

Significance
Posttranslational modification of cysteinyl thiolate to sulfenate has been found to play important roles in biology, such as redox signaling, and enzyme and gene regulation. Nitrile hydratase and thiocyanate hydrolase with cobalt and iron cofactors are the few known metalloenzymes requiring sulfenate coordination for reactivity. No other metal ions have been found to stably bind sulfenate in a biological context. Here we report a copper–sulfenate complex characterized in a protein environment, formed at the active site of a cavity mutant of Pseudomonas aeruginosa azurin. Computational studies strongly suggest that noncovalent interactions in the secondary coordination sphere are critical in stabilizing this species.
Keywords: bioinorganic chemistry, metalloprotein design, protein engineering, blue copper proteins, posttranslational modification
Abstract
Metal–sulfenate centers are known to play important roles in biology and yet only limited examples are known due to their instability and high reactivity. Herein we report a copper–sulfenate complex characterized in a protein environment, formed at the active site of a cavity mutant of an electron transfer protein, type 1 blue copper azurin. Reaction of hydrogen peroxide with Cu(I)–M121G azurin resulted in a species with strong visible absorptions at 350 and 452 nm and a relatively low electron paramagnetic resonance gz value of 2.169 in comparison with other normal type 2 copper centers. The presence of a side-on copper–sulfenate species is supported by resonance Raman spectroscopy, electrospray mass spectrometry using isotopically enriched hydrogen peroxide, and density functional theory calculations correlated to the experimental data. In contrast, the reaction with Cu(II)–M121G or Zn(II)–M121G azurin under the same conditions did not result in Cys oxidation or copper–sulfenate formation. Structural and computational studies strongly suggest that the secondary coordination sphere noncovalent interactions are critical in stabilizing this highly reactive species, which can further react with oxygen to form a sulfinate and then a sulfonate species, as demonstrated by mass spectrometry. Engineering the electron transfer protein azurin into an active copper enzyme that forms a copper–sulfenate center and demonstrating the importance of noncovalent secondary sphere interactions in stabilizing it constitute important contributions toward the understanding of metal–sulfenate species in biological systems.
The presence of cysteine sulfenic acid (Cys-SOH) as a product of posttranslational oxidation of the cysteine thiol side chain has been found to play important roles in biology (1–6) in the context of metal coordination (7), enzyme–protein regulation (8, 9), redox signaling (1, 5), and gene regulation (10–13). Unlike its higher oxidation state counterparts, i.e., sulfinic (Cys-SO2) and sulfonic (Cys-SO3) acids, the sulfenic acid is inherently unstable and highly reactive and thus requires stabilization to operate in a controlled context in biological systems (1–5). For example, a crystal structure of a sulfenic acid form of SarZ, a redox active global transcriptional regulator in Staphylococcus aureus, revealed that cysteine sulfenic acid is stabilized through two hydrogen bonds with surrounding residues, and its reversible oxidation–reduction allows redox-mediated virulence regulation in S. aureus (13).
Because of the inherent instability and high reactivity, few examples of cysteine sulfenic acid have been observed in metal-binding sites in proteins (7, 14). Nature has evolved proteins to allow metal ions to coordinate sulfenic acids, resulting in interesting functions. For example, nitrile hydratase, a metalloenzyme which has long found utility in the industrial synthesis of acrylamide (15), employs a posttranslationally modified cysteine sulfenate and a cysteine sulfinate in the active site, both coordinated to the metal center (Fe or Co) (16). In a similar evolutionary context is thiocyanate hydrolase (7), which uses a similar Co coordination environment to decompose thiocyanate to carbonyl sulfide, ammonia, and water. In both enzymes, the sulfenate and sulfinate groups are unambiguously required for activity (7, 16). It is commonly postulated that such oxidized forms of cysteine in a metal site encourage and stabilize the more oxidized form of redox-active metals, suppressing their potential toxicity in enzymes that do not require the metal center to participate in electron transfer (17, 18). In a more recent example, an iron-coordinated sulfenate was observed when a thiolate-containing substrate analog in isopenicillin N-synthase was unexpectedly oxidized to a coordinated sulfenato group in crystallo, and a connection to the biogenesis of nitrile hydratases and thiocyanate hydrolases was drawn (14).
