

Significance
The B-cell lymphoma-2 (Bcl-2) protein binds to the inositol 1,4,5-trisphosphate receptor (InsP3R), a ubiquitous intracellular Ca2+ channel, thereby promoting cell survival by preventing excessive Ca2+ elevation. We report here that this mechanism involves Bcl-2 interaction with the Ca2+-activated protein phosphatase calcineurin (CaN) and dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32), a CaN-regulated inhibitor of protein phosphatase 1. Our findings indicate that this complex constitutes a rapid negative feedback mechanism that is activated by Ca2+ elevation and decreases InsP3R phosphorylation, thus inhibiting InsP3R-mediated Ca2+ release. We posit that Bcl-2-overexpressing cancer cells may exploit this mechanism to inhibit apoptosis.
Keywords: calcium, lymphocyte, cancer, signaling, signal transduction
Abstract
Bcl-2 interacts with the inositol 1,4,5-trisphosphate receptor (InsP3R) and thus prevents InsP3-induced Ca2+ elevation that induces apoptosis. Here we report that Bcl-2 binds dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32), a protein kinase A (PKA)-activated and calcineurin (CaN)-deactivated inhibitor of protein phosphatase 1 (PP1). Bcl-2 docks DARPP-32 and CaN in a complex on the InsP3R, creating a negative feedback loop that prevents exaggerated Ca2+ release by decreasing PKA-mediated InsP3R phosphorylation. T-cell activation increases PKA activity, phosphorylating both the InsP3R and DARPP-32. Phosphorylated DARPP-32 inhibits PP1, enhancing InsP3R phosphorylation and Ca2+ release. Elevated Ca2+ activates CaN, which dephosphorylates DARPP-32 to dampen Ca2+ release by eliminating PP1 inhibition to enable it to dephosphorylate the InsP3R. Knocking down either Bcl-2 or DARPP-32 abrogates this feedback mechanism, resulting in increased Ca2+ elevation and apoptosis. This feedback mechanism appears to be exploited by high levels of Bcl-2 in chronic lymphocytic leukemia cells, repressing B-cell receptor-induced Ca2+ elevation and apoptosis.
Periodic elevations of intracellular Ca2+ serve as second messengers regulating mitochondrial metabolism, cell cycle entry, and cell survival (1, 2). Ca2+ signals are generated when inositol 1,4,5-trisphosphate receptors (InsP3Rs) open in response to InsP3, thus transferring Ca2+ from the endoplasmic reticulum into the cytoplasm and mitochondria. InsP3R-mediated Ca2+ release is highly regulated, as excessive Ca2+ release causes cellular dysfunction and death (3). A number of proteins bind to InsP3Rs and regulate channel opening, thus preventing excessive Ca2+ release (4). Among these is the antiapoptotic protein B-cell lymphoma-2 (Bcl-2) (5).
The Bcl-2 homology domain 4 (BH4) domain of Bcl-2 mediates its interaction with InsP3Rs (6, 7). This domain also binds the Ca2+/calmodulin-activated serine/threonine (Thr) protein phosphatase calcineurin (CaN) (8). The BH4 domain binds within the regulatory and coupling domain of the InsP3R, located between the InsP3 binding site near the N terminus and the channel domain near the C terminus (6). Protein kinase A (PKA) phosphorylates serine (Ser) 1589 and Ser1755 within this domain, increasing InsP3-mediated channel opening and Ca2+ release (9). Bcl-2 is reported to regulate InsP3R phosphorylation at Ser1755 and to thereby govern InsP3R-mediated Ca2+ release in murine embryonic fibroblasts, although the mechanism is not yet identified (10).
Bcl-2 plays an important role in regulating T-cell development and selection, processes that involve Ca2+ signaling and apoptosis regulation (11, 12). A 20-amino acid peptide (InsP3R-derived peptide, or IDP) corresponding to the Bcl-2 binding site on the InsP3R inhibits Bcl-2–InsP3R interaction, thus eliminating Bcl-2’s control over InsP3R-mediated Ca2+ elevation (6, 7). This peptide induces marked Ca2+ elevation and Ca2+-mediated apoptosis in primary chronic lymphocytic leukemia (CLL) cells and in B-cell lymphoma lines, indicating that Bcl-2–InsP3R interaction contributes to the apoptosis-resistance characteristic of these lymphoid malignancies (13, 14).
Here we report that Bcl-2 interacts with dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32), a potent inhibitor of protein phosphatase 1 (PP1) that is activated by PKA-mediated phosphorylation at Thr 34 and deactivated by CaN-mediated dephosphorylation at this site (15). Our findings indicate that Bcl-2 docks DARPP-32 and CaN on the InsP3R, creating a negative feedback loop that responds to InsP3R-mediated Ca2+ release by inhibiting InsP3R phosphorylation at Ser1755, thereby preventing excessive Ca2+ elevation capable of inducing cell death. Our findings implicate this Bcl-2–CaN–DARPP-32 feedback mechanism in regulating Ser1755 phosphorylation and apoptosis in primary human CLL cells.
