

Significance
Metal thiolates are common components of electron transfer proteins, enzymes, and redox sensors. Reduction of ferric thiolate hemes often involves protonation of the Cys ligand and ligand loss or substitution. These reactions contribute to enzyme inactivation and form the basis of sensing functions, but their mechanistic details are not well understood, particularly in proteins. A neutral thiol is a viable ligand to the heme; however, analysis of this interaction has been challenging. Herein, we describe a series of model proteins that offer detailed spectroscopic and thermodynamic characterization of ferric thiolate and ferrous thiol species as well as their interconversions. Results have shed light on the forces that control a particular heme ligation and redox reactivity of these key bioinorganic centers.
Keywords: metalloenzyme, folding, electron transfer
Abstract
Cysteine-bound hemes are key components of many enzymes and biological sensors. Protonation (deprotonation) of the Cys ligand often accompanies redox transformations of these centers. To characterize these phenomena, we have engineered a series of Thr78Cys/Lys79Gly/Met80X mutants of yeast cytochrome c (cyt c) in which Cys78 becomes one of the axial ligands to the heme. At neutral pH, the protonation state of the coordinated Cys differs for the ferric and ferrous heme species, with Cys binding as a thiolate and a thiol, respectively. Analysis of redox-dependent stability and alkaline transitions of these model proteins, as well as comparisons to Cys binding studies with the minimalist heme peptide microperoxidase-8, demonstrate that the protein scaffold and solvent interactions play important roles in stabilizing a particular Cys–heme coordination. The increased stability of ferric thiolate compared with ferrous thiol arises mainly from entropic factors. This robust cyt c model system provides access to all four forms of Cys-bound heme, including the ferric thiol. Protein motions control the rates of heme redox reactions, and these effects are amplified at low pH, where the proteins are less stable. Thermodynamic signatures and redox reactivity of the model Cys-bound hemes highlight the critical role of the protein scaffold and its dynamics in modulating redox-linked transitions between thiols and thiolates.
Iron–cysteine bonds are common in biological systems, especially in heme enzymes and sensors. P450 enzymes, with a negatively charged Cys thiolate coordinated to the heme, catalyze diverse oxidation reactions and are important targets both for therapeutic intervention and industrial catalysis (1–3). Heme–thiolate nitric oxide synthase (NOS) (4, 5) and cystathionine β-synthase (CBS) (6) are responsible for the formation of the signaling molecule nitric oxide and detoxification of homocysteine, respectively. Two other heme-thiolate enzymes, chloroperoxidase (7) and the SoxAX complex (8) play important roles in the synthesis of halogenated compounds and oxidation of thiosulfate and sulfide, respectively. Thiolate-bound hemes are also found in many sensor proteins (9), including those that regulate circadian rhythms in mammals (10).
The protonation state of the coordinated Cys is critical for the catalytic function of these enzymes and ligand lability of the sensors. The deprotonated Cys provides a strong thiolate “push” that enables heterolytic O—O bond cleavage by P450 enzymes (1). Protonation of the native thiolate ligand to a neutral thiol has been suggested as a mechanism of P450 deactivation yielding the infamous P420 species (11). The ferrous thiol is easily displaced by other ligands resulting in functional conformational changes in sensors (9, 12) but also, upon exposure to dioxygen (13), in deleterious effects of Cys oxidation.
Protonation (or deprotonation) reactions of a Cys ligand often accompany redox transformations of the Cys-bound hemes. Heme reduction increases the electron density on the iron increasing the effective pKa of the coordinated thiol. Although ferrous thiol coordination is frequently lost, model studies have indicated that neutral Cys is a viable ligand to the ferrous heme (12). For ferric hemes, where the metalloporphyrin dianion unit has a core charge of +1, a thiolate is the preferred ligand. Only a few examples of thiol-ligated ferric hemes are currently known (14–16), all with highly electron-rich systems.
Understanding redox-dependent stability and protonation of Cys-bound hemes is critical for establishing mechanistic principles of these redox centers. With native systems, their evolved function often limits the number of easily observed species. Small synthetic models (14, 17) are useful for detailed thermodynamic and kinetic investigations but may not capture all of the complexity of the protein framework. Finally, reduction of the heme iron and degradation of the porphyrin by thiyl radicals complicate studies at high concentration of thiols (18, 19).
In the present study, we have engineered a series of Thr78Cys/Lys79Gly/Met80X (X = Leu, Ile, or Phe) mutants of yeast cytochrome c (cyt c) in which the original Met80 ligand was mutated to noncoordinating residues and Cys78 becomes one of the axial ligands to the heme (Fig. 1). The strategic placement of a coordinating Cys in the hydrophobic interior of this protein has yielded a robust system for examining redox-dependent stability and interconversions of Cys-bound hemes. This model system allows a detailed thermodynamic characterization of ferric thiolate and ferrous thiol species and also provides access to kinetic intermediates. The results demonstrate the importance of the polypeptide scaffold for redox-dependent stability of Cys-bound heme proteins and highlight the role of protein motions in their redox reactions.
Fig. 1.

(A) Structure of yeast iso-1 cyt c (2YCC) (54) showing positions of mutated residues. (B) EPR spectra at 10 K of ferric Thr78Cys/Lys79Gly/Met80X variants of yeast iso-1 cyt c and Met80Cys variant of horse heart cyt c in a 100 mM sodium phosphate buffer at pH 7.4.
Results
Spectroscopic Characteristics of the Designed Protein Variants.
The electronic absorption (Fig. 2A and Table S1) and magnetic circular dichroism (MCD) spectra (Fig. 2A) of the ferric proteins revealed features of thiolate ligation (20, 21). The Soret band shifts to 418 nm compared with its position at 409 nm in the Met-bound wild-type WT* cyt c (the reference protein with two background mutations Lys72Ala and Cys102Ser, to prevent coordination to the heme of Lys72, the residue that is trimethylated when cyt c is isolated from yeast and is not trimethylated when cyt c is expressed in E. coli (22), and formation of cyt c dimers, respectively). The spectra also show a red-shifted Q band (538 nm) and a set of two weak charge transfer bands (Fig. 2A and Table S1). In the MCD spectra, the peak and trough in the Soret region are less intense and red-shifted compared with these features for the wild-type protein. In the visible region, the MCD trough corresponding to the Q bands is also dramatically red-shifted. The two low-energy bands in the 600- to 750-nm region (Table S1) are very similar in their positions to the thiolate-to-Fe(III) charge transfer bands of the imidazole adduct of P450 (23).
Fig. 2.

Electronic absorption (Upper) and MCD (Lower) spectra of (A) ferric and (B) ferrous WT* and the Thr78Cys/Lys79Gly/Met80Leu variant of yeast iso-1 cyt c in a 50 mM Tris buffer at pH 7.4 and 4 °C.
The 10 K EPR spectra of all of the ferric variants (Fig. 1 and Table S1) show low-spin signals with a narrow spread of the g values, typical of thiolate-ligated hemes. These signatures have been attributed to reduction in the tetragonality of the complex and/or strong delocalization of the spin density onto the thiolate ligand (18). Interestingly, the gz values of the Cys78 variants are shifted to higher field, compared with that of horse heart Met80Cys, suggesting a difference in the axial position of the thiolate ligand.
For the Phe mutant, two overlapping spectra (gz = 2.42 and gz = 2.51) are observed, indicating the presence of two ferric components. This heterogeneity did not disappear with repeated preparations of the mutant protein, suggesting that it is an intrinsic feature of the variant. The spectra vary with pH: the population of the species with gz = 2.42 increases at higher pH (Fig. S1). Similar sets of two overlapping EPR signals and their pH dependence have been described for other thiolate-ligated heme proteins (8, 24, 25). It has been hypothesized that they arise from variations in the protonation or hydrogen bonding of the thiolate or from two different orientations of this ligand (15, 25). The EPR spectra of the Leu and Ile variants show a single low-spin ferric signal (gz = 2.52 and gz = 2.51, respectively), which does not change with pH. A small population of the high-spin signal is also apparent in the spectra (Fig. 1B and Fig. S1), likely from the H2O-ligated species formed upon opening of the heme crevice.