Although Cys-SOH coordinated to cobalt and nonheme iron has been observed in the enzymes described above (7, 14) and the corresponding synthetic models reported (19), the Cys-SOH has not been observed to stably bind other metals in a biological context. Given the importance of the metal-coordinated sulfenic acids in biological functions and the inherent requirement to stabilize this form from further oxidation, it is important to explore the functional behavior of this group with tools that allow for exquisite control of the metal center and protein function. The creation of metal-binding sites within existing protein scaffolds by engineering the active sites has proven to be a powerful method to design functional metalloproteins (20, 21). Here we demonstrate generation of a copper-coordinated sulfenic acid complex in the type 1 blue copper protein, Pseudomonas aeruginosa azurin, which has shown to be an excellent scaffold for designing novel copper sites (22–27). These results and their implication in our understanding of the metal–sulfenate centers related to their biological functions are discussed.
Results and Discussion
Ultraviolet-Visible Absorption Spectroscopy and Crystallographic Characterization of Cu(II)–M121G Azurin.
M121G azurin was purified in the metal-free (apo-) form. Treatment of apo-M121G azurin with copper(II) sulfate resulted in a strong blue-colored solution. This color remained associated with the protein after size exclusion chromatography via a PD-10 column. The Ultraviolet-visible absorption (UV-vis) spectrum of the protein exhibited a strong absorption at 614 nm, with a shoulder at 453 nm—consistent with that of Cu(II)–M121G azurin reported previously (28) and similar to those of other type 1 blue copper proteins (29, 30)—characterized by a trigonal planar His–His–Cys ligation and a variable axial ligand with small hyperfine splittings in the parallel region of the electron paramagnetic resonance (EPR) spectrum.
To elucidate the structural changes of the M121G mutation on azurin, we solved its crystal structure. Analysis of the crystal structure reveals that the geometry and the bond distances around the copper-binding site are quite similar to those of wild-type (WT) azurin (Fig. 1 A and B). In each of the two proteins, an equatorial plane is established by two histidine imidazolyl ligands and one cysteine thiolate ligand. The major difference between the two structures is the presence of a crystallographically defined water molecule in the active site of M121G azurin, located 2.92 Å from the copper ion and occupying the Met-121 axial ligand position in WT azurin. This observation offers structural support for previously described resonance Raman (rR) spectroscopic data suggesting the presence of an axial water ligand in M121G azurin (26). Interestingly, no crystallographically defined water in the axial position to the copper ion was observed in M121A azurin (27), indicating that, in azurin, the subtle differences in size and hydrophobicity between axial Ala and Gly may dictate water access to the metal center.
Fig. 1.

X-ray crystal structures showing the active sites of WT azurin and M121G azurin. (A) Cu(II)–WT azurin [Protein Data Bank (PDB) code 4AZU]. (B) Cu(II)–M121G azurin (1.54 Å; PDB code 4MFH). (C) Surface accessibility maps generated with a probe sphere radius of 65:00 PM to simulate O-atom accessibility from crystal structures of Cu(II)–M121G azurin; the water channel to the active site is highlighted. (D) Hydrogen bonding network around the water channel to the active site. (Cu in orange, S in yellow, N in blue, C in cyan, and O in red).
It is noteworthy that a water channel is also observed in the crystal structure of Cu(II)–M121G azurin, shown in Fig. 1C. This channel contains four water molecules in a hydrogen-bonding network with the protein backbone and side chains (Fig. 1D), and is not present in the structure of WT azurin. We investigated whether this channel may allow the Cu(I) site of reduced M121G azurin to react with small molecules. Hydrogen peroxide (H2O2) is a small molecule capable of oxidizing or reducing metal centers (31). Interestingly, addition of five equivalents of H2O2 to Cu(I)–M121G azurin in 50 mM potassium phosphate buffer (pH 7.0) resulted in formation of a green-colored solution, with a visible electronic absorption spectrum dominated by spectral features at 452 nm, 614 nm, and a shoulder peak around 350 nm (Fig. 2A). The color of the green solution persisted for several hours at room temperature in the absence of air. In contrast, upon addition of five equivalents of H2O2 to apo-M121G azurin, Zn(II)–M121G azurin, or Cu(II)–M121G azurin under the same conditions, no significant change in the UV-vis spectrum was observed over 30 min (SI Appendix, Fig. S1 A–C). These results suggest that the species in the green-colored solution was a result of a process requiring the reduced Cu(I).When Cu(I)–WT azurin was treated with the same amount of H2O2, we observed only reappearance of the intense blue solution with absorbance spectrum typical of Cu(II)–WT azurin (SI Appendix, Fig. S1D). This result indicated that whereas Cu(I)–WT azurin was oxidized to Cu(II)–WT azurin without significant active site modification, treatment of Cu(I)–M121G azurin with H2O2 resulted in one or more active site modifications. The requirement for a reduced metal center and a small molecule access channel with a labile axial ligand suggests that Cu(I)-coordinated hydrogen peroxide can attack the S(thiolate) in a homolytic, heterolytic, or direct fashion. Further experimental data would be needed to discriminate among these possibilities.
