

Significance
The acute respiratory distress syndrome (ARDS) is a devasting clinical problem with high mortality, no drug therapy, and poorly understood pathogenesis. The hallmark of ARDS is persistent pulmonary edema, attributable in part to impaired Na+ and fluid transport across the alveolo-capillary barrier, undertaken by the epithelial sodium channel (ENaC). We describe a unique signaling pathway driven by TGF-β, which acutely dysregulates ENaC trafficking, blocking alveolar Na+ transport and edema resolution. This pathway represents a unique pathomechanism in ARDS, highlights potential “druggable” targets, and may represent a physiological means of acutely regulating ENaC in lungs and other organs.
Keywords: alveolar epithelium, fluid homeostasis
Abstract
TGF-β is a pathogenic factor in patients with acute respiratory distress syndrome (ARDS), a condition characterized by alveolar edema. A unique TGF-β pathway is described, which rapidly promoted internalization of the αβγ epithelial sodium channel (ENaC) complex from the alveolar epithelial cell surface, leading to persistence of pulmonary edema. TGF-β applied to the alveolar airspaces of live rabbits or isolated rabbit lungs blocked sodium transport and caused fluid retention, which—together with patch-clamp and flow cytometry studies—identified ENaC as the target of TGF-β. TGF-β rapidly and sequentially activated phospholipase D1, phosphatidylinositol-4-phosphate 5-kinase 1α, and NADPH oxidase 4 (NOX4) to produce reactive oxygen species, driving internalization of βENaC, the subunit responsible for cell-surface stability of the αβγENaC complex. ENaC internalization was dependent on oxidation of βENaC Cys43. Treatment of alveolar epithelial cells with bronchoalveolar lavage fluids from ARDS patients drove βENaC internalization, which was inhibited by a TGF-β neutralizing antibody and a Tgfbr1 inhibitor. Pharmacological inhibition of TGF-β signaling in vivo in mice, and genetic ablation of the nox4 gene in mice, protected against perturbed lung fluid balance in a bleomycin model of lung injury, highlighting a role for both proximal and distal components of this unique ENaC regulatory pathway in lung fluid balance. These data describe a unique TGF-β–dependent mechanism that regulates ion and fluid transport in the lung, which is not only relevant to the pathological mechanisms of ARDS, but might also represent a physiological means of acutely regulating ENaC activity in the lung and other organs.
The acute respiratory distress syndrome (ARDS) is a devastating syndrome characterized by alveolar flooding (edema), which impairs gas exchange, leading to respiratory failure (1). The high mortality rate of 35–45% observed in patients with ARDS and the lack of any pharmacological therapy (1) underscores the need to better understand the pathomechanisms of this lethal disease, in the hope of facilitating improved clinical management of affected patients.
Alveolar edema occurs as a consequence of increased fluid influx into the alveolar airspaces from the vasculature, across the thin alveolo-capillary barrier (2), as well as a failure of transepithelial Na+ and Cl− ion transport, which drives fluid clearance from the alveolar airspaces. Transepithelial sodium transport is undertaken by the concerted action of several ion transporters, namely the Na+/K+-ATPase (3) and the epithelial sodium channel (ENaC) (4, 5), which actively transport Na+ out of the fluid lining the alveolar airspaces (epithelial lining fluid, ELF). This process generates an osmotic gradient that clears water from the alveolar airspaces (6). This fluid clearance process is defective in ARDS patients with compromised alveolo-capillary barrier function, and it is widely believed that edema fluid must be cleared for patients with ARDS to survive (7, 8).
TGF-β is a key mediator of acute lung injury (ALI), where TGF-β is activated locally by integrin αvβ6 (9) in cooperation with protease-activated receptor-1 (10), to increase epithelial and endothelial permeability and promote alveolar flooding. In further support of a role for TGF-β in ALI, two studies have demonstrated increased TGF-β levels in lung fluids from patients with ALI/ARDS (11, 12), and in these patients lower TGF-β levels correlate with more ventilator-free and intensive care unit-free days (11). Some evidence has also implicated TGF-β in transepithelial ion transport in vitro, where TGF-β down-regulated gene expression of one of three ENaC subunits (13), temporally modulated gene expression of the Na+/K+-ATPase (14), and impacted Cl− transport (15). In animal models of ALI/ARDS, administration of a soluble type II TGF-β receptor, which sequesters free TGF-β, attenuated the degree of pulmonary edema (9), confirming a role for TGF-β in disturbed lung fluid dynamics associated with experimental ALI/ARDS, however, a role for TGF-β in regulating alveolar fluid reabsorption has not been established.
In this study, a unique TGF-β signaling pathway is described, whereby TGF-β—acting through the Tgfbr1/Smad2/3 axis (16)—recruits phosphoinositide-metabolizing enzymes and an NADPH oxidase to generate reactive oxygen species (ROS), which drive αβγENaC complex internalization from the lung epithelial cell surface and, hence, block the sodium-transporting capacity of alveolar epithelial cells. Using animal and isolated organ models of edema resolution, we demonstrate that TGF-β, applied at clinically relevant doses, rapidly blocked the transepithelial ion fluxes necessary for alveolar fluid reabsorption, and indeed alveolar fluid reabsorption itself. Given the rapid onset and progression of ARDS and the critical role played by ENaC-mediated alveolar fluid clearance in the survival of ARDS patients, the pathway described here has important implications for the understanding of the pathological mechanisms that promote formation or persistence of alveolar edema in ARDS patients. This idea is highlighted by the findings reported here that identify TGF-β, exclusively, as the factor in the lung fluids of ARDS patients responsible for promoting loss of ENaC from the lung epithelial cell surface. In addition to revealing an entirely unique TGF-β signaling pathway that regulates ion channel trafficking, these data point to a pathway that may be amenable to pharmacological manipulation in patients with ARDS, a devastating and lethal syndrome for which no pharmacological therapy currently exists.
Results
TGF-β Levels Are Elevated in Lavage Fluids of Patients with ARDS.
Levels of active TGF-β1 were elevated in bronchoalveolar lavage (BAL) fluids of mechanically ventilated patients with ARDS [137.1 ± 55.2 pg/mL by ELISA (Fig. S1A); 70.0 ± 38.8 pg/mL by bioassay (Fig. S1B); (n = 17)]. Active TGF-β1 could not be detected in BAL fluids from healthy volunteers (Fig. S1A) (ELISA detection limit ∼7.5 pg/mL), but was detected by bioassay in three of eight volunteers (2.7 ± 4.2 pg/mL) (Fig. S1B), approaching the bioassay detection limit (∼1 pg/mL). BAL fluid TGF-β levels were elevated in ARDS patients irrespective of disease etiology (Fig. S1 C and D). Using a BAL fluid to ELF conversion of 1:100 (17), TGF-β levels in ELF of ARDS patients are estimated at 7–14 ng/mL.
Exogenous TGF-β Application Blocks Fluid Reabsorption in Live, Anesthetized, and Ventilated Rabbits.
TGF-β was nebulized into the lungs of live anesthetized rabbits at a deposited concentration of 10 ng/mL in the ELF, followed 30 min later by a 1-mL fluid challenge. After 60 min, the ELF volume in TGF-β–treated rabbits was twofold increased, from 0.65 ± 0.02 mL in control rabbits to 1.12 ± 0.12 mL, indicating fluid accumulation in the lung (Fig. 1A). Similarly, pretreatment with amiloride (10 μM in ELF), an inhibitor of ENaC, caused a 1.5-fold increase in ELF volume, but the effects of pretreatment with amiloride and TGF-β together was not additive over each agent applied alone (Fig. 1A), suggesting that amiloride and TGF-β may share the same targets in the rabbit lung.
Fig. 1.

TGF-β blocks alveolar ion and fluid transport. (A) TGF-β or vehicle (Veh) was nebulized into the lungs of live, anesthetized rabbits. Changes in ELF volume was assessed 60 min later (n = 4, per group). The ELF volume (B) was assessed in isolated, ventilated, and perfused rabbit lungs pretreated with vehicle (Veh), low temperature (8 °C), ouabain (ouab; 10 μM), TGF-β, SB431542 (SB), amiloride (Amil), phalloidin oleate (PO), or combinations thereof, 60 min after application of a 2-mL fluid challenge (n = 8, per group; * indicates vs. vehicle). (C) 22Na+ clearance from isolated, ventilated, and perfused rabbit lungs, treated with vehicle (blue) or low temperature (8 °C, red), ouabain (10 μM, purple); TGF-β (10 ng/mL in ELF, green], SB431542 (SB, 10 μM in ELF, yellow), and TGF-β after SB431542 preapplication (brown). Data quantified from multiple experiments (n = 8, per group; * indicates vs. vehicle) for passive transport, determined from [3H]mannitol flux, and the active component of 22Na+ clearance. (D) Effects of amiloride (Amil, 10 μM in ELF, orange) and PO (1 μM in the ELF, yellow) alone, and amiloride (black) and PO (brown) applied 30 min before TGF-β application. Data quantified described for A. Data represent mean ± SD; *P < 0.05. NS, not significant.
