generation of a strong CD4+ T-cell response is crucial for protection against M.tb (13). We therefore assessed the potential effect of host genetic diversity on the protective coverage of the H65 vaccine, using open-source epitope binding prediction programs evaluating the binding of vaccine epitopes to class 2 HLA (DRB1) alleles. Binding prediction is a cost-effective method for assessment of TB vaccine candidates that can give information regarding a worldwide use that could not be obtained even from several clinical trials. We focused on epitope-binding predictions for high-frequency HLA alleles among TB high-burden populations, as these are the groups in which a vaccine is most needed. The H65 vaccine contains many predicted epitopes for the tested alleles and is therefore likely to be broadly recognized in the populations with the highest burden of TB. The lowest number of predicted H65 vaccine epitopes was seven for the alleles tested, which is clearly above the cutoff of four epitopes that has previously been used to identify alleles of concern (38). The presence of human T-cell epitopes in a TB vaccine is a prerequisite for human recognition, but they also represent an interesting caveat. Comparative analyses of 21 M.tb genomes revealed that the known human T-cell epitopes, including epitopes in Ag85, CFP10, and ESAT-6, are evolutionary hyperconserved (39). It has been speculated that the bacteria might benefit from a strong T-cell response because it could lead to necrosis, cavitation, and escape in humans (40). The implications for vaccine design are not clear, but the presence of conserved and highly immunodominant human T-cell epitopes in vaccines deserves further attention. Among the H65 Ags, only EsxG and EsxH epitopes were included in the comparative study, and EsxH was specifically shown to harbor a relatively high number of amino acid substitutions across the strains tested and was therefore not associated with this potential problem.
To summarize, our study presents a recombinant subunit vaccine, based on six secreted M.tb Ags not including the popular Ag85A/B, ESAT-6, or CFP10. The H65 vaccine is capable of protecting against infection with M.tb at a level comparable with M. bovis BCG and can be used without interfering with ESAT-6- and CFP10-based diagnostics. HLA allele binding predictions suggest that it contains ample CD4 epitopes for worldwide coverage.
Materials and Methods
Recombinant Proteins.
The five heterodimers and the H65 fusion were recombinantly expressed in E. coli BL21 AI and purified from inclusion bodies by a three-step process.
Animals, Immunizations, and Infection.
Groups of 6–8-wk-old female mice were immunized three times s.c. on days 1, 14, and 28, with CAF01 emulsified with 5 µg protein to a final volume of 200 μL. Ten weeks after the first immunization, the animals were challenged with ∼100 CFU M.tb strain Erdman per mouse.
CFU Measurements.
Lung homogenates of individual mice were plated as threefold serial dilutions on Middlebrook 7H11 Bacto agar and enumerated after 3 wk incubation at 37 °C.
Cytokine Secretion Assays.
Peripheral blood mononuclear cells, splenocytes, or lung mononuclear cells (2 × 105 per well) were cultured in the presence of 2 µg/mL Ag at 37 °C for 72 h, after which cytokine secretion was tested using ELISA.
Flow Cytometry.
Splenocytes or lung mononuclear cells (2 × 106 cells per well) were stimulated in vitro in the presence of recombinant Ag (2 µg/mL) for 1 h, and subsequently incubated for 5–6 h in the presence of 10 µg/mL brefeldin A. After overnight storage at 4 °C, cells were stained with antibodies against CD4, CD44, IFN-γ, TNF-α, or IL-2 and analyzed, using a flow cytometer.
Expression of M.tb Genes.
We determined the gene expression profile of 13 M.tb genes in in vitro cultures and during in vivo infection by real-time PCR, using total RNA isolated from lungs of individual mice.
Epitope Binding Predictions.
MHC-II binding predictions were done using IEDB Analysis Resources server for 34 HLA-DBR1 alleles. Binding affinities of IC50 values less than 500 nM were selected as cutoff.
For more details on material methods see SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Vivi Andersen, Merete Henriksen, and Linda Christensen for excellent technical assistance. The research leading to these results has received funding from the European Union's Seventh Framework Programme (FP7/2007-2013) under Grant Agreement 280873 ADITEC and Grant Agreement 241745 NEWTBVAC.
Footnotes
Conflict of interest statement: C.A. and P.A. are coinventors on a patent application covering the use of H65 as a vaccine. All rights have been assigned to Statens Serum Institut, a Danish nonprofit governmental institute.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1314973111/-/DCSupplemental.
References
- 1. World Health Organization (2013) Global Tuberculosis Report 2013 (World Health Organization, Geneva). Available at www.who.int/tb/publications/global_report/en/. Accessed December 17, 2013.