Fig. 2.

Time course of UV-vis spectra upon formation of the air-sensitive species and its decomposition upon air exposure. (A) Addition of five equivalents of H2O2 to 0.9 mM Cu(I)–M121G azurin in 50 mM potassium phosphate pH 7.0 under anaerobic condition. (B) Exposure of the resulting green solution to air. (C) Comparison of UV-vis spectra of the green species (in green), the product after air exposure (in red), and Cu(II)–M121G azurin (in blue). The absorbance was normalized to unchanging spectral contribution at 614 nm. (D) X-band EPR spectra of H2O2-treated Cu(I)–M121G azurin in 50 mM TIP7 buffer pH 7.0 for 30 min (black), simulations of the air-sensitive species (green), Cu(II)–M121G azurin (blue), and a type 2 copper site (red).
Interestingly, exposing the above green-colored solution [generated by treatment of Cu(I)–M121G azurin with H2O2] to air resulted in the rapid loss of the spectral features at 452 and 350 nm, whereas those at 614 nm were largely unaffected (Fig. 2B). The final spectrum was nearly identical in shape to that of Cu(II)–M121G azurin (Fig. 2C), albeit with significantly less intensity. This result demonstrated that the green-colored solution was a mixture of an air-sensitive yellow species and an air-stable blue species, the latter of which resembled authentic Cu(II)–M121G azurin.
EPR Investigation of the Air-Sensitive Species.
To investigate the nature of the copper-containing air-sensitive species, we collected EPR spectra of frozen reaction solutions at various time points after anaerobic addition of H2O2 to Cu(I)–M121G azurin. The EPR data revealed gradual formation of two major axial Cu(II) species besides Cu(II)–M121G azurin (SI Appendix, Fig. S3A). The exposure to air of the H2O2-treated Cu(I)–M121G azurin sample resulted in complete disappearance of one of the major axial Cu(II) species (SI Appendix, Fig. S3B).
Simulation of the above EPR data suggests formation of three different Cu(II) species from oxidation of Cu(I)–M121G azurin with H2O2: Cu(II)–M121G azurin, an air-sensitive Cu(II) species, and a normal type 2 copper species––classified as having tetragonal ligation with N- and O-donor ligands and large hyperfine splittings in the parallel region of the EPR spectrum (Fig. 2D; spectral parameters in SI Appendix, Table S1). The air-sensitive Cu(II) species associated with the 452-nm peak in the UV-vis spectra had a lower gz value of 2.169 compared with normal type 2 copper, indicating a highly covalent interaction. The rest of the type 2 copper signal is most likely due to overoxidation caused by extra equivalents of hydrogen peroxide.
Characterization of the Air-Sensitive Species Using rR Spectroscopy.