Exogenous TGF-β Application Blocks Fluid Reabsorption in Isolated, Ventilated, and Perfused Rabbit Lungs.
Similar to the observations in live rabbits, preapplication of TGF-β to an isolated, ventilated, and perfused rabbit lung led to a net gain in the steady-state lung mass of 1.35 ± 0.27 g 60 min after a 2-mL fluid challenge, indicating fluid retention in the lung, which was also seen when lungs were maintained at 8 °C (which block active ion and fluid transport) or treated with amiloride (Fig. S1F). Lung fluid retention driven by TGF-β was blocked by preapplication of the TGF-β signaling inhibitor SB431542 (10 μM in ELF) and by phalloidin oleate (PO; 1 μM in ELF), a cell membrane-permeable inhibitor of F→G actin conversion that blocks endocytosis. Trends in changes in the ELF volume of the isolated, ventilated, and perfused rabbit lungs (Fig. 1B) paralleled the observations made on net steady-state lung mass (Fig. S1F). TGF-β did not impact lung endothelial permeability, assessed by capillary filtration coefficient (Kf,c) (Fig. S1E), but oleic acid, used as a positive control, did. Taken together, these data demonstrate that TGF-β applied to the alveolar airspaces blocks alveolar fluid reabsorption.
TGF-β Blocks Active 22Na+ Efflux from the Alveolar Airspaces in Isolated, Ventilated, and Perfused Rabbit Lungs.
Transepithelial Na+ transport out of the alveolar airspaces drives alveolar fluid clearance (18). Application of TGF-β (10 ng/mL in ELF) reduced the active component of 22Na+ efflux from the alveolar airspaces by 80% (Fig. 1 C and D) in isolated, ventilated, and perfused rabbit lungs. This effect was blocked by preapplication of SB431542 or PO, consistent with our net steady-state lung mass and ELF volume data (Fig. 1B). Also consistent was the observation that the effects of TGF-β alone and TGF-β + amiloride applied together were not additive over TGF-β alone (Fig. 1 C and D), suggesting that both agents shared the same target in the lung. These treatments did not impact passive (paracellular) permeability, assessed by [3H]mannitol flux (Fig. 1 C and D), although TGF-β signaling was demonstrably active in isolated lungs (Fig. S1G). Taken together, these data demonstrate that TGF-β blocks active 22Na+ efflux from the alveolar airspaces in isolated, ventilated, and perfused rabbit lungs, but it does not promote overt alveolar flooding, because endothelial barrier permeability was not increased.
TGF-β Targets Amiloride-Sensitive Na+ Channels, but Not the Na+/K+-ATPase in Lung Epithelial Cells.
Transepithelial Na+ transport across the alveolar epithelium is undertaken by the concerted action of the Na+/K+-ATPase and ENaC. Although TGF-β signaling was active in both A549 and primary mouse alveolar type II (ATII) cells (Fig. S2A), TGF-β (0.01–10 ng/mL) did not impact ouabain-sensitive 86Rb+-uptake by either cell type within 150 min (Fig. S2B) of TGF-β treatment, thereby excluding the Na+/K+-ATPase as a target of TGF-β. However, in patch-clamp studies on primary mouse ATII cells, TGF-β clearly blocked ion currents, as revealed by the current density versus voltage relationship (Fig. 2 A and B) and whole-cell current (Fig. 2C). Consistent with previous reports (19) amiloride significantly inhibited the currents (Fig. 2B). TGF-β and amiloride applied together had no additive effect over TGF-β applied alone (Fig. 2B). Similarly, in A549 cells, TGF-β blocked both amiloride-sensitive (Fig. S3) and benzamil-sensitive (Fig. S4) currents. The amiloride-sensitivity is consistent with reports from another group (20), and the effects of amiloride or benzamil applied together with TGF-β were not additive, suggesting that TGF-β did target amiloride/benzamil-sensitive Na+ channels in A549 cells. Both SB431542 and PO inhibited the effects of TGF-β, confirming a role for the TGF-β/Tgfbr1/Smad2/3 signaling axis, as well as actin mobility, consistent with observations made in live rabbits and isolated rabbit lungs. Thus, an amiloride-sensitive Na+ channel, most likely ENaC, was targeted by TGF-β.
Fig. 2.

TGF-β blocks ion currents in primary ATII cells, and promotes loss of αENaC from the epithelial cell surface. (A) Mean current density versus voltage relationship for vehicle and TGF-β1– (10 ng/mL; 30 min) treated ATII cells in the absence and presence of amiloride (10 µM). (B) Summarized data for whole-cell current density elicited by a test potential at −100 mV in vehicle and after treatment with TGF-β, amiloride, or TGF-β + amiloride (n ≥ 7); **P < 0.01 for comparison with vehicle. (C) Typical whole-cell sodium currents recorded in ATII cells in the absence or after treatment with 10 ng/mL TGF-β. (D) TGF-β promoted the internalization of αENaC from the surface of ATII cells from mice (black bar; n = 10 mice) and humans (white bars, data from cells isolated from the lungs from two different patients is illustrated, with replica determinations from each patient). Internalization was assessed by flow cytometry. *P < 0.05, comparing mean fluorescence values from TGF-β–treated versus untreated cells. (E) Representative scatter plot of human ATII cells screened by flow cytometry surface αENaC. The ATII cells, from the same patient, were either untreated (green) or treated with TGF-β (10 ng/mL, 30 min, red).
TGF-β Drives Internalization of αβγENaC in Lung Epithelial Cells.
Stimulation of A549 cells and primary mouse ATII cells with TGF-β for 150 min did not alter steady-state mRNA levels of the genes encoding the three classic ENaC subunits: αENaC, βENaC, and γENaC (Fig. S2C). ENaC complexes have very low cell-surface abundance in primary mouse ATII and lung epithelial cell-lines (21), thus both human (A549) and mouse (MLE-12) lung epithelial cell lines were transfected with epitope-tagged ENaC subunits for surface abundance studies, and cells were pretreated with brefeldin A, to prevent retrograde ENaC trafficking to the cell surface after internalization (22). Several cross-reacting bands were evident when A549 cell extracts were probed with anti-FLAG or anti-V5 antibodies (Fig. 3). These cross-reacting bands conveniently served as loading controls (see untransfected and empty vector-transfected lanes in Fig. 3). Stimulation of transfected A549 cells with TGF-β (10 ng/mL) for 30 min or 150 min had no impact on the cell-surface abundance of human αENaC (Fig. 3 A and D) or γENaC (Fig. 3 B and D) when these two subunits were expressed alone. However, a rapid and dramatic loss of βENaC from the cell surface was noted (Fig. 4 C and D). The βENaC “smear” is typical because of posttranslational processing (23), and less loading (2-μg input) and shorter exposure time resulted in a single well-resolved band for the cell-surface fraction (Fig. S2D). Identical trends were observed for mouse βENaC in a mouse lung epithelium cell-line (Fig. 3D and Fig. S2E). Because ENaC subunits are assembled together in the endoplasmic reticulum before trafficking (24), all three ENaC subunits were expressed together (25), TGF-β reduced the surface abundance of the αENaC and γENaC subunits (assessed together) and the βENaC subunit (Fig. 3D and Fig. S2F). These data demonstrate that TGF-β can rapidly promote loss of ENaC complexes from the surface of lung epithelial cells. Because no αENaC was detected in cell-culture supernatants after TGF-β treatment (Fig. S2G), internalization—perhaps by endocytosis—rather than plasma membrane shedding of ENaC, was believed to be the underlying mechanism, an idea that is further supported by the PO data in rabbit lungs. It is important to emphasize that TGF-β would drive internalization of the entire αβγENaC into lung epithelial cells using the βENaC located in these αβγENaC. No change in the gene-expression levels of any ENaC subunit in response to TGF-β is suggested by our data (Fig. 3 A–C, input lanes, and Fig. S2C), thus the formation, for example, of αENaC homomeric complexes generating a nonselective 20 pS channel versus a selective 4 pS (26) is not anticipated or proposed here. Application of SB431542 (10 μM) (Fig. 4A and Fig. S5A) or PO (1 μM) (Fig. 4A and Fig. S5B) blocked the ability of TGF-β to promote ENaC loss from the lung epithelial cell surface, underscoring roles for Tgfbr1-mediated signaling and actin mobility. Furthermore, genetic ablation of Smad2 and Smad3 by siRNA also blocked the effects of TGF-β on βENaC surface abundance (Fig. 4A and Fig. S5C). To independently support this idea, the surface expression of αENaC, which can be detected on ATII cells by flow cytometry (Fig. S4F), was assessed in mouse (Fig. 2D) and human (Fig. 2 D and E) ATII cells before and after a 30-min exposure to TGF-β (10 ng/mL). These data clearly revealed that TGF-β promoted a significant loss of αENaC (a surrogate for αβγENaC) in primary mouse and human ATII cells. This observation is particularly important because the flow cytometry studies explored endogenous ENaC, where subunits were expressed without an epitope tag, and at endogenous levels, establishing confidence in the data generated using epitope-tagged ENaC subunits overexpressed in lung cell-lines.