Results
Bcl-2 Regulates InsP3R-1 Ser1755 Phosphorylation.
T-cell lines are used in the present work, as in previous studies (8, 13, 16). Jurkat is a human CD4+ T-cell line that expresses readily detectable levels of Bcl-2 (Fig. 1A). The murine WEHI7.2 T-cell line is CD4+/CD8+-positive and thus corresponds to an early developmental stage in which Bcl-2 levels are low. These cells are referred to as “Lo Bcl-2” cells to distinguish them from Bcl-2 overexpressing WEHI7.2 cells, referred to as “Hi Bcl-2” cells (Fig. 1A). Bcl-2 levels are undetectable in the S49 murine T-cell line (Fig. 1A). This line is useful because of two available mutants deficient in PKA activity: one lacking Gαs (Cyc−, historical naming) and the other lacking functional PKA (PKA−) (17).
Fig. 1.

Bcl-2 inhibits anti-CD3–induced elevation of P-Ser1755 InsP3R-1 and Ca2+. (A) Immunoblot showing relative Bcl-2 levels in T-cell lines. (B) Anti-CD3 (20 µg/mL)-induced Ca2+ elevation in WEHI7.2 cells; each trace is average cytoplasmic Ca2+ level in 85 cells in an individual experiment representative of four experiments. (C) Peak cytoplasmic Ca2+ levels before and after adding anti-CD3 to WEHI7.2 cells (mean ± SE, four experiments, 340 cells per experiment). (D) Immunoblot representative of four experiments with similar results, illustrating dynamic changes in P-Ser1755 InsP3R-1 before (0 min) and at 5 and 10 min after adding 20 µg/mL anti-CD3 to WEHI7.2 cells. (E) P-Ser1755 InsP3R-1 levels relative to total InsP3R-1 levels (mean ± SE, n = 4) 5 and 10 min after adding 20 µg/mL anti-CD3 antibody. Ratios representing the 0-min points (before adding anti-CD3) were normalized to a value of one.
In WEHI7.2 cells, Bcl-2 overexpression inhibits anti-CD3–induced Ca2+ elevation, as previously reported (6, 7, 18, 19) (Fig. 1 B and C). Before adding anti-CD3, basal InsP3R-1 phosphorylation at Ser1755 in Hi Bcl-2 WEHI7.2 cells is equal to or greater than phosphorylation at this site in Lo Bcl-2 WEHI7.2 cells. On average, the ratio of phosphorylated (P)-Ser1755 InsP3R-1 levels in Hi Bcl-2 to Lo Bcl-2 cells, normalized to actin, is 1.3 ± 0.3 (n = 7; P = 0.16). InsP3R-1 P-Ser1755 levels increase after anti-CD3 treatment in Lo Bcl-2 WEHI7.2 cells but decrease in Hi Bcl-2 WEHI7.2 cells (Fig. 1 D and E). The increase in Ser1755 phosphorylation in Lo Bcl-2 WEHI7.2 cells is inhibited by the PKA inhibitor H89 (N-[2-[[3-(4-Bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide), as is phosphorylation of cAMP response element-binding protein (CREB), a known PKA target (Fig. S1A). In addition, anti-CD3–mediated T cell receptor (TCR) activation increases InsP3R-1 Ser1755 phosphorylation in S49 cells, but not in Cyc− and PKA− S49 cells (Fig. S1C). These findings are consistent with evidence that TCR activation increases the activity of PKA (20) and indicate that anti-CD3 induced phosphorylation of InsP3R-1 at Ser1755 is PKA-mediated. Moreover, the findings indicate that InsP3R Ser1755 phosphorylation contributes to the increase in cytoplasmic Ca2+ concentration induced by anti-CD3 treatment, as H89 treatment or PKA deficiency are associated with a marked decrease in anti-CD3–induced Ca2+ elevation (Fig. S1 B and D).
In summary, anti-CD3–induced TCR activation increases PKA-mediated phosphorylation of Ser1755 in InsP3R-1, and this phosphorylation enhances anti-CD3–induced Ca2+ elevation. Bcl-2, in contrast, is associated with a decline in Ser1755 phosphorylation induced by anti-CD3 antibody treatment, and this decline correlates with Bcl-2-mediated inhibition of anti-CD3–induced Ca2+ elevation.
Bcl-2-Mediated Inhibition of Ser1755 Phosphorylation Involves Both CaN and PP1.