In an effort to gain more information about the air-sensitive species, the Cu(I)–M121G azurin solutions after reacting with H2O2 were frozen in liquid N2 and analyzed using rR spectroscopy. rR data using laser excitation into the 452-nm charge-transfer (CT) band of the species formed from the reaction of Cu(I)–M121G azurin with  and
and 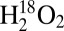 (pH 5.1) are given in Fig. 3 A and B (λex = 457.9 nm). Analogous data have been obtained at pH 7.0 and are given in SI Appendix, Fig. S4, but are of lower resolution due to an intense fluorescence background (however, no significant changes in frequencies or relative intensities were observed between pH 5.1 and 7.0). For
(pH 5.1) are given in Fig. 3 A and B (λex = 457.9 nm). Analogous data have been obtained at pH 7.0 and are given in SI Appendix, Fig. S4, but are of lower resolution due to an intense fluorescence background (however, no significant changes in frequencies or relative intensities were observed between pH 5.1 and 7.0). For  -treated Cu(I)–M121G azurin, three well-resolved resonance-enhanced vibrations were observed in the Cu–ligand stretching region at 374, 380, and 407 cm−1 (Fig. 3A and SI Appendix, Table S2), with the 407-cm−1 feature (c) being the most intense. Several features are also resonance enhanced in the higher energy region of the spectrum (∼700–875 cm−1; Fig. 3B). For type 1 Cu proteins, this region is known to contain the S(Cys)–Cβ stretch and overtone and combination bands of the fundamentals observed in the lower energy region. In particular, for the species formed from the reaction of Cu(I)–M121G azurin with
-treated Cu(I)–M121G azurin, three well-resolved resonance-enhanced vibrations were observed in the Cu–ligand stretching region at 374, 380, and 407 cm−1 (Fig. 3A and SI Appendix, Table S2), with the 407-cm−1 feature (c) being the most intense. Several features are also resonance enhanced in the higher energy region of the spectrum (∼700–875 cm−1; Fig. 3B). For type 1 Cu proteins, this region is known to contain the S(Cys)–Cβ stretch and overtone and combination bands of the fundamentals observed in the lower energy region. In particular, for the species formed from the reaction of Cu(I)–M121G azurin with 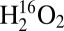 , the feature observed at 837 cm−1 is too high in energy to be related to the fundamental vibrations observed in the lower energy region. No higher energy vibrations are observed. When
, the feature observed at 837 cm−1 is too high in energy to be related to the fundamental vibrations observed in the lower energy region. No higher energy vibrations are observed. When 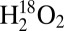 is used as the oxidant, the lower energy vibrations are observed at 371, 380, and 395 cm−1, which correspond to Δ (16–18)’s = 3, 0, and 12 cm−1, respectively. Also, the lower energy feature [labeled (a) in Fig. 3A] becomes the most intense. The experimental energies, intensities, and isotopic shifts of features (a) and (c) are consistent with a Fermi resonance. These values were used to determine the vibrational energies in the absence of this interaction. For the reaction with
is used as the oxidant, the lower energy vibrations are observed at 371, 380, and 395 cm−1, which correspond to Δ (16–18)’s = 3, 0, and 12 cm−1, respectively. Also, the lower energy feature [labeled (a) in Fig. 3A] becomes the most intense. The experimental energies, intensities, and isotopic shifts of features (a) and (c) are consistent with a Fermi resonance. These values were used to determine the vibrational energies in the absence of this interaction. For the reaction with 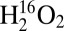 , features (a) and (c) are predicted to be at 399 and 381 cm−1, respectively, whereas for reaction with
, features (a) and (c) are predicted to be at 399 and 381 cm−1, respectively, whereas for reaction with  , (a) and (c) are predicted to be at 387 and 383 cm−1, respectively. In the higher energy region, the 837 cm−1 feature shifts down in energy to 807 cm−1 (Δ = −30 cm−1) when
, (a) and (c) are predicted to be at 387 and 383 cm−1, respectively. In the higher energy region, the 837 cm−1 feature shifts down in energy to 807 cm−1 (Δ = −30 cm−1) when  is used as the oxidant. The rR-enhanced vibrations observed here, combined with the EPR results, may either reflect an oxidized thiolate bound to Cu(II) or a Cu(II)–OOH species. These possibilities, as well as different Cu(II)–SOxHx species and their binding modes, are evaluated using density functional theory (DFT) and time-dependent DFT (TDDFT) calculations (vide infra).
is used as the oxidant. The rR-enhanced vibrations observed here, combined with the EPR results, may either reflect an oxidized thiolate bound to Cu(II) or a Cu(II)–OOH species. These possibilities, as well as different Cu(II)–SOxHx species and their binding modes, are evaluated using density functional theory (DFT) and time-dependent DFT (TDDFT) calculations (vide infra).
Fig. 3.

rR spectroscopy of the air-sensitive species. (A) rR spectra of  and
and  (blue) in the low-energy vibrational region (* indicates Raman scattering feature of ice) and (B) the high-energy vibrational region (λex = 457.9 nm). (C) Low-energy vibrational region using λex = 647.1 nm.
(blue) in the low-energy vibrational region (* indicates Raman scattering feature of ice) and (B) the high-energy vibrational region (λex = 457.9 nm). (C) Low-energy vibrational region using λex = 647.1 nm.