Fig. 3.

TGF-β drives internalization of βENaC in A549 cells. Cells expressed FLAG-tagged (A) αENaC, (B) γENaC, or (C) V5-tagged βENaC. Total ENaC levels are evident in input lanes, and cell-surface abundance was assessed by biotinylation, pull-down, and immunoblot (IB) on the same membrane. Cells were pretreated with brefeldin A (BFA; 10 μg/mL), and were untransfected (UT), transfected with empty vector (EV), or transfected (TF) with constructs expressing the epitope-tagged ENaC subunits. Input levels could be adjusted to obtain single bands for βENaC (Fig. S2D). (D) Data from multiple experiments (including those in Fig. S2 D–F) were quantified by densitometric analysis of immunoblot bands. Data represent mean ± SD (n = 3–8, per group). *P < 0.05.
Fig. 4.
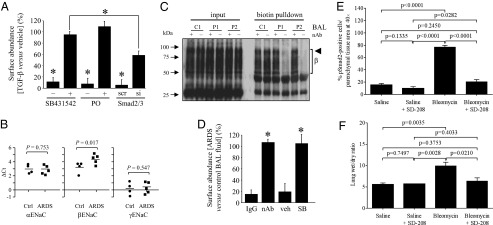
TGF-β drives ENaC internalization in a Tgfbr1/Smad2/3-dependent pathway, and is both the active principle in ARDS patient lungs that promotes ENaC internalization and plays a central role in pertubed lung fluid homeostasis in the bleomycin lung-injury model. (A) Effects of the Tgfbr1 inhibitor SB431542 (10 μM), PO (10 μM), an inhibitor of actin conversion, and Smad2/3 knockdown by small-interfering (si)RNA [or scrambled (scr) siRNA], on internalization of V5-tagged βENaC in A549 cells stimulated with TGF-β (10 ng/mL; 30 min). Presented are data from multiple experiments quantified by densitometry (n = 3, per group), with original representative experiments illustrated in Fig. S5. (B) Levels of mRNA transcripts encoding αENaC (SCNN1A), βENaC (SCNN1B), and γENaC (SCNN1G) assessed by real-time RT-PCR in lung tissues from mechanically ventilated patients with ARDS (n = 5), or control (Ctrl) patients who died from myocardial infarction without lung disease (n = 4). (C) Ability of a TGF-β1, -2, -3 neutralizing antibody (nAb; +) or nonspecific IgG (−) to block βENaC internalization in A549 cells that was driven by BAL fluid from one control volunteer (C1) and two patients with ARDS (P1 and P2). The bracket indicates the βENaC smear (β). (D) Data from multiple experiments illustrated in C, as well as studies using SB431542 (SB; 10 μM) or vehicle (PBS) alone (veh) (presented in Fig. S6B) were quantified by densitometry. (E) Activation of TGF-β signaling in the lungs of mice (n = 5, per group) that received intratracheal bleomycin to induce lung injury, and edema formation was assessed by phospho-Smad2 (pSmad2) abundance in the parenchyma. The impact of the orally active Tgfbr1 inhibitor SD-208 on Smad2 phosphorylation in the lung was assessed to demonstrate an attenuation of canonical TGF-β signaling in the lungs of SD-208–treated mice. (F) SD-208 administration normalized lung fluid balance in bleomycin-treated mice. Data represent mean ± SD. Specific P values are indicated, or an asterisk (*) indicates P < 0.05. UT; untransfected; EV, empty vector-transfected.
TGF-β Is the Active Principle in the Lung Fluids of ARDS Patients That Promotes Loss of ENaC from the Lung Epithelial Cell Surface, and TGF-β Mediates Lung Fluid Balance in Bleomycin-Induced Lung Injury.
In lung tissue from patients with ARDS, no increase in the expression of genes encoding αENaC or γENaC was observed, compared with healthy lung tissue, although βENaC expression was elevated (Fig. 4B). BAL fluids from ARDS patients could rapidly drive βENaC internalization, in comparison with BAL fluids harvested from healthy volunteers (Fig. S6A). This effect could be blocked when BAL fluids from ARDS patients were preincubated with a pan–TGF-β1, -2, -3 neutralizing antibody (nAb) (Fig. 4 C and D) or when A549 cells were pretreated with SB431542 (Fig. 4D and Fig. S6B). These data indicate that TGF-β is the active principle in BAL fluid from ARDS patients that can drive internalization of βENaC.
Lung injury induced by intratracheal instillation of bleomycin is a widely used model of lung injury characterized by alveolar edema in the early (>7 d postbleomycin administration) “exudative” phase of lung injury (27). TGF-β signaling was neutralized in vivo by administration of the orally bioactive SB431542 analog SD-208, initiated one day after bleomycin administration to mice (Fig. S5D). Smad2 phosphorylation in the lung parenchyma was used as a surrogate readout of TGF-β signaling, which was clearly dampened by SD-208 administration (Fig. 4E). The administration of SD-208, applied in a therapeutic regimen 24 h after bleomycin administration, led to normalization of lung fluid balance in bleomycin-treated mice on day 5 postbleomycin administration, as assessed by lung wet/dry ratio (Fig. 4F).
TGF-β Effects on ENaC Surface Abundance Depend on Phospholipase D1 and PIP5K1α.
A broad screen of candidate signaling molecules ruled out several key cell-signaling pathways that have been previously demonstrated to impact alveolar ion transport, including Ca2+/calmodulin (28), protein kinase C (PKC) (29), and phosphoinositide-3-kinase (15) pathways, where the following inhibitors did not inhibit TGF-β–stimulated internalization of βENaC: N-(p-amylcinnamoyl)anthranilic acid (25 μM; phospholipase A2 inhibitor), isotetrandrine (1 μg/mL; the phospholipase A inhibitor), BAPTA-AM (25 mM, Ca2+-chelator,), STO-609 (20 μg/mL; Ca2+/calmodulin-dependent protein kinase kinase inhibitor), bisindolylmaleimide I [1 and 10 μM; a PKC inhibitor, which discriminates between atypical (PKC-ι and PKC-ζ) and the remaining unique and classic PKC isoforms], wortmannin (100 nM, phosphoinositide-3-kinase inhibitor); and c-jun N-terminal kinase inhibitor II (50 μM) (Fig. S7). However, 0.1% (vol) l-butanol could block the impact of TGF-β–induced βENaC internalization, implicating phospholipase D (PLD) (Fig. S8A). Genetic ablation of PLD1 by siRNA blocked the ability of TGF-β to drive βENaC internalization (Fig. 5A). Expression of a dominant-negative PLD1 variant in A549 cells also markedly reduced the ability of TGF-β to drive βENaC internalization (Fig. S8C). TGF-β drove production of both phosphatidylbutanol (PBut, which is diagnostic for PLD activity) as well as phosphatidic acid (PA, a natural product of PLD activity) by A549 cells (Fig. S8B). Genetic ablation of Smad2/3 blocked TGF-β–induced PBut, formation (Fig. S8B), placing PLD activity downstream of Tgfbr1-induced Smad2/3 phosphorylation in this pathway.
Fig. 5.

TGF-β activation of PLD1 and PIP5K1α is required for βENaC internalization. (A) The impact of PLD1 gene ablation by siRNA [or scrambled (scr) siRNA] on βENaC internalization by A549 cells in response to TGF-β (10 ng/mL) assessed by biotin pull-down. Effect of PIP5K1A gene ablation by siRNA on (B) βENaC internalization and (C) [3H]phosphatidic acid (PA) production by A549 cells in response to TGF-β. Data represent mean ± SD *P < 0.05 (n = 3, per group). UT; untransfected; EV, empty vector-transfected, Lam, Lamin A/C.