The electronic absorption and MCD spectra of the ferrous variants (Fig. 2B) are very similar to those of the wild-type protein, consistent with a neutral coordinated ligand rather than an anionic thiolate. The spectra do not feature the red-shifted Soret band typical of ferrous six-coordinate thiolate complexes (26), suggesting that the thiolate is either protonated or replaced by another ligand upon heme reduction. Removal of Lys79 eliminates this residue from obvious candidates for ligand substitution. As we will show below, substitution by another Lys is possible in these variants but requires more basic conditions, pointing to the thiol as the ligand to the ferrous heme at neutral and acidic pH. Thioether Met-ligated WT* cyt c is a good model for thiol coordination in the ferrous proteins (Fig. 2), with only subtle differences apparent in the α-band region.
Redox Potentials and Redox-Dependent Stability of the Cys78 Proteins.
The reduction potentials of the three variants were determined by spectroelectrochemistry, with the heme iron midpoint potentials given in Table 1. The highly negative potentials reflect unfavorable reduction of the ferric species and are consistent with data on other thiolate-ligated heme proteins (4, 21).
Table 1.
Reduction potentials, pKa values for alkaline transitions, and thermodynamic parameters from thermal denaturation experiments for wild-type WT*, Met80Ala, and Thr78Cys/Lys79Gly/Met80X variants of yeast iso-1 cyt c
| Variant | E°m, mVa | Ferric |
Ferrous |
||||||
| pKaalk | ΔCp, kcal/mol Kb | Tm, Kc | ΔHD,vH, kcal/molc | pKaalk | ΔCp, kcal/mol Kb | Tm, Kc | ΔHD,vH, kcal/molc | ||
| WT*d | 290e | 8.8 ± 0.1f | 1.4 ± 0.1g | 327.7 ± 0.4 | 81.6 ± 2 | 11.9 ± 0.2 | 1.5 ± 0.1g | 355.3 ± 0.5 | 120.2 ± 2 |
| Met80Ala | 185h | 5.9 ± 0.2i | n.d.j | 324.3 ± 0.4 | 60.7 ± 0.7 | 9.4 ± 0.1k | n.d.j | 331.3 ± 0.6 | 71 ± 2 |
| X = Leu | −340 | 8.1 ± 0.2 | 1.1 ± 0.2 | 337.5 ± 0.3 | 51.9 ± 0.7 | 9.9 ± 0.1 | 1.3 ± 0.1 | 315.8 ± 0.4 | 42.2 ± 0.4 |
| X = Ile | −340 | 8.1 ± 0.1 | 1.0 ± 0.1 | 337.1 ± 0.5 | 51.2 ± 0.8 | 9.8 ± 0.2 | 1.3 ± 0.2 | 315.3 ± 0.6 | 41.7 ± 0.8 |
| X = Phe | −390 | 8.4 ± 0.2 | 1.1 ± 0.1 | 338.7 ± 0.3 | 53.1 ± 0.9 | 10.4 ± 0.2 | 1.4 ± 0.1 | 316.2 ± 0.3 | 43.0 ± 0.7 |
At pH 7.4, the differences in the free energy of unfolding between ferrous and ferric Cys78 variants (ΔΔGD, II-III) calculated using these potentials (Eq. S4) are 4.4 (X = Leu and Ile) and 5.5 (X = Phe) kcal/mol.
Determined from analysis of the ΔHD,vH dependence on Tm; the pH range is from 3.5 to 5.5.
In a sodium acetate buffer at pH 5.0; results at pH 7.4 are presented in Table S2.
Contains two background mutations, Lys72Ala and Cys102Ser, to prevent Lys72 coordination to the heme and formation of cyt c dimers, respectively.
For the protein with trimethylated Lys72 and Cys102Thr; from ref. 33.
For the Met80 to Lys79 (or Lys73) transition.
From ref. 26, for the protein with trimethylated Lys72 and Cys102Thr.
For semisynthetic horse heart Met80Ala; from ref. 50.
For the H2O to OH– transition.
Not determined.
From ref. 35.
Thermal denaturation of the engineered variants (Table 1) was examined with circular dichroism (CD) spectroscopy. Similar to wild-type cyt c (27), thermal stabilities of both ferric and ferrous proteins increase with increases in pH. The heat capacities of denaturation, ΔCp, were determined by the analysis of perturbations in the ΔHD,vH (the van’t Hoff enthalpy) and Tm (midpoint of unfolding transition) values with pH. For either ferric or ferrous proteins, the ΔCp values are very similar for all three variants, consistent with previous reports of minimal changes in this parameter with point mutations (28). The ΔCp values for the ferric set, however, are slightly lower, compared with the wild-type WT* and other mutant cyt c proteins with preserved Met80 coordination (27, 28), suggesting a decrease in the exposure of hydrophobic surfaces upon denaturation of the Cys78 variants relative to wild type. A more compact denatured state (29, 30) or defects in the polypeptide packing of folded proteins could explain such changes. At micromolar protein concentration and low pH, the formation of intermolecular disulfide adducts of denatured proteins is less likely, but a fraction of the ferric protein does retain the thiolate coordination to the heme, thus forming polypeptide loops (Fig. S2). The change in the polypeptide packing of folded proteins is a reasonable possibility, and the below discussion of alkaline transition and kinetic studies addresses this effect.
Consistent with their quite negative redox potentials (Table 1), the thermal stabilities of the ferric variants are greater than those of the ferrous ones by 4–5 kcal/mol (Table 2). The ferric Cys78 variants are also more stable than WT* cyt c (by ∼3 kcal/mol) pointing to a stabilizing role of the thiolate coordination introduced by the Cys78/Gly79/X80 sequence. Thermodynamic analysis (Table 2) suggests that entropic factors govern the increased stability of the ferric thiolate-ligated proteins compared with their ferrous counterparts or ferric Met-ligated proteins. Calculations at TREF near Tm minimize errors in thermodynamic estimates (28), because the analysis focuses on the temperature range closest to the unfolding transition and requires fewer parameters (one of the ΔCp values cancels out). Analysis at lower pH (pH 5.0, Tables 1 and 2) reduces the complications that arise from His misligation in the denatured state at more basic pH (pH 7.4, Table S2) (31).
Table 2.
Thermodynamic parameters for wild-type WT* and Thr78Cys/Lys79Gly/Met80X variants of yeast iso-1 cyt c from thermal denaturation experiments at pH 5.0
| Variant | Ferric |
Ferrous |
|||||||
| TREF, K | ΔHD, kcal/mol | TΔSD, kcal/mol | ΔGD, kcal/mol | TREF, K | ΔHD, kcal/mol | TΔSD, kcal/mol | ΔGD, kcal/mol | ||
| TREF = Tm(ferric)a | |||||||||
| WT*b | 327.65 | 81.6 ± 2.0 | 81.6 ± 0.6 | 0 | 327.65 | 72.8 ± 2 | 64.4 ± 0.5 | 8.4 ± 0.8 | |
| X = Leu | 337.45 | 51.9 ± 0.8 | 51.9 ± 0.5 | 0 | 337.45 | 72.6 ± 0.8 | 76.5 ± 0.7 | −3.9 ± 0.6 | |
| X = Ile | 337.05 | 51.2 ± 0.9 | 51.2 ± 0.5 | 0 | 337.05 | 72.2 ± 1.1 | 76.2 ± 0.6 | −4.0 ± 0.7 | |
| X = Phe | 338.65 | 53.1 ± 0.9 | 53.1 ± 0.4 | 0 | 338.65 | 75.2 ± 1.2 | 79.4 ± 0.5 | −4.2 ± 0.5 | |
| TREF = Tm(X = Leu)c | |||||||||
| WT*b | 337.45 | 95.3 ± 2.0 | 98.0 ± 0.7 | −2.7 ± 0.7 | 315.75 | 55.0 ± 2.0 | 44.5 ± 0.8 | 10.5 ± 0.8 | |
| X = Leu | 337.45 | 51.9 ± 1.0 | 51.9 ± 0.5 | 0 | 315.75 | 42.2 ± 0.7 | 42.2 ± 0.6 | 0 | |
| X = Ile | 337.45 | 51.6 ± 0.7 | 51.6 ± 0.5 | 0 | 315.75 | 42.4 ± 0.9 | 42.4 ± 0.6 | 0 | |
| X = Phe | 337.45 | 51.8 ± 1.0 | 51.6 ± 0.6 | 0.2 ± 0.5 | 315.75 | 43.1 ± 1.0 | 43.0 ± 0.5 | 0.1 ± 0.5 | |
Calculated using ΔCp values in Table 1.