- 2.Rodrigues LC, Mangtani P, Abubakar I. How does the level of BCG vaccine protection against tuberculosis fall over time? BMJ. 2011;343:d5974. doi: 10.1136/bmj.d5974. [DOI] [PubMed] [Google Scholar]
- 3.Kaufmann SH. Tuberculosis vaccine development: Strength lies in tenacity. Trends Immunol. 2012;33(7):373–379. doi: 10.1016/j.it.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Rowland R, McShane H. Tuberculosis vaccines in clinical trials. Expert Rev Vaccines. 2011;10(5):645–658. doi: 10.1586/erv.11.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogerson BJ, et al. Expression levels of Mycobacterium tuberculosis antigen-encoding genes versus production levels of antigen-specific T cells during stationary level lung infection in mice. Immunology. 2006;118(2):195–201. doi: 10.1111/j.1365-2567.2006.02355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bold TD, Banaei N, Wolf AJ, Ernst JD. Suboptimal activation of antigen-specific CD4+ effector cells enables persistence of M. tuberculosis in vivo. PLoS Pathog. 2011;7(5):e1002063. doi: 10.1371/journal.ppat.1002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egen JG, et al. Intravital imaging reveals limited antigen presentation and T cell effector function in mycobacterial granulomas. Immunity. 2011;34(5):807–819. doi: 10.1016/j.immuni.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kassa D, et al. Analysis of immune responses against a wide range of Mycobacterium tuberculosis antigens in patients with active pulmonary tuberculosis. Clin Vaccine Immunol. 2012;19(12):1907–1915. doi: 10.1128/CVI.00482-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andersen P, Andersen AB, Sørensen AL, Nagai S. Recall of long-lived immunity to Mycobacterium tuberculosis infection in mice. J Immunol. 1995;154(7):3359–3372. [PubMed] [Google Scholar]
- 10.Elhay MJ, Oettinger T, Andersen P. Delayed-type hypersensitivity responses to ESAT-6 and MPT64 from Mycobacterium tuberculosis in the guinea pig. Infect Immun. 1998;66(7):3454–3456. doi: 10.1128/iai.66.7.3454-3456.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pollock JM, Andersen P. Predominant recognition of the ESAT-6 protein in the first phase of interferon with Mycobacterium bovis in cattle. Infect Immun. 1997;65(7):2587–2592. doi: 10.1128/iai.65.7.2587-2592.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ravn P, et al. Human T cell responses to the ESAT-6 antigen from Mycobacterium tuberculosis. J Infect Dis. 1999;179(3):637–645. doi: 10.1086/314640. [DOI] [PubMed] [Google Scholar]
- 13.Kaufmann SH, Hussey G, Lambert PH. New vaccines for tuberculosis. Lancet. 2010;375(9731):2110–2119. doi: 10.1016/S0140-6736(10)60393-5. [DOI] [PubMed] [Google Scholar]
- 14.Uplekar S, Heym B, Friocourt V, Rougemont J, Cole ST. Comparative genomics of Esx genes from clinical isolates of Mycobacterium tuberculosis provides evidence for gene conversion and epitope variation. Infect Immun. 2011;79(10):4042–4049. doi: 10.1128/IAI.05344-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertholet S, et al. Identification of human T cell antigens for the development of vaccines against Mycobacterium tuberculosis. J Immunol. 2008;181(11):7948–7957. doi: 10.4049/jimmunol.181.11.7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertholet S, et al. A defined tuberculosis vaccine candidate boosts BCG and protects against multidrug-resistant Mycobacterium tuberculosis. Sci Transl Med. 2010;2(53):53ra74. doi: 10.1126/scitranslmed.3001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdallah AM, et al. Type VII secretion—mycobacteria show the way. Nat Rev Microbiol. 2007;5(11):883–891. doi: 10.1038/nrmicro1773. [DOI] [PubMed] [Google Scholar]
- 18.Fortune SM, et al. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc Natl Acad Sci USA. 2005;102(30):10676–10681. doi: 10.1073/pnas.0504922102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daleke MH, et al. Specific chaperones for the type VII protein secretion pathway. J Biol Chem. 2012;287(38):31939–31947. doi: 10.1074/jbc.M112.397596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skjøt RL, et al. Comparative evaluation of low-molecular-mass proteins from Mycobacterium tuberculosis identifies members of the ESAT-6 family as immunodominant T-cell antigens. Infect Immun. 2000;68(1):214–220. doi: 10.1128/iai.68.1.214-220.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gideon HP, et al. Hypoxia induces an immunodominant target of tuberculosis specific T cells absent from common BCG vaccines. PLoS Pathog. 2010;6(12):e1001237. doi: 10.1371/journal.ppat.1001237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoft DF. Tuberculosis vaccine development: Goals, immunological design, and evaluation. Lancet. 2008;372(9633):164–175. doi: 10.1016/S0140-6736(08)61036-3. [DOI] [PubMed] [Google Scholar]