The PA generated by PLD1 is a regulator of phosphoinositide signaling (30), because PA activates phosphatidylinositol-4-phosphate 5-kinase 1α (PIP5K1α), which generates phosphatidylinositol (4,5)-bisphosphate [PtdIns(4,5)P2], a phosphoinositide that has been implicated in the positive (31) and negative (32) regulation of ENaC activity. No specific inhibitor of PIP5K1α exists; however, knockdown of PIP5K1α expression in A549 cells prevented TGF-β–induced internalization of βENaC (Fig. 5B) but did not impact TGF-β–driven PA production (via PLD1) (Fig. 5C). Thus, PIP5K1α lies downstream of PLD1 in the TGF-β/βENaC internalization pathway. As expected, a dominant-negative PIP5K1α expressed in A549 cells also prevented βENaC internalization driven by TGF-β (Fig. S8D).
TGF-β Effects on ENaC Surface Abundance Depend on ROS Generated by NADPH Oxidase 4.
ROS and NO can regulate alveolar fluid clearance (33) and ENaC activity (34, 35). During a pathway screen, the cell-permeable nonspecific ROS quenchers polyethylene glycol (PEG)-complexed superoxide dismutase (Fig. S9A) and EU.K.-134 (Fig. S9C) both blocked the TGF-β–driven βENaC internalization, although l-NAME (100 mM; an inhibitor of NO generation by NO synthases) was without effect (Fig. S9D). TGF-β could drive ROS production in A549 cells using an H2O2-coupled assay (29) (Fig. S9B), which was blocked with SB431542 and Smad2/3 knockdown (Fig. S9F). Inhibitors of complexes I–III of the mitochondrial electron transport chain did not block TGF-β–induced ROS production, including 3-nitroproprionic acid (complex II), thenoyltrifluoroacetone (complex II), antimycin A (complex III), and rotenone (complex I) (Fig. S9G). However, NaN3 (an inhibitor of complex IV and NADPH oxidases) (Fig. S9G) and apocynin [a general ROS quencher (36) and NADPH oxidase inhibitor (Fig. S9 E and G)] did block TGF-β–driven ROS production and βENaC internalization. Genetic ablation of NADPH oxidase 4 (NOX4) blocked TGF-β–stimulated ROS production (Fig. S9B) without impacting baseline ROS levels (Fig. S9B, Inset) and also prevented the TGF-β–induced βENaC internalization (Fig. 6A). Thus, NOX4 is the source of TGF-β–induced ROS in A549 cells. Ablation of both PLD1 and PIP5K1α cells prevented TGF-β–stimulated ROS production (Fig. S9B), demonstrating that NOX4 lies downstream of both PLD1 and PIP5K1α in the TGF-β/βENaC internalization pathway. Thus, NOX4 represents the most distal branch of the TGF-β pathway that regulates ENaC trafficking. In support of a pathophysiological role for NOX4 in regulating fluid balance in injured lungs, lung injury was induced by bleomycin in mice in which Nox4 expression was ablated by gene deletion. The nox4−/− mice (37) were deficient in nox4 mRNA (Fig. S9H). The nox4−/− mice were fully protected against disturbances to lung fluid balance that are elicited by bleomycin application, which was revealed by nox4−/− mice having a comparable lung wet/dry ratio to control mice that received intratracheal administration of vehicle without bleomycin (Fig. 6B; compare with positive control data in Fig. 4F). These data point to a key role for Nox4 in mediating perturbed fluid balance in bleomycin-induced lung injury in mice.
Fig. 6.

TGF-β activation of NOX4 is required for βENaC internalization and for perturbed lung fluid balance in the bleomycin lung-injury model, and TGF-β signaling targets Cys43 of βENaC for oxidation. (A) The impact of NOX4 gene ablation by siRNA [or scrambled (scr) siRNA] on βENaC internalization by A549 cells in response to TGF-β (10 ng/mL) assessed by biotin pull-down. (B) Lung fluid balance assessed by wet/dry ratio in the lungs of nox4−/− mice that receved either intratracheal vehicle (saline) alone, or bleomycin, to induce lung injury. Data represent mean ± SD (n = 5, per group). (C) The impact of cysteine residue replacements on mouse βENaC (arrowhead) internalization by MLE-12 cells in response to TGF-β (10 ng/mL; 30 min) was assessed by biotin pull-down. As the biotin pull-down and input fractions were resolved on separate gels, an α-tubulin input loading control was also prepared. (D) The oxidation of Cys43 by TGF-β (10 ng/mL; 30 min) was demonstrated using a 4-(3-azidopropyl)cyclohexane-1,3-dione probe for oxidized cysteine residues followed by a phosphine-biotin detection, by biotin pull-down, performed on cells stimulated with TGF-β to drive βENaC oxidation. Blots were then probed for the V5 epitope to detect V5-tagged βENaC (arrowhead).
TGF-β Signaling Targets Cys43 of βENaC in Human and Mouse Cells.
Cysteine residues react with ROS and H2O2, and ethanol-induced oxidation of cysteine residues in αENaC can modulate ENaC transporting activity (38). Therefore, all cysteine residues in the cytosolic domains of βENaC (Fig. S10A) were converted to serine residues. Conversion of Cys10, Cys30, Cys557, and Cys595 mouse βENaC to a serine residue (Fig. 6C) did not impact TGF-β–driven βENaC internalization in mouse MLE-12 cells (Fig. 7C), but conversion of Cys43 completely blocked this effect. Identical trends were observed using human βENaC, where conversion of Cys43 (Fig. S10C) but not Cys30 (Fig. S10B) blocked TGF-β–induced βENaC internalization by human A549 cells. The conversion of Cys43 to a serine or alanine residue did not impair delivery to the cell surface, as is evident in the “no TGF-β” lanes in Fig. 6C. A surface half-life was assessed by pulse-chase surface labeling for the wild-type and Cys43 βENaC variants expressed alone in A549 cells at 94 ± 18 min and 79 ± 19 min, respectively. As such, the delivery and baseline stability of both βENaC variants does not appreciably differ (P = 0.70). Furthermore, although mutational studies have revealed that cysteine residues in the conserved cysteine-rich domains can impact channel expression at the cell surface (39), Cys43 is not among these cysteine residues. Additionally, although intracellular thiols are known to modulate ENaC activity (40), when all intracellular cysteine residues in αβγENaC were replaced simultaneously (including Cys43 in βENaC, together with seven other Cys residues), there was no appreciable impact on baseline activity assessed in Xenopus oocytes. Interestingly, Cys30and Cys43 are the only two intracellular cysteine residues in human βENaC, and their positions are exactly conserved comparing mouse and human ENaC. Thus, the conserved Cys43 appears to represent a direct or indirect target of TGF-β–induced ROS, generated by NOX4. In response to TGF-β stimulation, the oxidation of Cys43 was revealed by the OxyBlot protein oxidation-detection methodology, which identifies the oxidation of cysteine residues in protein (Fig. 6D).
Fig. 7.

Schematic illustration of the TGF-β/ENaC pathway in a generalized alveolar epithelial cell monolayer, described in this study. In healthy lungs, latent TGF-β is inactive, and the Na+/K+-ATPase and ENaC drive Na+ absorption, maintaining fluid influx and reabsorption in equilibrium, and epithelial lining fluid volume at an appropriate level. Activation of latent TGF-β by elastase- (72), integrin- (9), or protease-activated receptor (PAR)-dependent mechanisms (10) in acute lung injury has been described. TGF-β is implicated in chronic (gene regulatory) effects (green arrows), including loss of cell–cell junctions (9), down-regulation of SCNN1A (13), and epithelial-to-mesenchymal transition (73), leading to barrier failure. TGF-β–neutralizing antibodies (nAb) or soluble type II receptors (sTgfbr2) reduce the deleterious effects of TGF-β (9). We report here an acute effect of TGF-β on alveolar fluid reabsorption. In the unique signaling pathway delineated (dark gray arrows), TGF-β, acting through the type I TGF-β receptor, induces Smad2/3 phosphorylation, which activates PLD1, which generates PA from phosphatidylcholine (PC). PA activates PIP5K1α, which drives NOX4 activation, perhaps by PtdIns(4,5)P2 formation from phosphatidylinositol 4′-monophosphate (PI4P). Activated NOX4 generates ROS, which promote βENaC internalization that is dependent on Cys43, which leads to loss of Na+-absorbing capacity of the epithelial cell and alveolar flooding, promoting persistence of alveolar edema. The targets of reagents used in this study are also illustrated.