Lastly, the 614-nm absorption band was also investigated on the same samples discussed above (λex = 647.1 nm). Using this excitation wavelength, identical rR data (only with variable total intensities) were obtained upon reaction of Cu(I)–M121G azurin with 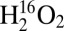 or
or 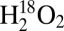 , or upon further exposure of the reaction mixture to air (Fig. 3C). These data are the same as those obtained by laser excitation into the Cu(II)–M121G azurin CT transition, and indicate that the electronic absorption contributions in the 610–630-nm region observed in the UV-vis experiments can be assigned to variable concentrations of authentic Cu(II)–M121G azurin, consistent with the EPR results above.
, or upon further exposure of the reaction mixture to air (Fig. 3C). These data are the same as those obtained by laser excitation into the Cu(II)–M121G azurin CT transition, and indicate that the electronic absorption contributions in the 610–630-nm region observed in the UV-vis experiments can be assigned to variable concentrations of authentic Cu(II)–M121G azurin, consistent with the EPR results above.
Capture of Sulfenic Acid with the Selective Labeling Agent, Dimedone.
Because the above results suggested the presence of an oxidized sulfur at the active site of the air-sensitive species, we tested the hypothesis that a sulfenic acid moiety coordinates a copper ion using dimedone, a well-established agent for the selective detection of sulfenic acid (32, 33). After reaction of sulfenic acids with dimedone, the total mass should increase by +138 Da due to irreversible and selective condensation, which can be observed via mass spectrometry (34). Addition of dimedone to the air-sensitive species under normal physiological conditions did not result in any modification of the protein, probably due to a lack of access by the dimedone to the sulfenic group in the folded protein. To overcome this constraint, we added guanidine hydrochloride to a final concentration of 6 M to the air-sensitive species in the presence of an excess of dimedone. This procedure resulted in an increase of +138 Da to the molecular mass of the M121G azurin [Fig. 4A, incubation time of Cu(I)–M121G azurin with H2O2 of 30 s], consistent with a dimedonylated protein. When the incubation time of Cu(I)–M121G azurin with H2O2 was increased from 30 s to 30 min, significantly enhanced intensity of dimedone-labeled product was observed (Fig. 4B), suggesting an increased yield of sulfenic group formation with time. A control experiment using CuI–WT azurin after treatment with H2O2 under the same condition showed no labeling peak, and this result was critical in demonstrating that the sulfenic moiety did not form through processes involving unfolded protein (SI Appendix, Fig. S5).
Fig. 4.

MS spectra of the air-sensitive species. Electrospray mass spectrum before (black) and after (red) dimedone labeling of the air-sensitive species. Cu(II)–sulfenate-M121G azurin is generated by treating Cu(I)–M121G azurin with 2 eq. H2O2 in 50 mM potassium phosphate pH 7.0 for (A) 30 s and (B) 30 min. For M121G azurin, cal. 13871.67 Da and obs. 13871 Da. The –SOH (+16 Da) is the sum of Cys112–sulfenate and other singly oxygenated byproducts (cal. 13,887.67 Da and obs. 13,887 Da). The –S-dimedone (+138 Da) peak is the dimedone-labeled M121G azurin (cal. 14,009.67 Da and obs. 14,010 Da). (C) Calculated and experimental isotopic distributions for product EGEQYMFF(C–SO3H)TFPGHSALGK tryptic peptides [(pep) + 2H+]+2 with progressive 18O labeling. Experimental spectra were obtained with 1 eq. additions of  or
or 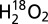 to Cu(I)–M121G and an aerobic workup.
to Cu(I)–M121G and an aerobic workup.
As azurin contains three native cysteine residues (two of which constitute a disulfide bond), we confirmed dimedone labeling on Cys-112 via tryptic digest and tandem MS/MS analysis of the dimedone-labeled material. The dimedone modified peptide NH2-LKEGEQYMFFCdimedoneTFPGHSALGK-COOH was robustly detected, and was further analyzed via MS/MS. A 241.0105-Da difference was observed between b10 and b11 ions, corresponding to the molecular mass of dimedone-labeled cysteine after dehydration (calculated: 241.0773 Da). This result confirms that the sulfenic acid modification is isolated to the Cys-112 residue of M121G azurin (SI Appendix, Fig. S6 and Table S3).
Isotopic Enrichment Study of Cys-SO3H Formation.