Because the BH4 domain of Bcl-2 binds CaN, we investigated a possible role of CaN in mediating the Bcl-2-associated decline in Ser1755 phosphorylation. Intracellular Ca2+ chelation by BAPTA-AM [1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)] reverses the inhibitory effect of Bcl-2 on anti-CD3–induced elevation of InsP3R-1 P-Ser1755 (Fig. 2A). In addition, the CaN inhibitor tacrolimus (FK506) (21) increases InsP3R-1 P-Ser1755 levels (Fig. 2 B and C) and reverses the inhibitory effect of Bcl-2 on anti-CD3–induced Ca2+ elevation (Fig. 2 D and E). These findings implicate CaN in the mechanism by which Bcl-2 decreases InsP3R-1 P-Ser1755 after TCR activation. However, PKA-mediated protein phosphorylation is typically regulated by PP1 (22), and the effects of CaN on the InsP3R are predicted to be indirect and secondary to the action of PP1 (23).
Fig. 2.

CaN and PP1 mediate P-Ser1755 InsP3R-1 regulation by Bcl-2. (A) Immunoblot representative of three experiments indicating that Ca2+ chelation reverses the inhibitory effect of Bcl-2 on anti-CD3 (20 µg/mL)-induced P-Ser1755 InsP3R-1. (B) Pretreatment with FK506 (0.5 µM for 2 h) reverses the inhibitory effect of Bcl-2 on anti-CD3-induced P-Ser1755 InsP3R-1. (C) P-Ser1755 InsP3R-1 levels relative to total InsP3R-1 levels (mean ± SE, n = 3) 5 min after anti-CD3 antibody, indicating that pretreatment with FK506 (0.5 µM for 2 h) reverses the inhibitory effect of Bcl-2 on anti-CD3-induced P-Ser1755 InsP3R-1. (D) Ca2+ traces representing the average cytoplasmic Ca2+ level in 85 cells in individual experiments (n = 3), indicating that pretreatment with FK506 (0.5 µM for 2 h) reverses the inhibitory effect of Bcl-2 on anti-CD3-induced Ca2+ elevation. (E) Summary of cytoplasmic Ca2+ concentrations before and after anti-CD3 addition to Hi Bcl-2 WEHI7.2 cells pretreated for 2 h with DMSO vehicle or FK506 (mean ± SE, n = 3 experiments). (F) In vitro phosphatase assays indicating that PP1, but not PP2A, dephosphorylates P-Ser1755 InsP3R-1 (Upper, representative immunoblots; Lower, P-Ser1755 InsP3R-1 phosphorylation levels relative to total InsP3R-1 levels determined by densitometry of immunoblots; mean ± SD). (G) Immunoblots from a representative in vitro phosphatase assay indicating that CaN does not directly dephosphorylate InsP3R-1 P-Ser1755; Ca2+-dependent dephosphorylation of NFAT-c2 serves as a positive control.
Therefore, in vitro phosphatase assays were used to clarify the respective roles of CaN and PP1 in regulating Ser1755 phosphorylation on InsP3R-1. WEHI7.2 cells were pretreated with forskolin to increase InsP3R-1 P-Ser1755 levels, and then InsP3R-1s were immunoprecipitated from cell lysates. Immune-captured InsP3R-1 was incubated with purified PP1 or PP2A enzymes in a phosphatase assay buffer (24). Under these conditions, recombinant PP1 and PP2A retain their substrate specificities (24). PP1, but not PP2A, causes a dose-dependent decrease of InsP3R-1 P-Ser1755 (Fig. 2F). Consistent with this conclusion, 2 µM okadaic acid, a concentration that inhibits PP1 (22, 25, 26), reverses the inhibitory effect of Bcl-2 on anti-CD3–induced elevation of P-Ser1755 InsP3R-1 and Ca2+ (Fig. S2). Moreover, an in vitro phosphatase assay was used to assess the effect of CaN on InsP3R-1 P-Ser1755 (21). Incubation with purified CaN does not alter the phosphorylation of immune-captured InsP3R-1, regardless of the presence of Ca2+ (Fig. 2G). CaN shifts the mobility of a typical CaN target, NFATc2, ensuring that CaN is active in the assay system (Fig. 2G).
These findings indicate a direct role for PP1 and an indirect role for CaN in Bcl-2-mediated regulation of InsP3R-1 phosphorylation at Ser1755, suggesting potential involvement of DARPP-32 in regulating InsP3R phosphorylation at Ser1755.
DARPP-32 and CaN Interact with Bcl-2 on the InsP3R-1 and Regulate Ca2+ Elevation and Cell Survival.
DARPP-32 is detected at equal levels in both Lo Bcl-2 and Hi Bcl-2 WEHI7.2 cells, along with InsP3R-1, PKA, PP1, and CaN, the levels of which are similar regardless of differing Bcl-2 levels (Fig. 3A). Bcl-2 immunoprecipitation from Lo Bcl-2 and Hi Bcl-2 cells pulls down both DARPP-32 and CaN, whereas these proteins are not pulled down by control IgG antibody (Fig. 3B). Bcl-2–DARPP-32 interaction is confirmed in the reciprocal experiment, where DARPP-32 is immunoprecipitated, pulling down both InsP3R-1 and Bcl-2 (Fig. 3C). DARPP-32 also coimmunoprecipitates with ΔBH4-Bcl-2, a previously described Bcl-2 mutant that lacks the BH4 domain and neither interacts with the InsP3R-1 or CaN nor regulates TCR-induced Ca2+ elevation (6, 16) (Fig. 3D).