The ubiquitination of ENaC by neural precursor cell expressed, developmentally down-regulated (Nedd)4-2 ubiquitin ligases is also recognized as a key regulatory mechanism of ENaC trafficking (25, 41–44), and specifically related to the lung, genetic ablation of Nedd4-2 in mice disturbs ENaC trafficking and causes premature fetal lung fluid clearance (45) and cystic fibrosis-like disease (46). Therefore, a role for Nedd4-2 in the regulation of ENaC trafficking by TGF-β and the impact of TGF-β on βENaC ubiquitination was also assessed. Knockdown of Nedd4-2 expression by siRNA had no impact on the ability of TGF-β to drive βENaC internalization (Fig. S11A), suggesting that Nedd4-2 did not participate in the TGF-β/ENaC trafficking pathway described here. Nedd4-2 has also been described as a Smad2/3 E3 ubiquitin ligase (47). Although hypoxia was able to promote increased ubiquitination of βENaC (48) (used here as a positive control), there was no appreciable impact of TGF-β on βENaC ubiquitination over a 150-min time course (Fig. S11B). As such, ubiquitination does not play a role in TGF-β–mediated trafficking of βENaC.
The preceding data, therefore, establish the sequence of this unique TGF-β signaling pathway, where (i) TGF-β, acting via the type I TGF-β receptor Tgfbr1, drives (ii) Smad2/3 phosphorylation, and (iii) PLD1, (iv) PIP5K1α, and (v) NOX4 activation, which generates ROS and promotes βENaC internalization through interaction or recognition of an oxidized Cys43. This pathway is illustrated schematically in Fig. 7.
Discussion
The data presented here demonstrate that TGF-β plays a key role in the acute regulation of ENaC activity, and hence can impact alveolar ion and fluid transport. These observations are relevant to pathological conditions characterized by a failure of fluid reabsorption, exemplified by alveolar edema in patients with ALI/ARDS. TGF-β has already been implicated as an important mediator of ALI/ARDS; however, to date all proposed roles for TGF-β in ALI/ARDS have been ascribed to long-term effects dependent upon transcriptional regulation (9, 10, 13). Further in vivo studies have demonstrated that intratracheal instillation of TGF-β into live rats impedes fluid reabsorption in the lung (13), and administration of a soluble type II TGF-β receptor, which sequesters free TGF-β during experimental lung injury, attenuated the degree of pulmonary edema (9). In a clinical setting, increased abundance of TGF-β mRNA and protein in lung tissue from ARDS patients (12) and increased active TGF-β1 levels in BAL fluids from ARDS patients have been described (11, 12). Notably, lower BAL fluid TGF-β levels in ARDS patients were correlated with improved survival in ARDS (11). In our study, we estimated active TGF-β levels in ARDS patients at 7–14 ng/mL in the ELF, approximately double that of plasma TGF-β levels in healthy subjects [4.1 ± 2.0 ng/mL (49)] but lower than TGF-β levels in the pleural fluid during thoracic empyema [40 ng/mL (50)].
The activity of ENaC may be regulated chronically (at the gene-transcription level), which may have pathological consequences. For example, SCNN1G (encoding γENaC) transcription is impaired by TNF-α in Crohn disease (51), leading to impaired sodium transport across the colon epithelium (52). Related to our study, TGF-β can down-regulate the SCNN1A gene (encoding αENaC) in alveolar epithelial cells after a 96-h exposure to 10 ng/mL TGF-β (13). However, TGF-β did not impact mRNA levels of genes that encode α-, β-, and γENaC subunits over the short (30 min to 2 h) time course in our study, ruling out gene regulatory effects.
The primary mechanisms regulating ENaC activity are acute, allowing the cell to rapidly respond to fast-changing needs in sodium absorption. These mechanisms include the regulation of channel open probability (Po) (23, 53, 54) or channel trafficking (55, 56), which alters channel cell-surface abundance, because only a small fraction of ENaC channels reside on the plasma membrane, the remainder being located in subapical compartments that are rapidly delivered to the plasma membrane by the appropriate stimulus (55). ENaC activity is negatively regulated by internalization of channel complexes into clathrin-coated pits (56), but the pathways directing ENaC internalization are unclear (56). In this study, we identify the β-subunit of ENaC as a target for TGF-β–driven internalization. Appropriately, the β-subunit of ENaC is the regulatory subunit responsible for stabilizing ENaC complexes in the plasma membrane (4). This finding represents a hitherto undescribed ENaC regulatory pathway that relies on ENaC trafficking.
Disturbances to ENaC trafficking, which lead to abnormal cell-surface stability of the αβγENaC complex, cause severe disease, such as Liddle Syndrome, where mutations in the SCNN1B and SCNN1G genes generate βENaC and γENaC variants, respectively, that are truncated at the C terminus (57), leading to loss of a critical PY recognition domain that prevents ubiquitination and subsequent internalization (58), leading to increased cell-surface stability and thus, hyperabsorption of sodium. In our study, however, a role for ubiquitination in the trafficking of βENaC in response to TGF-β, including ubiquitination mediated by the Nedd4-2 ubiquitin ligase, has been ruled out.
In this study, we describe what may be a unique ENaC trafficking defect, where TGF-β can drive abnormal internalization of the αβγENaC complex, leading to a pronounced reduction in αβγENaC complexes at the lung epithelial cell surface, and hence, reduced sodium and fluid reabsorption. Importantly, BAL fluids from healthy volunteers did not drive ENaC internalization, but BAL fluids from ARDS patients did, indicating that BAL fluids from ARDS patients contain a factor that drove ENaC internalization in alveolar epithelial cells. In this study, using neutralizing antibodies or a TGF-β inhibitor (SB431542), TGF-β was identified as being the factor in ARDS patient lungs that was exclusively responsible for driving ENaC internalization by alveolar epithelial cells. This observation makes a strong case for a role for TGF-β in the impaired alveolar fluid reabsorption observed in ARDS patients, thereby contributing to the rapid onset and dangerous persistence of alveolar edema in these patients. In the longer term, these effects would be exacerbated by the chronic effects of TGF-β on the transcriptional regulation of ion-transporting machinery and alveolo-capillary barrier permeability described by other investigators (9, 13, 14), an idea supported by the observation that live rats exhibit a reduction in distal airspace fluid clearance 24 h after intratracheal instillation of TGF-β (13).
We have gone on to clarify seven steps of an entirely unique TGF-β signaling pathway (Fig. 7) that underlie this ENaC trafficking defect. Unique for TGF-β signaling—which generally affects gene regulation—this pathway drives a rapid (within 30 min) and dramatic (>80%) reduction in the cell-surface abundance of ENaC on lung epithelial cells. This pathway is activated by TGF-β acting through Tgfbr1, which drives Smad2/3 phosphorylation, but then diverges from the classic TGF-β gene regulatory pathway at this point by activating PLD1, a phospholipid phosphohydrolase that generates PA (30). PA can activate PIP5K1α (59), which we confirmed in this study to play a role in the TGF-β/βENaC internalization pathway, contributing to a growing and complex discussion about how phosphoinositides regulate ENaC (53). PIP5K1α was located upstream of the ROS-producing oxidase, identified as NOX4, and it is speculated that PtdIns(4,5)P2 generated by PIP5K1α activated NOX4 (Fig. 7). The activation of NADPH oxidases (although not NOX4) by PIP5K1α enzymatic products is not without precedent, because PtdIns(4,5)P2 can regulate the subcellular distribution and ROS production by NOX5 (60). The proper cellular localization of NADPH oxidases is critical for NADPH oxidase function (61); therefore, PtdIns(4,5)P2 might directly activate NOX4, or might recruit or position NOX4 to drive βENaC internalization. This report, demonstrating that TGF-β can drive ROS production by NOX4 in the absence of transcriptional regulation of the NOX4 gene, is unique. The findings presented here documenting the acute negative regulation of ENaC activity by ROS are interesting, considering that neutralization of ROS in animal models of ARDS—by application of the membrane-permeable aminothiol N-acetylcysteine in an acute pancreatitis rat model (62), or application of the ROS quencher EU.K.-8 in a porcine LPS model (63)—attenuated alveolar edema. It is tempting to speculate that these results might have been in part because of neutralization of NOX4-generated ROS, which would drive ENaC internalization. The observation that nox4−/− mice are fully protected against the perturbations to lung fluid balance that are characteristic of bleomycin-induced lung injury highlight Nox4 as a candidate interventional target for the management of disturbed fluid balance.