To evaluate differences between ferric and ferrous species of each variant.
Contains two background mutations, Lys72Ala and Cys102Ser, to prevent Lys72 coordination to the heme and formation of cyt c dimers, respectively.
To allow comparison of stabilities among the variants in their ferric and ferrous oxidation states.
Alkaline Transitions.
An increase in pH alters the electronic spectra of both ferric and ferrous proteins (Fig. S3). Changes in the protonation state of thiol (thiolate)-bound hemes are associated with large shifts in the Soret spectral region (15, 32). Substitution of the ferric thiolate is also easy to identify owing to a characteristic red-shifted Soret band for this species not encountered with other protein ligands. For all three ferric Cys78 variants, the Soret band maximum remains at 418 nm even at pH as high as 13, indicating retention of thiolate ligation. However, the intensity of this band increases at higher pH, yielding a well-resolved transition with a pKa of 8.2 ± 0.1 and a number of protons transferred n = 1.1 ± 0.1. Without changes in the thiolate protonation state, this transition must involve another group (or residue) in close proximity to the heme.
For the ferrous proteins, the protonation state of the coordinated Cys78 thiol also does not change with pH but the ligand appears to be replaced at higher pH by another neutral residue. Under alkaline conditions, cyt c undergoes a conformational transition where Lys residues 72, 73, and 79 substitute the native Met80 ligand at the heme (22, 33, 34). The pKa of this transition in native and mutated cyt c proteins reflects the ligand’s propensity for heme coordination. In the absence of both Lys72 and Lys79 in our variants, the Cys78 thiol is presumably replaced by Lys73. Notably, the pKa values (Table 1) for the ferrous transitions of the Cys78 variants are lower than that of the Met80-coordinated wild-type WT* protein but greater than corresponding values for many of the Met80X cyt c variants with noncoordinating residues X (35). Similar to the results of the thermal stability measurements, the Phe variant has a higher pKa for the ferrous alkaline transition compared with the other two variants.
Photolysis of Ferrous CO Adducts.
All of the ferrous proteins readily form adducts with exogenous CO (Fig. S4), in contrast to Met-bound wild-type cyt c, which does not bind CO. The position of the Soret band in the CO adducts is typical of that of other cyt c variants with preserved His18 ligation and CO bound at the other coordination site (35). The quantum yields for CO photodissociation and rates of CO rebinding are listed in Table S3. The rebinding kinetics of the Leu and Ile variants are monoexponential and depend on the concentration of CO. For the Phe variant, a second CO-independent phase is apparent with a rate constant kobs = (2.0 ± 0.4) × 104 s−1 (Fig. S4C).
For all of the variants, the quantum yields are relatively low (similar to Met80X mutants of cyt c) (35), suggesting that the heme continues to be well surrounded by the polypeptide and there are effective pathways for geminate rebinding, likely occurring on the picosecond timescale. The second-order rate constants for CO rebinding, however, are larger than those of the Met80X mutants and more comparable to that of carboxymethyl cyt c (Cmcyt c, 1.6 × 106 s−1) (36). These findings suggest that access of CO to the heme is facile. Thus, at least for the ferrous proteins with displaced Cys78, the heme crevice is somewhat open.
We tentatively assign the second CO-independent phase in the transients of the Phe variant to rebinding of Cys78 to the heme. Photolysis of the ferrous proteins in the absence of CO did not yield detectable kinetics on the same timescale, suggesting that Cys78 rebinding in this case may happen much faster. However, with the heme pocket likely somewhat perturbed in the ferrous CO adduct (Fig. S4D), Cys rebinding may be slower, particularly if a bulky Phe residue obscures access to the heme.
Model Binding Studies.
To evaluate the energy consequences of Cys ligation to ferric and ferrous hemes, we have examined the binding of N-acetylcysteine (AcCys) to a model heme peptide N-acetylmicroperoxidase-8 (AcMP8). This eight-residue peptide, formed by proteolytic cleavage of cyt c, retains the c-type heme and preserves the His18 coordination; N-terminal acetylation of the peptide inhibits heme aggregation (37). The spectral features of microperoxidase-8 (MP8) and its acetylated form AcMP8 have been extensively characterized as have been their interactions with a number of different ligands (37, 38). Earlier attempts to examine the binding of Cys and other thiols to ferric MP8 (or AcMP8) have produced erratic results and were accompanied by reduction and destruction and of the heme chromophore (19, 39). Formation of Cys disulfides is another complication of binding studies with Cys.
Under stringent anaerobic conditions, we were able to achieve reproducible results with sets of spectra showing isosbestic points (Fig. 3). At pH 7.4, the spectrum of ferric AcMP8 features a Soret band at 397 nm and broad absorption bands at 500 nm, as well as a charge transfer band at 622 nm. Addition of AcCys gradually shifts the Soret band to 417 nm and a stronger Q band appears at 538 nm with a shoulder at 565 nm. Charge transfer absorption bands emerge at 641 and 767 nm. These features are similar to those of the Cys78 cyt c variants in this study and match even closer to that of the thiomethoxide adduct of Met80Ala (40). No detectable binding could be observed with similar concentrations of AcCys at a low pH of 4.5. After accounting for the pKa of AcCys of 9.5 (41), we calculate the binding constant of the AcCys thiolate to the ferric AcMP8 to be (2.5 ± 0.6) × 103.
Fig. 3.

Changes in the absorption spectra of ferric (A) and ferrous (B) AcMP8 upon addition of AcCys in a 100 mM sodium phosphate buffer at pH 7.4. Traces shown in red are of AcMP8 with no AcCys present. The concentration range of AcCys was 0–300 mM for ferric and 0–90 mM for ferrous titrations; the last traces in the titration series are shown in green. (Insets) Binding curves from the titration data and their fits. The apparent binding constants from these measurements are (1.7 ± 0.4) × 101 and (7.3 ± 2.9) × 101 for the ferric and ferrous AcMP8, respectively. Taking into account the pKa of 9.5 for AcCys, the binding constant of Cys thiolate to ferric AcMP8 is ∼2.5 × 103.
The titrations of ferrous AcMP8 with AcCys revealed changes similar to those previously seen with AcMet (37). The Soret and Q bands sharpen and the spectrum of the complex again resembles that of ferrous Cys78 variants and the Met-bound wild-type WT* cyt c. The binding constant is (7.3 ± 2.9) × 101, about sixfold lower than that of AcMet.
The measured binding constants of AcCys to the AcMP8 model suggest that, in the absence of the protein scaffold, the ferric thiolate adduct is ∼2 kcal/mol more stable than the corresponding ferrous thiol. Thus, the difference in stability of the two redox forms of our Cys78 variants (Tables 1 and 2) cannot be fully accounted for by the changes in the iron oxidation state and ligation.
Kinetics of Redox Interconversions.
The redox kinetics of the Cys78 variants were examined using stopped-flow and manual mixing techniques. Fig. 4A shows a square scheme illustrating interconversions between ferric thiolate and ferrous thiol species. The observed kinetics were biexponential both for oxidation and reduction reactions.
Fig. 4.

(A) A square scheme showing redox interconversions and protonation processes. (B) Electronic absorption spectra of ferric Thr78Cys/Lys79Gly/Met80Leu (black, thiolate-ligated), the intermediate species during oxidation of Thr78Cys/Lys79Gly/Met80Leu (red), Met80Ala at pH 7.4 (green, hydroxide-bound), and wild-type WT* at pH 7.4 (blue, Met-bound) and pH 9.0 (pink, Lys-bound). (Inset) Zoom of the Q-bands region of the spectra. (C) Changes in the electronic absorption spectra during oxidation of Thr78Cys/Lys79Gly/Met80Leu with 1.0 mM Ru(NH3)63+ in a sodium phosphate buffer at pH 7.4. (Inset) Time dependences of kinetic progress curves at selected wavelengths.