Fig. 3.

Bcl-2 recruits DARPP-32 and CaN to InsP3R-1, regulating DARPP-32 phosphorylation at Thr34 after anti-CD3-mediated TCR activation. (A) Immunoblots comparing levels of InsP3R-1 and other proteins in Lo Bcl-2 and Hi Bcl-2 WEHI7.2 cells and in WEHI7.2 cells expressing Bcl-2 6,7mut, which does not interact with InsP3R-1. (B) Coimmunoprecipitation (coip) of DARPP-32 and CaN with Bcl-2 (confirmed in four experiments). CaN input is best seen on prolonged exposure, whereas CaN coip is best compared in Lo vs. Hi Bcl-2 cells on short exposure. (C) coip of InsP3R-1 and Bcl-2 with DARPP-32 (confirmed in four experiments). (D) coip of DARPP-32 with either Bcl-2 or Bcl-2 6,7mut and with ΔBH4Bcl-2, which lacks the BH4 domain (confirmed in three experiments). (E) Immunoblots demonstrating an increase in P-Thr34 DARPP-32 levels induced by anti-CD3 in Lo Bcl-2 WEHI7.2 cells and in WEHI7.2 cells expressing Bcl-2 6,7mut. In contrast, P-Thr34 DARPP-32 levels decline in Hi Bcl-2 WEHI7.2 cells after anti-CD3 addition (confirmed in three experiments).
These findings indicate that DARPP-32 interaction with Bcl-2 by itself is insufficient to regulate Ca2+ and that CaN interaction with the BH4 domain of Bcl-2 and Bcl-2 interaction with the InsP3R are required to form a functional Bcl-2–CaN–DARPP-32 complex. This conclusion is substantiated by experiments in WEHI7.2 cells expressing Bcl-2 6,7mut, a previously described dicodon mutant in which arginine at position 6 and serine at position 7 in the BH4 domain of Bcl-2 are replaced by glycine (6). Immunoprecipitation of this mutant form of Bcl-2 pulls down DARPP-32 (Fig. 3D). Because this Bcl-2 mutant neither interacts with InsP3Rs nor inhibits anti-CD3–induced Ca2+ elevation (6), the coimmunoprecipitation of DARPP-32 with Bcl-2 6,7mut confirms that the Bcl-2–DARPP-32 complex must be localized to the InsP3R to function in regulating InsP3R-mediated Ca2+ release. Consistent with these observations, TCR activation induces PKA-mediated DARPP-32 phosphorylation at Thr34 in cells expressing low levels of Bcl-2 or Bcl-2 6,7mut, whereas in cells expressing high levels of wild-type Bcl-2, basal P-Thr34 levels are already elevated and decline after TCR activation (Fig. 3E).
These findings suggest that the decline in InsP3R Ser1755 phosphorylation observed in Bcl-2-positive WEHI7.2 cells is mediated by a Ca2+/CaN-induced dephosphorylation of DARPP-32 at Thr34, thus eliminating its inhibition of PP1 and resulting in PP1-mediated InsP3R Ser1755 dephosphorylation.
Reversal of Bcl-2’s Action by Knocking Down Bcl-2 or DARPP-32.
The Bcl-2 level in Jurkat T-cells is sufficient to attenuate anti-CD3–induced Ca2+ elevation (6). Here we find that siRNA-mediated Bcl-2 knockdown in Jurkat cells reverses the inhibitory effect of Bcl-2 on InsP3R Ser1755 phosphorylation (Fig. 4 A and B). Thus, an anti-CD3–induced elevation of InsP3R Ser1755 phosphorylation is observed in cells in which Bcl-2 is knocked down, but not in control cells in which Bcl-2 is present and increased after anti-CD3 treatment. As reported previously (6), knocking down Bcl-2 results in increased Ca2+ elevation in response to anti-CD3 (Fig. 4C). Knocking down DARPP-32 (Fig. 4D) also increases InsP3R Ser1755 phosphorylation (Fig. 4 E and F). Moreover, DARPP-32 knockdown increases anti-CD3–induced Ca2+ elevation (Fig. 4G) and apoptosis (Fig. 4H) in Jurkat cells, suggesting a role for this feedback mechanism in preserving cell survival after TCR activation.
Fig. 4.