ROS, NO, and H2O2 are second messengers (33), and both ROS and NO regulate ENaC activity (33). Additionally, reports exist that indicate that oxidative stress, including exogenously applied H2O2, can both negatively (64) and positively (65, 66) regulate ENaC expression and activity, demonstrating that the regulation of ENaC by oxidizing agents is complex, and that the source and compartmentalization of the oxidative agent likely influence the outcome of oxidative reactions on ENaC function. We demonstrate here that NOX4-generated ROS (measured as H2O2) regulate ENaC internalization in response to TGF-β. Although H2O2 is a mild oxidant, H2O2 has signaling activity that is attributed to chemo-selective oxidation of cysteine residues (67). A critical role for a conserved cysteine residue, Cys43, in both mouse and human ENaC was demonstrated in our study, as was the oxidation of Cys43 in response to TGF-β. This finding suggests that the TGF-β/ENaC internalization pathway is a conserved means of ENaC regulation across many species [Cys43 is also conserved in rabbits (GenBank accession no. NP001076197) and dogs (GenBank accession no. XP534912), for example]. This βENaC Cys43 oxidation by NOX4-generated ROS could serve as a trigger for βENaC internalization. Some studies have reported that intracellular cysteine residues regulate channel properties of ENaC expressed in Xenopus oocytes (39, 40); however, channel trafficking was not assessed in those studies. Our data clearly point to Cys43 as the cysteine residue that exclusively mediates TGF-β–driven channel trafficking. The Cys43 residue appears to be an excellent candidate as a target for reaction with ROS or H2O2 because of the adjacent acidic residue (Glu), which confers special reactivity conducive to oxidative modification (67). Interestingly, of all cysteine residues, only Cys43 was conserved between human and mouse βENaC, where an adjacent acidic residue was also present (Fig. S10A). Alternatively, NOX4-generated ROS might target an intermediate signaling molecule that recognizes the oxidized Cys43 in βENaC and promotes βENaC internalization. These observations contribute to a growing body of evidence implicating NOX4 (61, 68) and thiol regulation of ENaC function as an important player in lung pathophysiology.
In summary, a unique TGF-β signaling pathway that acutely regulates the activity of ENaC in the alveolar epithelium is described herein. This regulatory pathway appears to be conserved across several species, and may also represent a normal ENaC regulatory mechanism in healthy tissues. Given the pathological roles played by TGF-β in conditions associated with a failure of fluid reabsorption, such as ARDS, this pathway represents a candidate pathomechanism at play in affected lungs. Delineation of this signaling pathway revealed several candidate enzyme systems that might be targeted in an attempt to normalize alveolar fluid clearance in affected patients, including targeting phosphoinositide metabolism and ROS. Additionally, this pathway may also be operative under physiological and pathological conditions in other organs where ENaC plays an important role, including the collecting tubules of the kidney and the colon.
Materials and Methods
Human Patient Material.
Investigations involving human subjects received institutional approval by the Ethik-Kommission of the Faculty of Medicine of the University of Giessen (the equivalent of an Institutional Review Board in Germany) (under approvals 84/83 and 29/01 for BAL and lung autopsy material, and 10/06 for lung tissue from patients that underwent lobectomy), and written informed consent was obtained from all patients or their next-of-kin. All ARDS patients required mechanical ventilation. BAL fluid was obtained by flexible fiberoptic bronchoscopy within the first 72 h after initiation of mechanical ventilation. The patient groups consisted of: (i) eight control patients (44.9 ± 4.6 y; five male/three female) who were healthy volunteers without history of cardiac or pulmonary disease; (ii) eight patients with ARDS with nonpulmonary origins (sepsis, n = 5; pancreatitis, n = 1; other, n = 2; 53.7 ± 7.7 y; five male/three female; PaO2/FiO2, 157.7 ± 21.9); and (iii) nine patients with ARDS resulting from pneumonia (47.8 ± 5.0 y; five male/four female; PaO2/FiO2, 159.2 ± 20.1). This patient population has been fully described in another report from the authors (69). Lung tissue was also harvested at autopsy from five ARDS patients. All patients met all of the clinical American-European Consensus Conference criteria, and died in the early phase, with a mean duration of mechanical ventilation of 92 h. The clinical characteristics of these patients are illustrated in Table 1. Control lung specimens were obtained at autopsy from four patients who died of myocardial infarction, with no evidence of pulmonary disease.
Table 1.
Characteristics of ARDS patients from which lung mRNA material was obtained
| Patient | Age | Sex | Background | Modified APACHE II | PaO2/FiO2 |
| 1 | 40 | F | Pneumonia | 11 | 83.0 |
| 2 | 51 | F | Trauma | 6 | 181.5 |
| 3 | 48 | F | Pancreatitis | 20 | 127.2 |
| 4 | 59 | F | Trauma | 16 | 137.1 |
| 5 | 67 | M | Sepsis | 20 | 109.0 |
APACHE, Acute Physiology and Chronic Health Evaluation; F, female; FiO2, fraction of inspired oxygen; M, male; PaO2, partial pressure of oxygen in arterial blood.
Ventilation of Live Anesthetized Rabbits.
Animal investigations received government approval from the responsible Regierungspräsidium, which houses the equivalent of an Institutional Animal Care and Use Committee in Germany (Regierungspräsidium Darmstadt under approval number B2/330). New Zealand White rabbits (Bauer; 3 ± 0.5 kg) were anesthetized by a bolus of 0.3 mL xylazine (Rompun, 20 mg/mL) and 0.2 mL ketamine (100 mg/mL). Animals were artificially ventilated via a tracheal cannula with room air (tidal volume 6 mL/kg; 30 breaths per minute, I:E 1:1). Deep anesthesia was maintained by 25 mg/mL ketamine/1% xylazine in 0.9% NaCl infused at 8 mL/h via the left ear vein. Arterial and venous blood pressures were monitored via sensors in the left carotid artery and right ear vein. After a 30-min equilibration period, 0.5 mL saline alone or containing TGF-β (yielding 10 ng/mL in the ELF) was nebulized into the lungs. After 30 min, 1 mL saline as a fluid challenge, with or without amiloride (yielding 10 μM in the ELF), was nebulized to the lungs. Ventilation was maintained for 60 min, after which the bronchoalveolar space was lavaged with 50 mL iso-osmolar mannitol, and the ELF volume was measured as described previously (70, 71).
Other Techniques.
Radioactive tracer analysis in isolated, ventilated, and perfused rabbit lungs, 86Rb+ uptake, patch-clamp, real-time RT-PCR (primers described in Table S1), immunoblotting, site-directed mutagenesis, transient transfections, cell-surface biotinylation, PLD activity assays, siRNA interference, half-life measurements, H2O2 assays, and in vivo neutralization of TGF-β or application of nox4−/− mice in the bleomycin model of lung injury were all conducted exactly as previously described, and are outlined in detail in the SI Materials and Methods.
Statistics.
Values represent mean ± SD. Means of unpaired experiments were compared by the nonparametric Mann–Whitney U test. For the comparison of dependent means, a paired Student t test was used. For electrophysiological studies, values are mean SE, where intergroup differences were assessed by a factorial analysis of variance with post hoc analysis with Fisher’s least-significant difference test. Statistical significance was defined as P < 0.05.
Supplementary Material
Acknowledgments
The authors thank Thomas R. Kleyman for epitope-tagged mouse α-, β-, and γ-epithelial sodium channel-expressing constructs; David Lambeth for advice on dominant-negative NADPH oxidase 4 (NOX4) variants; Karl-Heinz Krause for the wild-type NOX4-expressing construct; Michael A. Frohman for wild-type and dominant-negative phospholipase D1 (PLD1)-expressing constructs; Kyota Aoyagi for wild-type and dominant-negative PIP5K1 α-, β-, and γ-ENaC expressing constructs; Nancy Grant for advice on PLD1 detection; Sylvain G. Bourgoin for anti-PLD antibodies and recombinant PLD protein; Daniela Rotin for providing an HA-tagged α-epithelial sodium channel expression construct; and Florian Veit, Zbigniew Zasłona, Simone Becker, Susanne Tannert-Otto, Markus Queisser, and Viktor Malec for assistance. This study was supported by von Behring-Röntgen Foundation Grant 51-0031 (to R.E.M. and S.W.); University Medical Center Giessen and Marburg Grants 62580935 (to R.E.M.) and 62589064 (to I.V.); the Federal Ministry of Higher Education, Research and the Arts of the State of Hessen Landes-Offensive zur Entwicklung Wissenschaftlich-ökonomischer Exzellenz Programme (I.V., M.W., S.H., K.M., A.G., S.W., and R.E.M.); the German Center for Lung Research (all authors); the German Research Foundation through Excellence Cluster 147 “Cardio-Pulmonary System” (ECCPS) (I.V., R.P.B., A.G., W.S., O.E., and R.E.M.) and Mo 1789/1 (to R.E.M.); and a Clinical Scientist Career Program grant from the ECCPS (to I.V. and S.H.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 885.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1306798111/-/DCSupplemental.