Oxidation of ferrous proteins by Ru(NH3)63+ (E° = 0.05 V) (42), as well as with Co(phen)33+ (E° = 0.37 V) (43), yielded a distinct new ferric species (Fig. 4B) that slowly converts to the ferric thiolate. This transient low-spin species differs from Lys-coordinated cyt c and is readily detectable at pH as low as 6.0, where heme coordination by Lys must be inhibited. If Cys78 coordination is preserved upon heme oxidation, the likely heme ligand in the transient is thiol. With a neutral sulfur ligand, the ferric thiol-coordinated heme may be expected to have a spectrum very similar to that of the ferric Met-coordinated heme. Indeed, the spectra of WT* and the transient are very similar, but the latter species lacks a detectable charge transfer band near 700 nm. Although frequently viewed as a diagnostic for Met ligation in cyt c proteins, this band is sometimes missing even if the protein has a Met ligand (44). With uncertainty about the exact origin of this band for cyt c (44), the lack of a similar charge transfer band does not rule out the thiol coordination in the transient.
Alternatively, if the Cys78 thiol rapidly dissociates from the heme upon oxidation, a His or a hydroxide can take its place. All three His residues (apart from coordinated His18) are distant from the heme and would require major changes in the protein structure to coordinate to the heme, making His an unlikely ligand. The visible spectrum of the transient species also deviates from that of the hydroxide-coordinated Met80Ala. Finally, the EPR spectra of the frozen reaction mixture have revealed the presence of a heme component with a g value of 3.2 that disappeared at longer times after mixing. The position of this signal is distinct from that of the hydroxide-coordinated Met80Ala (g = 2.6) but similar to that for the Met-bound cyt c (g = 3.1) (45), further supporting that the thiol heme adduct is likely among the trapped intermediate species.
The kinetics of the conversion of this ferric intermediate to the thiolate is the same with the two different oxidants we have tested, but depend on the solution pH. The trend of the pH dependence (Fig. 5A), however, is the opposite of the one expected if simply the proton concentration was governing the rates. Furthermore, there is only a minor hydrogen/deuterium solvent isotope effect 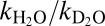 = 1.4 ± 0.1 on this step. Together, these results suggest that there is another process that influences the rate of the thiolate formation.
= 1.4 ± 0.1 on this step. Together, these results suggest that there is another process that influences the rate of the thiolate formation.
Fig. 5.

Kinetics of Thr78Cys/Lys79Gly/Met80Leu oxidation. (A) pH dependence of the rate constant k3, SH-S− and of the midpoint of the unfolding transition Tm for the ferric protein. (B) Dependence of the rate constant k3, SH-S− and the midpoint of the unfolding transition Tm for the ferric protein on the solution viscosity modulated by addition of glycerol.
The interior location of Cys78 promotes its coordination to the heme but limits the access of this residue to proton acceptors. We hypothesize that kinetics of the ferric thiolate formation depends on protein rearrangements. Previous studies of yeast cyt c have illustrated that, as the stability of the protein decreases, the rates of the protein rearrangements involving heme ligands and surrounding residues within the pocket increase (34). The decrease in the folding stability of our variants with decrease in pH parallels the pH-dependent increase in the rate constant k3, SH-S− (Fig. 5A). The correlation between stability and rearrangement rate constant k3, SH-S− of different mutants (Phe variant versus Leu and Ile variants, Table 1 and Table S3) also supports this argument. The viscosity dependence of k3, SH-S− (Fig. 5B) provides a strong confirmation of the role of protein conformational rearrangements. Glycerol slows down protein motions, thereby decreasing the rate.
The kinetics of reduction was explored with two low-potential compounds, dithionite (E° = −0.46 V) (46) and Eu2+-EGTA (E° = −0.88 V) (47). Again, the kinetics was biexponential, but no observable intermediate has been detected in these measurements. The rate constant for the slower process kslow depends on the concentration of the reductant (Fig. S5), but the rate for the faster process kfast does not. Furthermore, the fast process was the same with both reductants and very similar for all of the variants, suggesting that it is not an electron transfer process but rather a protonation reaction or conformational gating step.
The rate constants kfast in reduction reactions are more than an order of magnitude faster than the k3, SH-S− values for the oxidation reactions (Table S3). With the slow apparent rate of Cys78 deprotonation k3, SH-S−, it is likely that the rate of the ferric thiolate protonation, if dependent on similar rearrangements, is also slow. However, the residue(s) donating protons in the reduction reaction may be different from the proton acceptor in the oxidation reaction, explaining the difference in the timescale. To examine this possibility, we have modeled the kinetic progress curves for the reduction route involving the ferric intermediate (Fig. 4A, route RedA). With the experimentally observed rate constants, the ferric intermediate should be readily observable under our experimental conditions if the reaction proceeds through this route (Fig. S6). The experimental results suggest otherwise, thus arguing against this mechanistic scheme.
If the reaction proceeds through the route RedB (Fig. 4A), conversion of the ferric thiolate to the ferrous thiol would occur without significant buildup of the intermediate when the first step (electron transfer, kET, S−) is slower than the second one (protonation, k2, S−-SH). The CO photolysis experiments support this mechanism—the openness of the ferrous heme pocket in the Cys78 variants suggests that protons from bulk solvent could easily access the heme crevice. Two reduction channels, both having a very fast protonation step k2, S−-SH, can explain the observed biexponential kinetics, with the faster channel gated by protein conformational rearrangement and thus exhibiting reductant-independent kfast. Supporting this interpretation, two reduction pathways have previously been described for cyt c reaction with sodium dithionite (48). One of these pathways has been suggested to involve the opening of the heme crevice. If kfast is similarly associated with the opening of the heme crevice, the increase in protein conformational dynamics at lower pH may ease this process accounting for the observed pH dependence of kfast (Fig. S5C).
Discussion
The set of engineered cyt c mutants described herein offers a robust system for examining the thermodynamic stability, protonation, and redox transformations of Cys-bound hemes. The protein scaffold not only favors the ferric thiolate coordination and stabilizes the elusive ferrous thiol species, but also provides an easy access to two kinetic intermediates, including the rarely seen ferric thiol.
The Cys Position in Model Proteins.
The placement of a Cys within the protein interior was critical for the successful coordination of the residue to the heme. The Lys79Cys mutant has the Cys side chain oriented toward the solvent, evidenced by the ease of thiol-labeling reactions (49). Earlier work has described the preparation of horse heart Met80Cys by semisynthesis (21, 50). However, our attempts to recombinantly make yeast Met80Cys variants have yielded proteins with an oxidized Cys80 side chain, which did not ligate as a thiolate to the heme upon oxidation. The properties of the yeast and horse heart proteins are very similar, but the former protein is less stable (34, 51), with the Met80 loop that is more mobile and unfolds more easily (Fig. S2). Recombinant expression of horse heart cyt c yielded some thiolate-bound species; however, the protein amounts were insufficient for detailed characterization of this derivative. In contrast, large quantities of thiolate-ligated Cys78 variants can be readily obtained, even without the need for anaerobic conditions during protein purification.
The highly conserved Thr78 residue is a part of the hydrogen-bonding network (Fig. S2) that has been suggested to play a role in closing up the heme crevice in cyt c (52–54). Previous modifications to Val, Asn, and α-aminobutyric acid at this site have destabilized the protein and altered the heme reduction potential (52, 55). However, introduction of a Cys for ligand interactions with the heme instead increases the stability of ferric variants compared with the wild-type WT* protein (Table 2).
Ferric Thiolate Versus Ferrous Thiol: Forces in Play.
Our thermodynamic analysis reveals that stabilization of the ferric variants is mainly entropic, suggesting the role of the protein scaffold for this ligation. The slow kinetics of protein conformational changes during oxidation reactions points to the rigidity of the heme pocket in the ferric proteins. However, Lys displacement of Cys78 as the ligand to ferrous heme at high pH and ease of CO rebinding to the ferrous heme in flash photolysis experiments (Fig. S4) as well as rapid Cys78 protonation in reduction reactions, all indicate that the heme pocket in the ferrous proteins is somewhat open to the solvent and readily accommodates polypeptide rearrangements. Evidently, these differences in the polypeptide conformational dynamics for the two redox forms do not corroborate the entropic trends.