- 23.Ali H, Zeynudin A, Mekonnen A, Abera S, Ali S. Smear Posetive Pulmonary Tuberculosis (PTB) Prevalence Amongst Patients at Agaro Teaching Health Center, South West Ethiopia. Ethiop J Health Sci. 2012;22(1):71–76. [PMC free article] [PubMed] [Google Scholar]
- 24.Batra S, et al. Childhood tuberculosis in household contacts of newly diagnosed TB patients. PLoS ONE. 2012;7(7):e40880. doi: 10.1371/journal.pone.0040880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banerji D. The World Health Organization and public health research and practice in tuberculosis in India. Int J Health Serv. 2012;42(2):341–357. doi: 10.2190/HS.42.2.k. [DOI] [PubMed] [Google Scholar]
- 26.Daku M, Gibbs A, Heymann J. Representations of MDR and XDR-TB in South African newspapers. Soc Sci Med. 2012;75(2):410–418. doi: 10.1016/j.socscimed.2012.02.039. [DOI] [PubMed] [Google Scholar]
- 27.Ilghari D, et al. Solution structure of the Mycobacterium tuberculosis EsxG·EsxH complex: Functional implications and comparisons with other M. tuberculosis Esx family complexes. J Biol Chem. 2011;286(34):29993–30002. doi: 10.1074/jbc.M111.248732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Basaraba RJ, et al. Increased expression of host iron-binding proteins precedes iron accumulation and calcification of primary lung lesions in experimental tuberculosis in the guinea pig. Tuberculosis (Edinb) 2008;88(1):69–79. doi: 10.1016/j.tube.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raviglione M, et al. Scaling up interventions to achieve global tuberculosis control: Progress and new developments. Lancet. 2012;379(9829):1902–1913. doi: 10.1016/S0140-6736(12)60727-2. [DOI] [PubMed] [Google Scholar]
- 30.Millet JP, et al. Factors that influence current tuberculosis epidemiology. Eur Spine J. 2013;22(Suppl 4):539–548. doi: 10.1007/s00586-012-2334-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reiley WW, et al. Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proc Natl Acad Sci USA. 2010;107(45):19408–19413. doi: 10.1073/pnas.1006298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henao-Tamayo MI, et al. Phenotypic definition of effector and memory T-lymphocyte subsets in mice chronically infected with Mycobacterium tuberculosis. Clin Vaccine Immunol. 2010;17(4):618–625. doi: 10.1128/CVI.00368-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindenstrøm T, et al. Tuberculosis subunit vaccination provides long-term protective immunity characterized by multifunctional CD4 memory T cells. J Immunol. 2009;182(12):8047–8055. doi: 10.4049/jimmunol.0801592. [DOI] [PubMed] [Google Scholar]
- 34.Kannanganat S, Ibegbu C, Chennareddi L, Robinson HL, Amara RR. Multiple-cytokine-producing antiviral CD4 T cells are functionally superior to single-cytokine-producing cells. J Virol. 2007;81(16):8468–8476. doi: 10.1128/JVI.00228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beveridge NE, et al. Immunisation with BCG and recombinant MVA85A induces long-lasting, polyfunctional Mycobacterium tuberculosis-specific CD4+ memory T lymphocyte populations. Eur J Immunol. 2007;37(11):3089–3100. doi: 10.1002/eji.200737504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Darrah PA, et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med. 2007;13(7):843–850. doi: 10.1038/nm1592. [DOI] [PubMed] [Google Scholar]
- 37.Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: Implications for vaccine design. Nat Rev Immunol. 2008;8(4):247–258. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- 38.Davila J, McNamara LA, Yang Z. Comparison of the predicted population coverage of tuberculosis vaccine candidates Ag85B-ESAT-6, Ag85B-TB10.4, and Mtb72f via a bioinformatics approach. PLoS ONE. 2012;7(7):e40882. doi: 10.1371/journal.pone.0040882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Comas I, et al. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet. 2010;42(6):498–503. doi: 10.1038/ng.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orme IM. 2013. A new unifying theory of the pathogenesis of tuberculosis. Tuberculosis (Edinb), 10.1016/j.tube.2013.07.004.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.