References
- 1.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 2.Weibel ER. What makes a good lung? Swiss Med Wkly. 2009;139(27–28):375–386. doi: 10.4414/smw.2009.12270. [DOI] [PubMed] [Google Scholar]
- 3.Vadász I, Raviv S, Sznajder JI. Alveolar epithelium and Na,K-ATPase in acute lung injury. Intensive Care Med. 2007;33(7):1243–1251. doi: 10.1007/s00134-007-0661-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol. 2009;71:403–423. doi: 10.1146/annurev.physiol.010908.163250. [DOI] [PubMed] [Google Scholar]
- 5.Matalon S, O’Brodovich H. Sodium channels in alveolar epithelial cells: Molecular characterization, biophysical properties, and physiological significance. Annu Rev Physiol. 1999;61:627–661. doi: 10.1146/annurev.physiol.61.1.627. [DOI] [PubMed] [Google Scholar]
- 6.Matthay MA, Clerici C, Saumon G. Invited review: Active fluid clearance from the distal air spaces of the lung. J Appl Physiol (1985) 2002;93(4):1533–1541. doi: 10.1152/japplphysiol.01210.2001. [DOI] [PubMed] [Google Scholar]
- 7.Matthay MA. Alveolar fluid clearance in patients with ARDS: Does it make a difference? Chest. 2002;122(6) Suppl:340S–343S. doi: 10.1378/chest.122.6_suppl.340s. [DOI] [PubMed] [Google Scholar]
- 8.Sznajder JI. Alveolar edema must be cleared for the acute respiratory distress syndrome patient to survive. Am J Respir Crit Care Med. 2001;163(6):1293–1294. doi: 10.1164/ajrccm.163.6.ed1801d. [DOI] [PubMed] [Google Scholar]
- 9.Pittet JF, et al. TGF-β is a critical mediator of acute lung injury. J Clin Invest. 2001;107(12):1537–1544. doi: 10.1172/JCI11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jenkins RG, et al. Ligation of protease-activated receptor 1 enhances α(v)β6 integrin-dependent TGF-β activation and promotes acute lung injury. J Clin Invest. 2006;116(6):1606–1614. doi: 10.1172/JCI27183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Budinger GR, et al. Active transforming growth factor-beta1 activates the procollagen I promoter in patients with acute lung injury. Intensive Care Med. 2005;31(1):121–128. doi: 10.1007/s00134-004-2503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fahy RJ, et al. The acute respiratory distress syndrome: A role for transforming growth factor-beta 1. Am J Respir Cell Mol Biol. 2003;28(4):499–503. doi: 10.1165/rcmb.2002-0092OC. [DOI] [PubMed] [Google Scholar]
- 13.Frank J, et al. Transforming growth factor-β1 decreases expression of the epithelial sodium channel alphaENaC and alveolar epithelial vectorial sodium and fluid transport via an ERK1/2-dependent mechanism. J Biol Chem. 2003;278(45):43939–43950. doi: 10.1074/jbc.M304882200. [DOI] [PubMed] [Google Scholar]
- 14.Willis BC, et al. Modulation of ion conductance and active transport by TGF-β 1 in alveolar epithelial cell monolayers. Am J Physiol Lung Cell Mol Physiol. 2003;285(6):L1192–L1200. doi: 10.1152/ajplung.00379.2002. [DOI] [PubMed] [Google Scholar]
- 15.Roux J, et al. Transforming growth factor β1 inhibits cystic fibrosis transmembrane conductance regulator-dependent cAMP-stimulated alveolar epithelial fluid transport via a phosphatidylinositol 3-kinase-dependent mechanism. J Biol Chem. 2010;285(7):4278–4290. doi: 10.1074/jbc.M109.036731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rennard SI, et al. Estimation of volume of epithelial lining fluid recovered by lavage using urea as marker of dilution. J Appl Physiol (1985) 1986;60(2):532–538. doi: 10.1152/jappl.1986.60.2.532. [DOI] [PubMed] [Google Scholar]
- 18.Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev. 2002;82(3):569–600. doi: 10.1152/physrev.00003.2002. [DOI] [PubMed] [Google Scholar]
- 19.Kemp PJ, Kim KJ, Borok Z, Crandall ED. Re-evaluating the Na(+) conductance of adult rat alveolar type II pneumocytes: Evidence for the involvement of cGMP-activated cation channels. J Physiol. 2001;536(Pt 3):693–701. doi: 10.1111/j.1469-7793.2001.t01-1-00693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazrak A, Samanta A, Matalon S. Biophysical properties and molecular characterization of amiloride-sensitive sodium channels in A549 cells. Am J Physiol Lung Cell Mol Physiol. 2000;278(4):L848–L857. doi: 10.1152/ajplung.2000.278.4.L848. [DOI] [PubMed] [Google Scholar]
- 21.Planès C, et al. Hypoxia and beta 2-agonists regulate cell surface expression of the epithelial sodium channel in native alveolar epithelial cells. J Biol Chem. 2002;277(49):47318–47324. doi: 10.1074/jbc.M209158200. [DOI] [PubMed] [Google Scholar]
- 22.Althaus M, et al. Carbon monoxide rapidly impairs alveolar fluid clearance by inhibiting epithelial sodium channels. Am J Respir Cell Mol Biol. 2009;41(6):639–650. doi: 10.1165/rcmb.2008-0458OC. [DOI] [PubMed] [Google Scholar]
- 23.Kleyman TR, Carattino MD, Hughey RP. ENaC at the cutting edge: Regulation of epithelial sodium channels by proteases. J Biol Chem. 2009;284(31):20447–20451. doi: 10.1074/jbc.R800083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Canessa CM, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367(6462):463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 25.Lu C, Pribanic S, Debonneville A, Jiang C, Rotin D. The PY motif of ENaC, mutated in Liddle syndrome, regulates channel internalization, sorting and mobilization from subapical pool. Traffic. 2007;8(9):1246–1264. doi: 10.1111/j.1600-0854.2007.00602.x. [DOI] [PubMed] [Google Scholar]
- 26.Jain L, Chen XJ, Ramosevac S, Brown LA, Eaton DC. Expression of highly selective sodium channels in alveolar type II cells is determined by culture conditions. Am J Physiol Lung Cell Mol Physiol. 2001;280(4):L646–L658. doi: 10.1152/ajplung.2001.280.4.L646. [DOI] [PubMed] [Google Scholar]
- 27.Wygrecka M, et al. Cellular origin of pro-coagulant and (anti)-fibrinolytic factors in bleomycin-injured lungs. Eur Respir J. 2007;29(6):1105–1114. doi: 10.1183/09031936.00097306. [DOI] [PubMed] [Google Scholar]
- 28.Vadász I, et al. AMP-activated protein kinase regulates CO2-induced alveolar epithelial dysfunction in rats and human cells by promoting Na,K-ATPase endocytosis. J Clin Invest. 2008;118(2):752–762. doi: 10.1172/JCI29723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dada LA, et al. Hypoxia-induced endocytosis of Na,K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-ζ. J Clin Invest. 2003;111(7):1057–1064. doi: 10.1172/JCI16826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van den Bout I, Divecha N. PIP5K-driven PtdIns(4,5)P2 synthesis: Regulation and cellular functions. J Cell Sci. 2009;122(Pt 21):3837–3850. doi: 10.1242/jcs.056127. [DOI] [PubMed] [Google Scholar]
- 31.Ma HP, Chou CF, Wei SP, Eaton DC. Regulation of the epithelial sodium channel by phosphatidylinositides: Experiments, implications, and speculations. Pflugers Arch. 2007;455(1):169–180. doi: 10.1007/s00424-007-0294-3. [DOI] [PubMed] [Google Scholar]
- 32.Weixel KM, et al. Phosphatidylinositol 4-phosphate 5-kinase reduces cell surface expression of the epithelial sodium channel (ENaC) in cultured collecting duct cells. J Biol Chem. 2007;282(50):36534–36542. doi: 10.1074/jbc.M703970200. [DOI] [PubMed] [Google Scholar]
- 33.Iles KE, Song W, Miller DW, Dickinson DA, Matalon S. Reactive species and pulmonary edema. Expert Rev Respir Med. 2009;3(5):487–496. doi: 10.1586/ers.09.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Althaus M, et al. Nitric oxide inhibits highly selective sodium channels and the Na+/K+-ATPase in H441 cells. Am J Respir Cell Mol Biol. 2011;44(1):53–65. doi: 10.1165/2009-0335OC. [DOI] [PubMed] [Google Scholar]
- 35.Matalon S, et al. Regulation of ion channel structure and function by reactive oxygen-nitrogen species. Am J Physiol Lung Cell Mol Physiol. 2003;285(6):L1184–L1189. doi: 10.1152/ajplung.00281.2003. [DOI] [PubMed] [Google Scholar]