The likely difference in solvation of ferric and ferrous proteins is another factor to consider. Coordination of the short Cys side chain to the heme could tighten the polypeptide structure and thus help to exclude water from the protein core. The low-stability loop containing residues 76–84 is flexible and readily adjusts to changes in the heme coordination site induced by small molecules as well as endogenous ligands (56, 57). Interestingly, a conserved water molecule next to Thr78 has been detected in the cyt c crystal structures (Fig. S2C) (53). This water is close to the heme propionate HP-6, a group with pKa ≥ 9 (44). Considering the clear pH effects on the heme absorption spectra and the location of HP-6 in close proximity to Thr78 (Cys78), the pH transition in our titrations of ferric variants may be associated with this propionate. The downshift of its pKa to 8.2 ± 0.2 is consistent with the change in solvation in the Cys78 variants.
Although this scenario explains the stabilizing entropic effects of ferric variants, different forces are important for the ferrous variants. The low-affinity thiol ligand does not seem to drive the refolding and tightening of the heme pocket, but rather its coordination to the heme is favored by the Cys placement within the protein scaffold. The lack of success in making yeast Met80Cys is consistent with this hypothesis. The loss of Cys coordination in several sensors upon heme reduction further illustrates the importance of the surrounding polypeptide in preserving thiol interactions with the ferrous heme. Interestingly, studies by Rousseau and coworkers (11) have found that following CO photolysis thiol ligation is maintained in inducible NOS but not in a thiol model complex, again highlighting the role of the protein scaffold in keeping the ligand close to the heme.
The CO photolysis experiments in our work have shed light on the protein structure that surrounds the ferrous heme. The enclosed, but still solvent-accessible, heme crevice of the ferrous Cys78 variants is reminiscent of that in ferrous Cmcyt c (35, 36), the derivative that lacks the coordination link to the heme. However, in contrast to carboxymethylated Met80, Cys78 supported by the cyt c polypeptide structure, binds to the heme. Within this scaffold, there are opportunities for interactions of the ferrous heme with the solvent. Possible hydrogen-bonding interactions with the Cys78 thiol proton could also favor its solvation. The increased solvation of the protein core contributes to the decrease in the total entropy of stabilization (increase in the entropy of denaturation ΔSD) compared with that for the ferric variants (Table 2).
Our model studies of Cys binding to AcMP8, as well as earlier results with a variety of organic thiols (12), convincingly demonstrate that a neutral Cys thiol does ligate to the ferrous heme. However, because this interaction is weak, the protein has a role in promoting and preserving this ligation. The strategy of templating the ferrous–thiol interaction within the cyt c scaffold has successfully yielded a set of ferrous thiol proteins. Importantly, this design has yielded ferrous thiol variants that can be reversibly converted back to ferric thiolates.
Interconversions Between Thiols and Thiolates.
One of the most exciting results of this study is the observation of the ferric intermediate. Although not directly observed, the ferrous thiolate intermediate is the likely player during reduction of the Cys78 variants, because these reactions bypass the ferric thiol intermediate. Even with a powerful reductant such as Eu2+-EGTA, this intermediate does not accumulate, but it may be resolved by using faster flash photolysis (58) or pulse-radiolysis methods (59); design of these experiments is currently in progress. Thus, the designed cyt c protein scaffold could potentially provide an access to all of the four species within the square scheme (Fig. 4A).
Sets of ferric thiolate/ferrous thiolate or ferric thiolate/ferrous thiol are typical of native heme proteins. The protonation state of the coordinated Cys is critical for enzymatic activity (P450 enzymes) and signal transduction (sensors). However, what governs a particular protonation state of the Cys ligand and could these protein systems also support heme coordination by the other protonation form of this ligand?
In a couple of P450 enzymes from Mycobacterium tuberculosis, the transition from ferrous thiol to thiolate takes place at near neutral pH (32), but the factors controlling this behavior are currently not fully understood. Furthermore, the Cys thiol has been suggested as a ligand to the heme in the inactive ferrous P420 species (11). Analysis of the cyt c model systems here highlights the increased solvent accessibility of the heme pocket for the ferrous thiols, hinting to the role of polypeptide packing in mediating the thiolate-to-thiol transition.
Protein Dynamics and Redox Reactions.
The rates of redox interconversion in the Cys78 variants depend on the protein rearrangements. With a redox-dependent change in the Cys protonation state, protein motions control access of the Cys-coordinated heme to proton accepting and donating groups. Notably, owing to the Cys78 location in the protein interior, the protein rearrangements associated with Cys78 deprotonation take more than a minute to complete.
Despite very similar redox potentials (Table 1), the Phe variant is reduced more slowly than the other two variants. Greater stability, higher pKa of the ferrous alkaline transition, and distinct CO photolysis kinetics all suggest that the heme pocket in this variant is more tightly packed. These parallels imply that reduction of the Cys78 variants by both dithionite and the Eu2+-EGTA complex may require contact with the heme and again suggest the role of conformational dynamics in the protein redox mechanism. Perhaps during this rearrangement water can enter the heme pocket, accounting for the fast rates of protonation and the decrease in entropy of stabilization of the Cys78 variants upon reduction.
Conclusions
Our studies of the minimalist heme model MP8 and a series of engineered cyt c proteins demonstrate that the protein scaffold and solvent interactions play important roles in stabilizing a particular Cys–heme coordination. Increased stability of ferric thiolate, compared with ferrous thiol or Met-bound wild-type WT*, arises mainly from entropic factors associated with better polypeptide packing. In contrast, the low-affinity thiol ligand does not seem to drive the refolding of the heme pocket but rather its coordination to the heme is favored by the Cys placement within the protein structure. The protein also controls the protonation (deprotonation) kinetics during heme redox interconversions. The model system provides access to all four forms of Cys-bound hemes, most notably to the ferric thiol. This latter species could play an important role in the redox-dependent recovery of the heme thiolate ligation.
Similar to our cyt c models, the Cys/His ligand set is present in a number of native enzymes and sensors (60). The Cys protonation (deprotonation) reactions are intimately linked to their redox transformations, and in the case of sensors, control functional ligand switching and heme dissociation. The coordination of His as the sixth ligand should increase the effective pKa of the coordinated thiol, but interactions in the second coordination sphere may further modulate the properties of the Cys ligand and solvent accessibility of the heme. Interestingly, in human CBS where Cys52 forms ionic interactions with Arg266 and a hydrogen bond with Trp54, a thiolate is coordinated to the ferrous heme but thermal denaturation (and presumably the loss of these critical contacts) converts the protein to the CBS424 form with a neutral ligand set to the heme (61).
For years, vibrant discussion has focused on the role of hydrogen bonds to the thiolate ligand in the P450 enzymes (4, 62). These bonds provided by the protein scaffold have been suggested to stabilize the ferrous thiolates by decreasing the electron-donating properties of this group and to protect the thiolate from protonation. A small and robust cyt c template with a pliable loop that surrounds the Cys-bound heme offers exciting opportunities for engineering these interactions and correlating them to the heme electronic structure and reactivity; some of these further studies are currently underway.
Materials and Methods
Site-Directed Mutagenesis, Protein Expression, and Purification.
The plasmid Rbs (WT*) containing genes CYC1 and CYC3 that encode the yeast iso-1 cyt c and yeast cyt c heme lyase, respectively, has served as a basis for molecular biology work. The parent pseudo–wild-type construct WT* contained two additional mutations Lys72Ala and Cys102Ser, to prevent Lys72 coordination to the heme and formation of cyt c dimers, respectively. The new Thr78Cys/Lys79Gly/Met80X (X = Leu, Ile, and Phe) and Met80Ala plasmids were constructed by point mutations using a QuikChange Kit (Stratagene). The horse heart Met80Cys was constructed based on the pBTR plasmid. DNA was extracted using a Miniprep Kit (Qiagen) and sequenced at the Molecular Biology and Proteomics Core Facility (Dartmouth College, Hanover, NH).
All of the mutants were expressed and purified according to the published procedures (51) with the following modifications. Two liters of the Terrific Broth medium, instead of one liter, were used in each 2.8-L flask to increase the protein yields (63), and no DTT was added to the buffers during protein purification. Fresh purification of proteins before measurements was important for achieving reproducible results.
Cmcyt c was prepared from horse heart cyt c (Sigma-Aldrich) as described (64).
Preparation of MP8.