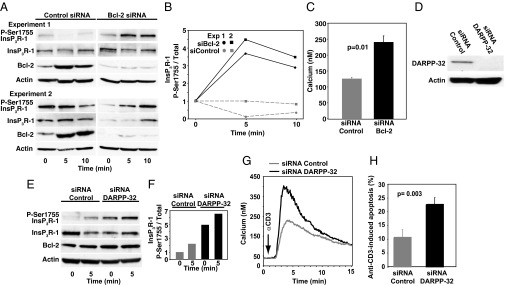
Knockdown of Bcl-2 or DARPP-32 reverses Bcl-2’s inhibition of InsP3R-1 phosphorylation, Ca2+ elevation, and apoptosis. (A) Immunoblots from two experiments in which siRNA Bcl-2 knockdown reversed Bcl-2’s inhibition of anti-CD3-induced P-Ser1755 InsP3R-1 elevation. (B) P-Ser1755 InsP3R-1 levels relative to total InsP3R-1 levels at 5 and 10 min after anti-CD3 addition; ratios representing the 0-min point (before anti-CD3) were normalized to a value of one (two experiments). (C) Symbols represent peak Ca2+ elevation (mean ± SEM, n = 3 experiments, 85 cells per treatment per experiment) induced by anti-CD3 in Jurkat cells and measured by digital imaging. Cells were pretreated with control siRNA or Bcl-2 siRNA to knockdown Bcl-2. (D) Immunoblot documenting DARPP-32 knockdown. (E) Immunoblot representative of two experiments with the same result, showing the effect of DARPP-32 knockdown on P-Ser1755 InsP3R levels before and 5 min after adding anti-CD3 to Jurkat cells. (F) P-Ser1755 InsP3R-1 levels relative to total InsP3R-1 levels 5 min after anti-CD3 addition (same experiments as in E; ratios representing the 0-min point before anti-CD3 addition were normalized to one). (G) Ca2+ elevation induced by anti-CD3 addition (arrow) in Jurkat cells pretreated with control or DARPP-32 siRNA. Traces represent average Ca2+ levels in 85 cells in a single experiment representative of three experiments. (H) Percentage of cells undergoing apoptosis 8–18 h after anti-CD3 addition (background apoptosis, before anti-CD3 addition, subtracted; mean ± SEM, six experiments).
Regulation of P-Ser1755 InsP3R-1 Involving Bcl-2 and DARPP-32 in CLL Cells.
To investigate the potential relevance of these findings in pathological conditions, we investigated InsP3R phosphorylation in CLL, a common form of leukemia invariably associated with elevated Bcl-2 levels, combined with tonic B-cell receptor signaling expected to generate InsP3R-mediated Ca2+ elevation (27). In recent studies, we found that treating primary human CLL cells with the peptide TAT-IDPDD/AA, which inhibits Bcl-2–InsP3R interaction, induces Ca2+ elevation and, consequently, apoptosis (13). Here we detect the expression of DARPP-32 in primary human CLL cells (Fig. S3A) and find that treating these cells with TAT-IDPDD/AA increases P-Ser1755 InsP3R-1 levels (Fig. S3 B and C), followed by Ca2+ elevation (Fig. S2D). These findings indicate that peptide-mediated disruption of Bcl-2–InsP3R interaction overcomes Bcl-2-imposed repression of P-Ser1755 InsP3R-1 levels and InsP3R-1-mediated Ca2+ release. These findings suggest that CLL cells exploit the Bcl-2–CaN–DARPP-32-mediated negative feedback mechanism to prevent Ca2+ elevation and cell death.
Discussion
The findings reported here indicate that Bcl-2 provides a molecular platform bringing CaN and DARPP-32 together on InsP3R-1, producing a rapid feedback mechanism that senses InsP3R-1-mediated Ca2+ release and responds by inhibiting PKA-mediated InsP3R-1 phosphorylation at Ser1755 (Fig. 5). In a two-step process, CaN initiates the feedback process by sensing Ca2+ elevation and responds by dephosphorylating DARPP-32, thereby eliminating its inhibition of PP1. The Ca2+ elevation that triggers CaN activation in this step, we posit, is localized at the mouth of the InsP3R, where CaN is positioned in close proximity to Bcl-2. Thus, this Ca2+ signal is not detected by the global measurement of intracellular Ca2+ used in this study. In the second step, PP1 dephosphorylates P-Ser1755 InsP3R-1, decreasing the InsP3R-mediated Ca2+ release that is readily detected by global Ca2+ measurements (e.g., Fig. 1B). In creating a negative feedback loop, Bcl-2 exploits the properties of CaN and DARPP-32 by targeting them to InsP3R-1, where they respond rapidly to the Ca2+ exiting the InsP3R-1 channel (Fig. 5). The requirement for Bcl-2 to assemble CaN and DARPP-32 in close proximity to InsP3R-1, whether all four proteins are in the same Bcl-2 complex or in close proximity in distinct pools of Bcl-2, is supported experimentally by evidence that the feedback mechanism is not operational when Bcl-2 is substituted with mutant forms of Bcl-2 that do not interact with InsP3R-1. Moreover, as PKA phosphorylates Ser1588 with similar effects on InsP3R-1-mediated Ca2+ release (9), it is likely that the Bcl-2-mediated feedback mechanism would also regulate phosphorylation at this site, contributing to the effects of Bcl-2 on InsP3R-mediated Ca2+ release.