- 36.Heumüller S, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51(2):211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 37.Zhang M, et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci USA. 2010;107(42):18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Downs CA, et al. Ethanol alters alveolar fluid balance via Nadph oxidase (NOX) signaling to epithelial sodium channels (ENaC) in the lung. PLoS ONE. 2013;8(1):e54750. doi: 10.1371/journal.pone.0054750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Firsov D, Robert-Nicoud M, Gruender S, Schild L, Rossier BC. Mutational analysis of cysteine-rich domains of the epithelium sodium channel (ENaC). Identification of cysteines essential for channel expression at the cell surface. J Biol Chem. 1999;274(5):2743–2749. doi: 10.1074/jbc.274.5.2743. [DOI] [PubMed] [Google Scholar]
- 40.Kellenberger S, Gautschi I, Pfister Y, Schild L. Intracellular thiol-mediated modulation of epithelial sodium channel activity. J Biol Chem. 2005;280(9):7739–7747. doi: 10.1074/jbc.M409955200. [DOI] [PubMed] [Google Scholar]
- 41.Abriel H, et al. Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle’s syndrome. J Clin Invest. 1999;103(5):667–673. doi: 10.1172/JCI5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fotia AB, et al. The role of individual Nedd4-2 (KIAA0439) WW domains in binding and regulating epithelial sodium channels. FASEB J. 2003;17(1):70–72. doi: 10.1096/fj.02-0497fje. [DOI] [PubMed] [Google Scholar]
- 43.Kamynina E, Debonneville C, Bens M, Vandewalle A, Staub O. A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. FASEB J. 2001;15(1):204–214. doi: 10.1096/fj.00-0191com. [DOI] [PubMed] [Google Scholar]
- 44.Snyder PM, Olson DR, McDonald FJ, Bucher DB. Multiple WW domains, but not the C2 domain, are required for inhibition of the epithelial Na+ channel by human Nedd4. J Biol Chem. 2001;276(30):28321–28326. doi: 10.1074/jbc.M011487200. [DOI] [PubMed] [Google Scholar]
- 45.Boase NA, et al. Respiratory distress and perinatal lethality in Nedd4-2-deficient mice. Nat Commun. 2011;2:287. doi: 10.1038/ncomms1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kimura T, et al. Deletion of the ubiquitin ligase Nedd4L in lung epithelia causes cystic fibrosis-like disease. Proc Natl Acad Sci USA. 2011;108(8):3216–3221. doi: 10.1073/pnas.1010334108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao S, et al. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol Cell. 2009;36(3):457–468. doi: 10.1016/j.molcel.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gille T, et al. Hypoxia-induced inhibition of ENaC in the lung: Role of Nedd4-2 and the ubiquitin-proteasome pathway. Am J Respir Cell Mol Biol. 2013 doi: 10.1165/rcmb.2012-0518OC. in press. [DOI] [PubMed] [Google Scholar]
- 49.Wakefield LM, et al. Transforming growth factor-β1 circulates in normal human plasma and is unchanged in advanced metastatic breast cancer. Clin Cancer Res. 1995;1(1):129–136. [PubMed] [Google Scholar]
- 50.Sasse SA, Jadus MR, Kukes GD. Pleural fluid transforming growth factor-beta1 correlates with pleural fibrosis in experimental empyema. Am J Respir Crit Care Med. 2003;168(6):700–705. doi: 10.1164/rccm.2202043. [DOI] [PubMed] [Google Scholar]
- 51.Zeissig S, et al. Altered ENaC expression leads to impaired sodium absorption in the noninflamed intestine in Crohn’s disease. Gastroenterology. 2008;134(5):1436–1447. doi: 10.1053/j.gastro.2008.02.030. [DOI] [PubMed] [Google Scholar]
- 52.Sandle GI, et al. Cellular basis for defective electrolyte transport in inflamed human colon. Gastroenterology. 1990;99(1):97–105. doi: 10.1016/0016-5085(90)91235-x. [DOI] [PubMed] [Google Scholar]
- 53.Pochynyuk O, Bugaj V, Stockand JD. Physiologic regulation of the epithelial sodium channel by phosphatidylinositides. Curr Opin Nephrol Hypertens. 2008;17(5):533–540. doi: 10.1097/MNH.0b013e328308fff3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rossier BC, Stutts MJ. Activation of the epithelial sodium channel (ENaC) by serine proteases. Annu Rev Physiol. 2009;71:361–379. doi: 10.1146/annurev.physiol.010908.163108. [DOI] [PubMed] [Google Scholar]
- 55.Bhalla V, Hallows KR. Mechanisms of ENaC regulation and clinical implications. J Am Soc Nephrol. 2008;19(10):1845–1854. doi: 10.1681/ASN.2008020225. [DOI] [PubMed] [Google Scholar]
- 56.Butterworth MB, Edinger RS, Frizzell RA, Johnson JP. Regulation of the epithelial sodium channel by membrane trafficking. Am J Physiol Renal Physiol. 2009;296(1):F10–F24. doi: 10.1152/ajprenal.90248.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tamura H, et al. Liddle disease caused by a missense mutation of β subunit of the epithelial sodium channel gene. J Clin Invest. 1996;97(7):1780–1784. doi: 10.1172/JCI118606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Staub O, et al. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997;16(21):6325–6336. doi: 10.1093/emboj/16.21.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moritz A, De Graan PN, Gispen WH, Wirtz KW. Phosphatidic acid is a specific activator of phosphatidylinositol-4-phosphate kinase. J Biol Chem. 1992;267(11):7207–7210. [PubMed] [Google Scholar]
- 60.Kawahara T, Lambeth JD. Phosphatidylinositol (4,5)-bisphosphate modulates Nox5 localization via an N-terminal polybasic region. Mol Biol Cell. 2008;19(10):4020–4031. doi: 10.1091/mbc.E07-12-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 62.Leme AS, Lichtenstein A, Arantes-Costa FM, Landucci EC, Martins MA. Acute lung injury in experimental pancreatitis in rats: Pulmonary protective effects of crotapotin and N-acetylcysteine. Shock. 2002;18(5):428–433. doi: 10.1097/00024382-200211000-00007. [DOI] [PubMed] [Google Scholar]
- 63.Gonzalez PK, et al. Role of oxidant stress in the adult respiratory distress syndrome: Evaluation of a novel antioxidant strategy in a porcine model of endotoxin-induced acute lung injury. Shock. 1996;6(Suppl 1):S23–S26. [PubMed] [Google Scholar]
- 64.Xu H, Chu S. ENaC alpha-subunit variants are expressed in lung epithelial cells and are suppressed by oxidative stress. Am J Physiol Lung Cell Mol Physiol. 2007;293(6):L1454–L1462. doi: 10.1152/ajplung.00248.2007. [DOI] [PubMed] [Google Scholar]
- 65.Downs CA, Kumar A, Kreiner LH, Johnson NM, Helms MN. H2O2 regulates lung ENaC via ubiquitin-like protein Nedd8. J Biol Chem. 2013;228(12):8136–8145. doi: 10.1074/jbc.M112.389536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goodson P, et al. Nadph oxidase regulates alveolar epithelial sodium channel activity and lung fluid balance in vivo via O⁻₂ signaling. Am J Physiol Lung Cell Mol Physiol. 2012;302(4):L410–L419. doi: 10.1152/ajplung.00260.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Forman HJ, Fukuto JM, Torres M. Redox signaling: Thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol. 2004;287(2):C246–C256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 68.Carnesecchi S, et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal. 2011;15(3):607–619. doi: 10.1089/ars.2010.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schmidt R, et al. Alteration of fatty acid profiles in different pulmonary surfactant phospholipids in acute respiratory distress syndrome and severe pneumonia. Am J Respir Crit Care Med. 2001;163(1):95–100. doi: 10.1164/ajrccm.163.1.9903029. [DOI] [PubMed] [Google Scholar]
- 70.Vadász I, et al. Oleic acid inhibits alveolar fluid reabsorption: A role in acute respiratory distress syndrome? Am J Respir Crit Care Med. 2005;171(5):469–479. doi: 10.1164/rccm.200407-954OC. [DOI] [PubMed] [Google Scholar]
- 71.Vadász I, et al. Thrombin impairs alveolar fluid clearance by promoting endocytosis of Na+,K+-ATPase. Am J Respir Cell Mol Biol. 2005;33(4):343–354. doi: 10.1165/rcmb.2004-0407OC. [DOI] [PubMed] [Google Scholar]
- 72.Buczek-Thomas JA, et al. Elastase mediates the release of growth factors from lung in vivo. Am J Respir Cell Mol Biol. 2004;31(3):344–350. doi: 10.1165/rcmb.2003-0420OC. [DOI] [PubMed] [Google Scholar]
- 73.Zhou G, et al. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol. 2009;297(6):L1120–L1130. doi: 10.1152/ajplung.00007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.