MP8 was prepared via proteolytic degradation of cyt c as previously described (65). Horse heart cyt c (250 mg; Sigma-Aldrich) was cleaved by treatment with pepsin and then trypsin. Crude MP8 was precipitated through the addition of ammonium sulfate, and the bright red product was collected by centrifugation. The resulting red solid was dissolved in water, and excess ammonium sulfate was removed by overnight dialysis using a 1,000 molecular weight cutoff (MWCO) tubing (Spectrum Labs). The peptide solution was concentrated with an Amicon ultrafiltration cell (YM-1 membrane) and purified on a GE Healthcare Akta FPLC using a Source 15RPC column [buffer A: 0.1% (vol/vol) TFA in water; buffer B: 10% buffer A and 90% (vol/vol) acetonitrile; 10–55% gradient of buffer B].
AcMP8 was prepared by slow addition of 500-fold excess acetic anhydride to a solution containing MP8 in 0.1 M sodium bicarbonate, pH 9.0. Excess bicarbonate was removed by dialysis against water and AcMP8 was concentrated with an Amicon ultrafiltration cell. AcMP8 was purified again on a Source 15RPC column and the product was confirmed by MALDI-TOF (m/z = 1,549). Stock solutions were prepared in methanol and their concentrations were determined spectrophotometrically (ε396 = 1.57 mM−1⋅cm−1) (65).
Spectroscopic Measurements.
Absorption spectra were acquired on an Agilent 8453 diode-array, Cary 400, or JASCO V-630 scanning spectrophotometers. CD spectra and CD temperature melts were recorded on a JASCO-J815 CD spectropolarimeter equipped with a variable temperature Peltier cell device (JASCO). MCD spectra were measured at a magnetic field strength of 1.41 T on a JASCO-J815 spectropolarimeter equipped with a JASCO MCD-1B electromagnet and interfaced with a Silicon Solutions PC through a JASCO IF-815-2 interface unit. MCD data acquisitions and manipulations were carried out using JASCO software (66).
EPR spectra were recorded at 10 K on a Bruker EMX 300 X-band EPR spectrometer using the following conditions: microwave frequency of 9.49 GHz, microwave power of 3.21 mW, modulation frequency of 100 kHz, modulation amplitude of 1.00 G, and time constant of 20.48 ms.
Protein extinction coefficients were determined by the pyridine hemochrome method (67). All experiments with ferrous proteins were done under anaerobic conditions. Oxygen was removed from buffers by stirring solutions under vacuum and repeated pump–purge cycles with nitrogen. Reduction of the heme iron was accomplished by adding sodium dithionite to the protein and excess of the reducing agent was removed by running the protein solution through a PD-10 column (GE Healthcare) under a nitrogen atmosphere in a nitrogen-filled glove box (COY Laboratory Products).
Details of thermal denaturation, pH dependence, and AcMP8 binding studies are described in SI Materials and Methods.
Spectroelectrochemistry.
The electrochemical experiments were performed using a spectroelectrochemistry kit (Pine Research Instrumentation) in a glove box (COY Laboratory Products) under a nitrogen atmosphere. The path length of the optical cell was 1.7 mm. A honeycomb electrode was immersed in the optical cell as the working electrode. A platinum electrode and Ag/AgCl gel (saturated KCl solution) electrode were used as auxiliary and reference electrodes, respectively. The potential was controlled by a WaveNow USB potentiostat and the time courses of the current during the reaction were monitored using the Aftermath software. The electronic absorption spectra were measured on a Shimadzu UV-1201 spectrophotometer.
Redox titrations were performed in a buffer containing 50 mM Tris and 100 mM NaCl at pH 7.0. The final concentration of the protein was 25 μM. The following mediators (100 μM of each, all potentials against SHE) were added into the sample solution to facilitate electrical communication between protein and electrode: anthraquinone 2-sulfonate (Em = −230 mV), benzyl viologen (Em = −350 mV) and methyl viologen (Em = −440 mV) (59). The gel reference electrode was calibrated from the redox titration of 1 mM K3Fe(CN)6 (E° = 436 mV) (68) in 100 mM KNO3. The absorbance contribution of the mediators was subtracted by measuring the height of the peak at 549 nm relative to the straight line connecting the two isosbestic points (540 and 557 nm) that flank the α band of the mutants (69).
Kinetics of Redox Reactions.
Fast kinetics was examined by performing mixing experiments on a BioLogic SFM-300 stopped-flow instrument inside a nitrogen-filled glove box (Cleatech). Slow oxidation reactions (lasting longer than 60 s) were initiated by the injection of the oxidant (through a septum cap of a sealed anaerobic cuvette) and kinetics was monitored using an Agilent 8453 spectrophotometer. In both cases, full visible heme spectra during the conversion were recorded by diode-array instruments (MMS-VIS model 060-20/4 in the stopped-flow setup and Agilent 8453 in the manual mixing experiments). Preparation of reductants and oxidants as well as experimental details and data analyses are described in SI Materials and Methods.
Laser Photolysis of Carbon Monoxide Adducts of Ferrous Proteins.
Ferrous-CO adducts were formed by the addition of a stock, CO (Airgas)-saturated buffer (a 100 mM sodium phosphate buffer at pH 7.4) into the solutions of ferrous proteins prepared by addition of dithionite. The desired CO concentration was achieved by the dilution of CO saturated buffer (1 mM at 1 atm). The preparation of the samples was carried out in the glove box (COY Laboratory Products) under a nitrogen atmosphere. Bound CO was photolysed using 5-ns pulses at 532 nm from a Continuum Minilite II Q-switched frequency-doubled Nd:YAG laser. Kinetics of the CO rebinding was monitored with transient absorption measurements at 429 nm. The transients were detected using a modified Proteus system (Ultrafast Systems). A 75-W Xe lamp (Oriel; model 66912) provided a probe light, and a Tektronix DPO 3032 digital phosphor oscilloscope and a ThorLabs DET10A photodiode were adapted to form the detection system. Quantum yields for CO photodissociation were measured as described (35). The laser intensity was attenuated using a built-in dielectric polarizer. The final concentrations of CO and proteins in these experiments were 100 and 5 μM, respectively.
Supplementary Material
Acknowledgments
We are grateful to Dean E. Wilcox for the numerous discussions that have strengthened this study. We thank Bruce E. Bowler and Kara L. Bren for providing the Rbs(WT*) and pBTR plasmids, respectively, Emily Johnson for her help during the MCD data collection, Ricardo Louro for his advice on spectroelectrochemistry, Aimin Liu and Ian Davis for their help with freeze-quench trapping of the ferric intermediate, and Soyeun Yang for preparation of Met80Ala. This work was supported by National Science Foundation CAREER Grant CHE-0953693 (to E.V.P.) and National Institutes of Health Grant GM 26730 (to J.H.D.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1317173111/-/DCSupplemental.