Fig. 5.

Diagram of how the Bcl-2–CaN–DARPP-32 signaling complex functions as a negative feedback loop controlling InsP3R-1-mediated Ca2+ release. (A) Bcl-2 serves as a platform docking CaN and DARPP-32 to InsP3R-1. For simplicity, DARPP-32-Thr34 and InsP3R-1-Ser1755 are depicted as being in a dephosphorylated state before TCR activation, although levels of Ser1755-InsP3R and Thr34-DARPP-32 phosphorylation are variably increased in Bcl-2 overexpressing cells (see Discussion). (B) TCR stimulation by anti-CD3 increases activities of both phospholipase C (PLC), to increase InsP3 synthesis, and adenylyl cyclase, to increase cAMP. The latter activates PKA, which phosphorylates Thr34-DARPP-32. PKA also phosphorylates Ser1755-InsP3R-1, and this phosphorylation step is enhanced by the PP1 inhibitory activity of P-Thr34-DARPP-32. (C) InsP3 activates InsP3R-1 channel opening, which is enhanced by its phosphorylation at Ser1755. Ca2+ released from the endoplasmic reticulum activates CaN, which dephosphorylates P-Thr34-DARPP-32, alleviating inhibition of PP1 and initiating the feedback process. (D) Continuing the feedback process, PP1 dephosphorylates P-Ser1755, decreasing InsP3R-1 channel opening and thus reducing Ca2+ release, accompanied by the inactivation of InsP3 either through hydrolysis to InsP2 or phosphorylation to InsP4.
In summarizing the findings in Fig. 5, we emphasize the dynamic changes in P-Ser1755-InsP3R-1 and P-Thr34-DARPP-32 that constitute the rapid feedback mechanism that takes place after TCR activation. For simplicity, this diagram does not show an increased basal phosphorylation of Ser1755-InsP3R-1 and Thr34-DARPP-32, although it is detected in certain experiments (Figs. 1D and 3E). Because PKA associates with InsP3R-1 (4), we hypothesize that an elevation of basal P-Ser1755 InsP3R-1 in cells with elevated Bcl-2, when observed, is secondary to constitutive TCR activity mediating basal PKA activity that increases P-Thr34-DARPP-32 levels, in turn inhibiting PP1 and thus raising the basal level of P-Ser1755 InsP3R-1.
DARPP-32 has been investigated extensively in the brain, where it localizes to regions enriched in dopaminergic nerve terminals (15). The possibility that DARPP-32 may mediate effects of CaN on InsP3R-1-induced Ca2+ release had been suggested previously (22, 23). Moreover, Tang and colleagues (22) discovered a direct association between PP1 and InsP3R-1 and established that the association with PP1 facilitates dephosphorylation of PKA-phosphorylated InsP3R-1. These investigators established the role of AKAP9, a multifunctional PKA anchoring protein, in docking PKA and PP1 to InsP3R-1 (28) and in experiments with medium spiny neurons from DARPP-32 knock-out mice, demonstrated a regulatory role of DARPP-32 in dopamine-induced Ca2+ oscillations (29). Although these results advance our understanding of crosstalk between cAMP and InsP3-mediated Ca2+ signaling pathways in the brain, much less is known about the role of DARPP-32 in peripheral tissues, including lymphocytes, although DARPP-32 has been shown to increase the phosphorylation and activity of various ion channels (30). The findings reported here are an indication of an interaction between Bcl-2 and DARPP-32 and of a role of this interaction in regulating Ca2+ signaling and cell survival.
Our findings indicate that the negative feedback mechanism conferred through Bcl-2 interaction with DARPP-32 and CaN contributes to the antiapoptotic function of the Bcl-2 protein. By regulating PKA-mediated InsP3R phosphorylation at Ser1755, the Bcl-2–CaN–DARPP-32 complex sets a threshold level that Ca2+ cannot exceed, thereby preventing Ca2+-induced cell death. Knocking down either Bcl-2 or DARPP-32 abrogates this protective mechanism, elevating Ser1755 phosphorylation and thus increasing Ca2+ elevation after TCR activation, leading to loss of cell viability. Moreover, IDPDD/AA-mediated inhibition of Bcl-2–InsP3R interaction also abrogates this feedback mechanism, leading to cell death in primary human CLL cells. Thus, the Bcl-2–CaN–DARPP-32-mediated feedback mechanism may be essential to block proapoptotic Ca2+ signals and thereby prolong survival of CLL cells and possibly other Bcl-2-positive malignancies.