To confirm that the oxygen atom on the cysteine residue of M121G was derived from H2O2, we compared the isotope distribution of oxygen found in the oxidized cysteine after treatment of Cu(I)–M121G azurin with either H2O2 or 18O-labeled H2O2
 . We exposed the green-colored solution to air before demetallation and tryptic digest. The peptide containing Cys112, NH2-EGEQYMFFCTFPGHSALGK-COOH, was analyzed by high-resolution MS. The data show that the peptide exhibited a mass increase consistent with addition of three oxygen atoms, suggesting formation of a cysteine sulfonic acid, the most oxidized organic form of sulfur. After comparing the experimental data with the calculated isotopic distribution, a nearly pure isotopic spectrum of Cys-S18O16O2H is formed (Fig. 4C), suggesting that under these conditions H2O2-mediated cysteine oxidation stopped at sulfenic acid, and further oxidation was from air. We note that higher equivalents of H2O2 may produce sulfinic and sulfonic acid directly by further oxidizing Cys-SOH produced in the reaction.
. We exposed the green-colored solution to air before demetallation and tryptic digest. The peptide containing Cys112, NH2-EGEQYMFFCTFPGHSALGK-COOH, was analyzed by high-resolution MS. The data show that the peptide exhibited a mass increase consistent with addition of three oxygen atoms, suggesting formation of a cysteine sulfonic acid, the most oxidized organic form of sulfur. After comparing the experimental data with the calculated isotopic distribution, a nearly pure isotopic spectrum of Cys-S18O16O2H is formed (Fig. 4C), suggesting that under these conditions H2O2-mediated cysteine oxidation stopped at sulfenic acid, and further oxidation was from air. We note that higher equivalents of H2O2 may produce sulfinic and sulfonic acid directly by further oxidizing Cys-SOH produced in the reaction.
DFT Calculations.
Several potential structural models for the air-sensitive species have been computationally evaluated; their structural and spectroscopic properties are compared with experiment. As a starting point, a small model of Cu(II)–M121G azurin was created from the X-ray crystal structure. This small computational model is referred to as S–M121G–H2O in this section. Optimization of this S–M121G–H2O with two N(Imidazole (Im)) and one S(Ethyl (Et)) equatorial ligands and one axial H2O ligand resulted in a structure in reasonable agreement with crystallography (SI Appendix, Fig. S7 and Table S4). The axial H2O–Cu distance is ∼0.4 Å too short in the DFT structure (2.5 vs. 2.9 Å). This difference in distance likely reflects the absence of several H-bonding partners to the Cu–OH2 bond, which are present in the X-ray crystal structure. For S–M121G–H2O, the calculated Cu–S frequencies (408 and 413 cm−1; SI Appendix, Table S5) are in good agreement with experiment (Fig. 3C). Note that the Cu–S vibration mixes with N(Im) and S(Et) ligand modes at similar energy. Furthermore, the TDDFT-calculated absorption spectrum of S–M121G–H2O (SI Appendix, Fig. S9A) agrees well with experiment, with one intense CT band at ∼560 nm (∼17,850 cm−1).
Several models of the air-sensitive species have been considered (1–7). All of these contain oxidized thiolate ligands. The relevant optimized bond distances are given in SI Appendix, Table S4 and Fig. S7. Mulliken spin densities are also given in SI Appendix, Table S4 for comparison. Specifically, 1 is a pseudoside-on sulfenate (Cu(II)–SO−) species; 2–4 have protonated sulfenates (Cu(II)–SOH) bound to Cu(II) in a pseudoside-on fashion (2), through S (3) or OH (4); 5–7 contain a sulfinate  group bound to Cu(II) via S (5), two Os (6), or one O (7). To produce a structural model for the air-sensitive species, both TDDFT and frequency calculations have been carried out on 1–7 for correlation to experimental data. The TDDFT-calculated absorption spectra are shown in SI Appendix, Fig. S9 A and B (divided for clarity). The best agreement between the experimental and calculated absorption spectrum of the air-sensitive species is given by 1 (red line, SI Appendix, Fig. S9A), which exhibits an intense CT transition at higher energy (∼475 nm; ∼21,050 cm−1) than that predicted for S–M121G–H2O. However, 2 (blue line, SI Appendix, Fig. S9A) and 3 (green line, SI Appendix, Fig. S9A) both exhibit higher-energy CT transitions and are therefore also potential candidates based on TDDFT calculations alone.