References
- 1.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-containing oxygenases. Chem Rev. 1996;96(7):2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 2.Julsing MK, Cornelissen S, Bühler B, Schmid A. Heme-iron oxygenases: Powerful industrial biocatalysts? Curr Opin Chem Biol. 2008;12(2):177–186. doi: 10.1016/j.cbpa.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 3.Sevrioukova IF, Poulos TL. Understanding the mechanism of cytochrome P450 3A4: Recent advances and remaining problems. Dalton Trans. 2013;42(9):3116–3126. doi: 10.1039/c2dt31833d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hannibal L, Somasundaram R, Tejero J, Wilson A, Stuehr DJ. Influence of heme-thiolate in shaping the catalytic properties of a bacterial nitric-oxide synthase. J Biol Chem. 2011;286(45):39224–39235. doi: 10.1074/jbc.M111.286351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Groves JT, Wang CC-Y. Nitric oxide synthase: Models and mechanisms. Curr Opin Chem Biol. 2000;4(6):687–695. doi: 10.1016/s1367-5931(00)00146-0. [DOI] [PubMed] [Google Scholar]
- 6.Su Y, et al. Comparative study of enzyme activity and heme reactivity in Drosophila melanogaster and Homo sapiens cystathionine β-synthases. Biochemistry. 2013;52(4):741–751. doi: 10.1021/bi300615c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hofrichter M, Ullrich R. Heme-thiolate haloperoxidases: Versatile biocatalysts with biotechnological and environmental significance. Appl Microbiol Biotechnol. 2006;71(3):276–288. doi: 10.1007/s00253-006-0417-3. [DOI] [PubMed] [Google Scholar]
- 8.Cheesman MR, Little PJ, Berks BC. Novel heme ligation in a c-type cytochrome involved in thiosulfate oxidation: EPR and MCD of SoxAX from Rhodovulum sulfidophilum. Biochemistry. 2001;40(35):10562–10569. doi: 10.1021/bi0100081. [DOI] [PubMed] [Google Scholar]
- 9.Shelver D, et al. Identification of two important heme site residues (cysteine 75 and histidine 77) in CooA, the CO-sensing transcription factor of Rhodospirillum rubrum. Biochemistry. 1999;38(9):2669–2678. doi: 10.1021/bi982658j. [DOI] [PubMed] [Google Scholar]
- 10.Dioum EM, et al. NPAS2: A gas-responsive transcription factor. Science. 2002;298(5602):2385–2387. doi: 10.1126/science.1078456. [DOI] [PubMed] [Google Scholar]
- 11.Sabat J, Stuehr DJ, Yeh SR, Rousseau DL. Characterization of the proximal ligand in the P420 form of inducible nitric oxide synthase. J Am Chem Soc. 2009;131(34):12186–12192. doi: 10.1021/ja901016a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perera R, et al. Neutral thiol as a proximal ligand to ferrous heme iron: Implications for heme proteins that lose cysteine thiolate ligation on reduction. Proc Natl Acad Sci USA. 2003;100(7):3641–3646. doi: 10.1073/pnas.0737142100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smulevich G, Bjerrum MJ, Gray HB, Spiro TG. Resonance raman spectra and the active site structure of semisynthetic Met80Cys horse heart cytochrome c. Inorg Chem. 1994;33(21):4629–4634. [Google Scholar]
- 14.Collman JP, Sorrell TN, Hodgson KO, Kulshrestha AK, Strouse CE. Molecular dynamics in the solid state. A dynamic model of the low-spin iron(III) to high-spin iron(III) transformation in P450 enzymes. J Am Chem Soc. 1977;99(15):5180–5181. doi: 10.1021/ja00457a049. [DOI] [PubMed] [Google Scholar]
- 15.Sono M, Andersson LA, Dawson JH. Sulfur donor ligand binding to ferric cytochrome P-450-CAM and myoglobin. Ultraviolet-visible absorption, magnetic circular dichroism, and electron paramagnetic resonance spectroscopic investigation of the complexes. J Biol Chem. 1982;257(14):8308–8320. [PubMed] [Google Scholar]
- 16.Sono M, Dawson JH, Hager LP. The generation of a hyperporphyrin spectrum upon thiol binding to ferric chloroperoxidase. Further evidence of endogenous thiolate ligation to the ferric enzyme. J Biol Chem. 1984;259(21):13209–13216. [PubMed] [Google Scholar]
- 17.McGuire DG, Khan MA, Ashby MT. Discontinuum between a thiolate and a thiol ligand. Inorg Chem. 2002;41(8):2202–2208. doi: 10.1021/ic011096g. [DOI] [PubMed] [Google Scholar]
- 18.Bayer E, Hill HAO, Roder A, Williams RJP. The interaction between haem-iron and thiols. J Chem Soc D. 1969;3:109. [Google Scholar]
- 19.Sadeque AJM, Shimizu T, Hatano M. Thiol-coordinated heme octapeptides of cytochrome c; model compounds of cytochrome P-450. Inorg Chim Acta. 1987;135(2):109–113. [Google Scholar]
- 20.Dawson JH, et al. Magnetic circular dichroism of purified forms of rabbit liver cytochromes P-450 and P-420. Biochemistry. 1978;17(1):33–42. doi: 10.1021/bi00594a006. [DOI] [PubMed] [Google Scholar]
- 21.Raphael AL, Gray HB. Semisynthesis of axial-ligand (position 80) mutants of cytochrome c. J Am Chem Soc. 1991;113(3):1038–1040. [Google Scholar]
- 22.Pollock WBR, Rosell FI, Twitchett MB, Dumont ME, Mauk AG. Bacterial expression of a mitochondrial cytochrome c. Trimethylation of Lys72 in yeast iso-1 cytochrome c and the alkaline conformational transition. Biochemistry. 1998;37(17):6124–6131. doi: 10.1021/bi972188d. [DOI] [PubMed] [Google Scholar]
- 23.McKnight J, Cheesman MR, Thomson AJ, Miles JS, Munro AW. Identification of charge-transfer transitions in the optical spectrum of low-spin ferric cytochrome P-450 Bacillus megaterium. Eur J Biochem. 1993;213(2):683–687. doi: 10.1111/j.1432-1033.1993.tb17808.x. [DOI] [PubMed] [Google Scholar]
- 24.Blanke SR, et al. Probing the heme iron coordination structure of alkaline chloroperoxidase. Biochemistry. 1996;35(46):14537–14543. doi: 10.1021/bi961512m. [DOI] [PubMed] [Google Scholar]
- 25.Reynolds MF, et al. EPR and electronic absorption spectroscopies of the CO-sensing CooA protein reveal a cysteine-ligated low-spin ferric heme. J Am Chem Soc. 1998;120(35):9080–9081. [Google Scholar]
- 26.Dawson JH, Andersson LA, Sono M. The diverse spectroscopic properties of ferrous cytochrome P-450-CAM ligand complexes. J Biol Chem. 1983;258(22):13637–13645. [PubMed] [Google Scholar]
- 27.Cohen DS, Pielak GJ. Entropic stabilization of cytochrome c upon reduction. J Am Chem Soc. 1995;117(6):1675–1677. [Google Scholar]
- 28.Liggins JR, Lo TP, Brayer GD, Nall BT. Thermal stability of hydrophobic heme pocket variants of oxidized cytochrome c. Protein Sci. 1999;8(12):2645–2654. doi: 10.1110/ps.8.12.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Betz SF, Pielak GJ. Introduction of a disulfide bond into cytochrome c stabilizes a compact denatured state. Biochemistry. 1992;31(49):12337–12344. doi: 10.1021/bi00164a007. [DOI] [PubMed] [Google Scholar]
- 30.Herrmann LM, Bowler BE. Thermal denaturation of iso-1-cytochrome c variants: Comparison with solvent denaturation. Protein Sci. 1997;6(3):657–665. doi: 10.1002/pro.5560060315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Godbole S, Hammack B, Bowler BE. Measuring denatured state energetics: Deviations from random coil behavior and implications for the folding of iso-1-cytochrome c. J Mol Biol. 2000;296(1):217–228. doi: 10.1006/jmbi.1999.3454. [DOI] [PubMed] [Google Scholar]
- 32.Dunford AJ, et al. Rapid P450 heme iron reduction by laser photoexcitation of Mycobacterium tuberculosis CYP121 and CYP51B1. Analysis of CO complexation reactions and reversibility of the P450/P420 equilibrium. J Biol Chem. 2007;282(34):24816–24824. doi: 10.1074/jbc.M702958200. [DOI] [PubMed] [Google Scholar]
- 33.Rosell FI, Ferrer JC, Mauk AG. Proton-linked protein conformational switching: Definition of the alkaline conformational transition of yeast Iso-1-ferricytochrome c. J Am Chem Soc. 1998;120(44):11234–11245. [Google Scholar]
- 34.Cherney MM, Bowler BE. Protein dynamics and function: Making new strides with an old warhorse, the alkaline conformational transition of cytochrome c. Coord Chem Rev. 2011;255(7-8):664–677. [Google Scholar]