Although the present report focuses on Ca2+ signaling in lymphocytes, one can speculate that the Bcl-2–CaN–DARPP-32 feedback mechanism may also function in neuronal cells and that defects in this pathway may contribute to neuropsychiatric illnesses. Bcl-2 plays an important role in the development and survival of neuronal cells (31). Moreover, the neuroprotective effect of Bcl-2 in primary neuronal cells involves shuttling CaN to InsP3Rs to regulate Ca2+ elevation (32). Exaggerated Ca2+ signals contribute to neuronal dysfunction in patients with bipolar disorder, schizophrenia, and Alzheimer disease (reviewed in ref. 33). In bipolar disorder, exaggerated Ca2+ signals correlate with single nucleotide polymorphisms associated with decreased Bcl-2 expression levels (34, 35). Moreover, the mechanism of action of mood stabilizers effective in treating this disorder involves, at least in part, elevation of Bcl-2 and stabilization of Ca2+ signaling (36). DARPP-32, in contrast, regulates many pathways in the central nervous system coupled with long-term plasticity and control of behavior (15). Altered DARPP-32 function has been implicated in the pathogenesis of schizophrenia (15), and DARPP-32 expression is deficient in leukocytes from patients with schizophrenia and bipolar disorder (37). Therefore, future studies are needed to investigate a potential functional link between unregulated Ca2+ signaling in these disorders and potential alterations in a Bcl-2–CaN–DARPP-32 feedback pathway required to prevent excessive Ca2+ signaling.
Materials and Methods
Reagents, primary cells, and cell lines have been described previously (6, 13). Also described are methods of peptide synthesis, immunoblotting and immunoprecipitation (6), T-cell stimulation by anti-CD3 antibodies and Ca2+ imaging (6), RNA interference (6), and in vitro phosphatase assays (24). Details of these methods and statistical analysis are in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Susann Brady-Kalnay, Zhenghe Wang, Shigemi Matsuyama, Humbert De Smedt, and Geert Bultynck for helpful discussions and Paolo Caimi, Ashley Rosko, Brenda Cooper, and Erica Campagnaro for providing CLL samples from their patients. We also thank David Yule for providing the DT40 cells used in verifying phospho-specific InsP3R antibodies. This work was supported by National Institutes of Health Grants RO1 CA085804 (to C.W.D.), R25 CA148052 (to M.-J.C.), 5T32HL007147 (to A.R.L.) and 5T32GM007250 (to A.R.L.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1323098111/-/DCSupplemental.
References
- 1.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: Dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4(7):517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 2.Mikoshiba K. IP3 receptor/Ca2+ channel: From discovery to new signaling concepts. J Neurochem. 2007;102(5):1426–1446. doi: 10.1111/j.1471-4159.2007.04825.x. [DOI] [PubMed] [Google Scholar]
- 3.Joseph SK, Hajnóczky G. IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis. 2007;12(5):951–968. doi: 10.1007/s10495-007-0719-7. [DOI] [PubMed] [Google Scholar]
- 4.Vanderheyden V, et al. Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim Biophys Acta. 2009;1793(6):959–970. doi: 10.1016/j.bbamcr.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Distelhorst CW, Bootman MD. Bcl-2 interaction with the inositol 1,4,5-trisphosphate receptor: Role in Ca(2+) signaling and disease. Cell Calcium. 2011;50(3):234–241. doi: 10.1016/j.ceca.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rong YP, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol Cell. 2008;31(2):255–265. doi: 10.1016/j.molcel.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monaco G, et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012;19(2):295–309. doi: 10.1038/cdd.2011.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shibasaki F, Kondo E, Akagi T, McKeon F. Suppression of signalling through transcription factor NF-AT by interactions between calcineurin and Bcl-2. Nature. 1997;386(6626):728–731. doi: 10.1038/386728a0. [DOI] [PubMed] [Google Scholar]
- 9.Wagner LE, 2nd, Li W-H, Yule DI. Phosphorylation of type-1 inositol 1,4,5-trisphosphate receptors by cyclic nucleotide-dependent protein kinases: A mutational analysis of the functionally important sites in the S2+ and S2- splice variants. J Biol Chem. 2003;278(46):45811–45817. doi: 10.1074/jbc.M306270200. [DOI] [PubMed] [Google Scholar]
- 10.Oakes SA, et al. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102(1):105–110. doi: 10.1073/pnas.0408352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andjelić S, Jain N, Nikolić-Zugić J. Immature thymocytes become sensitive to calcium-mediated apoptosis with the onset of CD8, CD4, and the T cell receptor expression: A role for bcl-2? J Exp Med. 1993;178(5):1745–1751. doi: 10.1084/jem.178.5.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siegel RM, et al. Inhibition of thymocyte apoptosis and negative antigenic selection in bcl-2 transgenic mice. Proc Natl Acad Sci USA. 1992;89(15):7003–7007. doi: 10.1073/pnas.89.15.7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong F, et al. Induction of Ca2+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood. 2011;117(10):2924–2934. doi: 10.1182/blood-2010-09-307405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akl H, et al. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis. 2013;4:e632. doi: 10.1038/cddis.2013.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walaas SI, Hemmings HC, Jr, Greengard P, Nairn AC. Beyond the dopamine receptor: Regulation and roles of serine/threonine protein phosphatases. Front Neuroanat. 2011;5:50. doi: 10.3389/fnana.2011.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rong YP, et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci USA. 2009;106(34):14397–14402. doi: 10.1073/pnas.0907555106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan L, Herrmann V, Hofer JK, Insel PA. β-adrenergic receptor/cAMP-mediated signaling and apoptosis of S49 lymphoma cells. Am J Physiol Cell Physiol. 2000;279(5):C1665–C1674. doi: 10.1152/ajpcell.2000.279.5.C1665. [DOI] [PubMed] [Google Scholar]
- 18.Chen R, et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol. 2004;166(2):193–203. doi: 10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong F, Davis MC, McColl KS, Distelhorst CW. Bcl-2 differentially regulates Ca2+ signals according to the strength of T cell receptor activation. J Cell Biol. 2006;172(1):127–137. doi: 10.1083/jcb.200506189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mosenden R, Taskén K. Cyclic AMP-mediated immune regulation—overview of mechanisms of action in T cells. Cell Signal. 2011;23(6):1009–1016. doi: 10.1016/j.cellsig.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 21.Fruman DA, Klee CB, Bierer BE, Burakoff SJ. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci USA. 1992;89(9):3686–3690. doi: 10.1073/pnas.89.9.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang TS, Tu H, Wang Z, Bezprozvanny I. Modulation of type 1 inositol (1,4,5)-trisphosphate receptor function by protein kinase a and protein phosphatase 1alpha. J Neurosci. 2003;23(2):403–415. doi: 10.1523/JNEUROSCI.23-02-00403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bultynck G, et al. Calcineurin and intracellular Ca2+-release channels: Regulation or association? Biochem Biophys Res Commun. 2003;311(4):1181–1193. doi: 10.1016/j.bbrc.2003.08.084. [DOI] [PubMed] [Google Scholar]
- 24.Ammosova T, et al. Protein phosphatase-1 activates CDK9 by dephosphorylating Ser175. PLoS ONE. 2011;6(4):e18985. doi: 10.1371/journal.pone.0018985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen P, Holmes CF, Tsukitani Y. Okadaic acid: A new probe for the study of cellular regulation. Trends Biochem Sci. 1990;15(3):98–102. doi: 10.1016/0968-0004(90)90192-e. [DOI] [PubMed] [Google Scholar]
- 26.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: A Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87(7):1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 27.Stevenson FK, Krysov S, Davies AJ, Steele AJ, Packham G. B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2011;118(16):4313–4320. doi: 10.1182/blood-2011-06-338855. [DOI] [PubMed] [Google Scholar]
- 28.Tu H, Tang TS, Wang Z, Bezprozvanny I. Association of type 1 inositol 1,4,5-trisphosphate receptor with AKAP9 (Yotiao) and protein kinase A. J Biol Chem. 2004;279(18):19375–19382. doi: 10.1074/jbc.M313476200. [DOI] [PubMed] [Google Scholar]
- 29.Tang TS, Bezprozvanny I. Dopamine receptor-mediated Ca(2+) signaling in striatal medium spiny neurons. J Biol Chem. 2004;279(40):42082–42094. doi: 10.1074/jbc.M407389200. [DOI] [PubMed] [Google Scholar]
- 30.Svenningsson P, et al. DARPP-32: An integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–296. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- 31.Merry DE, Veis DJ, Hickey WF, Korsmeyer SJ. bcl-2 protein expression is widespread in the developing nervous system and retained in the adult PNS. Development. 1994;120(2):301–311. doi: 10.1242/dev.120.2.301. [DOI] [PubMed] [Google Scholar]
- 32.Erin N, Lehman RAW, Boyer PJ, Billingsley ML. In vitro hypoxia and excitotoxicity in human brain induce calcineurin-Bcl-2 interactions. Neuroscience. 2003;117(3):557–565. doi: 10.1016/s0306-4522(02)00934-x. [DOI] [PubMed] [Google Scholar]
- 33.Berridge MJ. Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion. 2013;7(1):2–13. doi: 10.4161/pri.21767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warsh JJ, Andreopoulos S, Li PP. Role of intracellular calcium signaling in the pathophysiology and pharmacology of bipolar disorder. Current status. Clin Neurosci Res. 2004;4:201–213. [Google Scholar]
- 35.Machado-Vieira R, et al. The Bcl-2 gene polymorphism rs956572AA increases inositol 1,4,5-trisphosphate receptor-mediated endoplasmic reticulum calcium release in subjects with bipolar disorder. Biol Psychiatry. 2011;69(4):344–352. doi: 10.1016/j.biopsych.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rapoport SI, Basselin M, Kim HW, Rao JS. Bipolar disorder and mechanisms of action of mood stabilizers. Brain Res Brain Res Rev. 2009;61(2):185–209. doi: 10.1016/j.brainresrev.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torres KC, et al. The leukocytes expressing DARPP-32 are reduced in patients with schizophrenia and bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(2):214–219. doi: 10.1016/j.pnpbp.2008.10.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.