- 35.Silkstone G, Stanway G, Brzezinski P, Wilson MT. Production and characterisation of Met80X mutants of yeast iso-1-cytochrome c: Spectral, photochemical and binding studies on the ferrous derivatives. Biophys Chem. 2002;98(1-2):65–77. doi: 10.1016/s0301-4622(02)00085-6. [DOI] [PubMed] [Google Scholar]
- 36.Wilson MT, Brunori M, Rotilio GC, Antonini E. Properties of modified cytochromes. II. Ligand binding to reduced carboxymethyl cytochrome c. J Biol Chem. 1973;248(23):8162–8169. [PubMed] [Google Scholar]
- 37.Tezcan FA, Winkler JR, Gray HB. Effects of ligation and folding on reduction potentials of heme proteins. J Am Chem Soc. 1998;120(51):13383–13388. [Google Scholar]
- 38.Wang JS, Tsai AL, Heldt J, Palmer G, Van Wart HE. Temperature- and pH-dependent changes in the coordination sphere of the heme c group in the model peroxidase N alpha-acetyl microperoxidase-8. J Biol Chem. 1992;267(22):15310–15318. [PubMed] [Google Scholar]
- 39.Marques HM, Rousseau A. Reactions of ferric porphyrins and thiols. The reaction of the haem octapeptide, N-acetylmicroperoxidase-8, with cysteine. Inorg Chim Acta. 1996;248(1):115–119. [Google Scholar]
- 40.Lu Y, Casimiro DR, Bren KL, Richards JH, Gray HB. Structurally engineered cytochromes with novel ligand-binding properties. Expression of S. cerevisiae Met80Ala iso-1-cytochrome c. Proc Natl Acad Sci USA. 1993;90(24):11456–11459. doi: 10.1073/pnas.90.24.11456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Danehy JP, Parameswaran KN. Acidic dissociation constants of thiols. J Chem Engineer Data. 1968;13(3):386–389. [Google Scholar]
- 42.Matsubara T, Ford PC. Some applications of cyclic voltammetry to the reactions and properties of ruthenium ammine complexes. Reduction potentials and rate studies. Inorg Chem. 1976;15(5):1107–1110. [Google Scholar]
- 43.Taniguchi VT, Sailasuta-Scott N, Anson FC, Gray HB. Thermodynamics of metalloprotein electron transfer reactions. Pure Appl Chem. 1980;52(10):2275–2281. [Google Scholar]
- 44.Moore GR, Pettigrew GW. Cytochromes c: Evolutionary, Structural, and Physicochemical Aspects. New York: Springer; 1990. [Google Scholar]
- 45.Silkstone GG, Cooper CE, Svistunenko D, Wilson MT. EPR and optical spectroscopic studies of Met80X mutants of yeast ferricytochrome c. Models for intermediates in the alkaline transition. J Am Chem Soc. 2005;127(1):92–99. doi: 10.1021/ja045719b. [DOI] [PubMed] [Google Scholar]
- 46.Latimer WM. The Oxidation States of the Elements and Their Potentials in Aqueous Solution. New York: Prentice-Hall; 1952. [Google Scholar]
- 47.Vincent KA, et al. Instantaneous, stoichiometric generation of powerfully reducing states of protein active sites using Eu(II) and polyaminocarboxylate ligands. Chem Commun (Camb) 2003;(20):2590–2591. doi: 10.1039/b308188e. [DOI] [PubMed] [Google Scholar]
- 48.Creutz C, Sutin N. Reduction of ferricytochrome c by dithionite ion: Electron transfer by parallel adjacent and remote pathways. Proc Natl Acad Sci USA. 1973;70(6):1701–1703. doi: 10.1073/pnas.70.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeWeerd K, Grigoryants V, Sun Y, Fetrow JS, Scholes CP. EPR-detected folding kinetics of externally located cysteine-directed spin-labeled mutants of iso-1-cytochrome c. Biochemistry. 2001;40(51):15846–15855. doi: 10.1021/bi011414n. [DOI] [PubMed] [Google Scholar]
- 50.Wallace CJ, Clark-Lewis I. Functional role of heme ligation in cytochrome c. Effects of replacement of methionine 80 with natural and non-natural residues by semisynthesis. J Biol Chem. 1992;267(6):3852–3861. [PubMed] [Google Scholar]
- 51.Hanske J, et al. Conformational properties of cardiolipin-bound cytochrome c. Proc Natl Acad Sci USA. 2012;109(1):125–130. doi: 10.1073/pnas.1112312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wallace CJ, et al. Substitutions engineered by chemical synthesis at three conserved sites in mitochondrial cytochrome c. Thermodynamic and functional consequences. J Biol Chem. 1989;264(26):15199–15209. [PubMed] [Google Scholar]
- 53.Berghuis AM, et al. The role of a conserved internal water molecule and its associated hydrogen bond network in cytochrome c. J Mol Biol. 1994;236(3):786–799. doi: 10.1006/jmbi.1994.1189. [DOI] [PubMed] [Google Scholar]
- 54.Berghuis AM, Brayer GD. Oxidation state-dependent conformational changes in cytochrome c. J Mol Biol. 1992;223(4):959–976. doi: 10.1016/0022-2836(92)90255-i. [DOI] [PubMed] [Google Scholar]
- 55.ten Kortenaar PB, Adams PJ, Tesser GI. Semisynthesis of horse heart cytochrome c analogues from two or three fragments. Proc Natl Acad Sci USA. 1985;82(24):8279–8283. doi: 10.1073/pnas.82.24.8279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bren KL, Gray HB, Banci L, Bertini I, Turano P. Paramagnetic 1H NMR spectroscopy of the cyanide derivative of Met80Ala-iso-1-cytochrome c. J Am Chem Soc. 1995;117(31):8067–8073. [Google Scholar]
- 57.Assfalg M, et al. Structural model for an alkaline form of ferricytochrome C. J Am Chem Soc. 2003;125(10):2913–2922. doi: 10.1021/ja027180s. [DOI] [PubMed] [Google Scholar]
- 58.Pascher T, Chesick JP, Winkler JR, Gray HB. Protein folding triggered by electron transfer. Science. 1996;271(5255):1558–1560. doi: 10.1126/science.271.5255.1558. [DOI] [PubMed] [Google Scholar]
- 59.Nakajima H, et al. Redox properties and coordination structure of the heme in the co-sensing transcriptional activator CooA. J Biol Chem. 2001;276(10):7055–7061. doi: 10.1074/jbc.M003972200. [DOI] [PubMed] [Google Scholar]
- 60.Shimizu T. Binding of cysteine thiolate to the Fe(III) heme complex is critical for the function of heme sensor proteins. J Inorg Biochem. 2012;108(1):171–177. doi: 10.1016/j.jinorgbio.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 61.Pazicni S, et al. The heme of cystathionine β-synthase likely undergoes a thermally induced redox-mediated ligand switch. Biochemistry. 2005;44(51):16785–16795. doi: 10.1021/bi051305z. [DOI] [PubMed] [Google Scholar]
- 62.Galinato MG, Spolitak T, Ballou DP, Lehnert N. Elucidating the role of the proximal cysteine hydrogen-bonding network in ferric cytochrome P450cam and corresponding mutants using magnetic circular dichroism spectroscopy. Biochemistry. 2011;50(6):1053–1069. doi: 10.1021/bi101911y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ying T, et al. Evolutionary alkaline transition in human cytochrome c. J Bioenerg Biomembr. 2009;41(3):251–257. doi: 10.1007/s10863-009-9223-9. [DOI] [PubMed] [Google Scholar]
- 64.Schejter A, George P. Production of a “cytochrome c” with myoglobin-like properties by alkylating the cyanide complex with bromoacetate. Nature. 1965;206(989):1150–1151. doi: 10.1038/2061150a0. [DOI] [PubMed] [Google Scholar]
- 65.Low DW, Winkler JR, Gray HB. Photoinduced oxidation of microperoxidase-8—generation of ferryl and cation-radical porphyrins. J Am Chem Soc. 1996;118(1):117–120. [Google Scholar]
- 66.Pond AE, Roach MP, Thomas MR, Boxer SG, Dawson JH. The H93G myoglobin cavity mutant as a versatile template for modeling heme proteins: Ferrous, ferric, and ferryl mixed-ligand complexes with imidazole in the cavity. Inorg Chem. 2000;39(26):6061–6066. doi: 10.1021/ic0007198. [DOI] [PubMed] [Google Scholar]
- 67.Berry EA, Trumpower BL. Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal Biochem. 1987;161(1):1–15. doi: 10.1016/0003-2697(87)90643-9. [DOI] [PubMed] [Google Scholar]
- 68.Pandurangachar M, et al. Electrochemical investigations of potassium ferricyanide and dopamine by 1-butyl-4-methylpyridinium tetrafluoro borate modified carbon paste electrode: A cyclic voltammetric study. Int J Electrochem Sci. 2010;5(8):1187–1202. [Google Scholar]
- 69.Louro RO, Catarino T, LeGall J, Turner DL, Xavier AV. Cooperativity between electrons and protons in a monomeric cytochrome c(3): The importance of mechano-chemical coupling for energy transduction. ChemBioChem. 2001;2(11):831–837. doi: 10.1002/1439-7633(20011105)2:11<831::AID-CBIC831>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.