group bound to Cu(II) via S (5), two Os (6), or one O (7). To produce a structural model for the air-sensitive species, both TDDFT and frequency calculations have been carried out on 1–7 for correlation to experimental data. The TDDFT-calculated absorption spectra are shown in SI Appendix, Fig. S9 A and B (divided for clarity). The best agreement between the experimental and calculated absorption spectrum of the air-sensitive species is given by 1 (red line, SI Appendix, Fig. S9A), which exhibits an intense CT transition at higher energy (∼475 nm; ∼21,050 cm−1) than that predicted for S–M121G–H2O. However, 2 (blue line, SI Appendix, Fig. S9A) and 3 (green line, SI Appendix, Fig. S9A) both exhibit higher-energy CT transitions and are therefore also potential candidates based on TDDFT calculations alone.
The DFT-calculated frequencies and 16Ox and 18Ox shifts and the vibrational assignments for 1-7 are given in SI Appendix, Table S5. Out of these candidates, 1 gives the best agreement with the experimental rR data on the air-sensitive species. For 1, two Cu–SO-based vibrations are predicted at 442 and 418 cm−1, which shift down in energy to 431 and 412 cm−1, respectively, upon 18O substitution. A S–O vibration is also predicted at 832 cm−1, which shifts down in energy to 803 cm−1 upon 18O substitution [Δ = 29 cm−1; exp, 837 cm−1 (Δ = 30 cm−1)]. The calculated ν(16O)/ν(18O) ratio for 1 is 1.037, in excellent agreement with the harmonic value for 32S16O and 32S18O (1.039).
Although the air-sensitive species does not form upon addition of H2O2 to Cu(II)–M121G azurin, the experimental frequencies and isotope shifts would also be reasonably consistent with a Cu(II)–OOH species (35). We therefore also considered this possibility computationally. Relevant geometric parameters of the optimized Cu(II)–OOH structure are given in SI Appendix, Table S4, and the calculated vibrations are given in SI Appendix, Table S5. The Cu–ligand vibrations and their isotope shifts [Cu–S: 374(1) cm−1; Cu–O: 459(12) cm−1] are consistent with experiment; however, the calculated O–OH frequency and isotope shift [891(49) cm−1] are both too high. Furthermore, two intense CT transitions, split in energy by ∼4,000 cm−1, are predicted by TDDFT (SI Appendix, Fig. S9B). This is not consistent with the experimental absorption spectrum of the air-sensitive species in Fig. 2C. On the basis of these discrepancies with experiment, a Cu(II)–OOH species can be eliminated. Thus, 1, a pseudoside-on bound Cu(II)–SO− complex, can be considered a likely model of the air-sensitive species.
Note that, in addition to 1 [Cu(II)-SO−], two other geometric and electronic isomers exist as energetic minima for a Cu(II)–SO− species. Instead of the pseudoside-on binding, the SO group can bind through O or S in a monodentate fashion (8 and 9, respectively; SI Appendix, Fig. S7 and Table S4). In both cases the SO group reduces Cu to form a Cu(I)–SO• species, in contrast with what is observed experimentally by EPR. Here 8 is lower in energy than 1 by ∼6 kcal/mol, whereas 9 is higher in energy than 1 by ∼3 kcal/mol. Clearly, 8 and 9 are not viable candidates for the air-sensitive species due to reduction of the Cu active site. However, SO binding through O with reduction of Cu is favored in these small models. We have therefore considered the role of the protein environment in tuning the relative energies of 1, 8, and 9, as well as the influence of the protein matrix on their respective ground-state wave functions [i.e., Cu(II)–SO− vs. Cu(I)–SO•] (36).
To consider the second sphere coordination environment, a large active site model of Cu(II)–M121G azurin was constructed. This large computational model is referred to as L-M121G–H2O. The following second sphere interactions have been included: (i) a negatively oriented dipole near Cu; (ii) two amide backbone H bonds to S (or SO); and (iii) a negatively oriented dipole near S (SI Appendix, Fig. S8). The calculated geometric structure (SI Appendix, Table S4), Cu–S(Cys) vibrational frequency (SI Appendix, Table S6), and absorption spectrum (SI Appendix, Fig. S9C) of L-M121G–H2O are in good agreement with experiment. We have therefore used this model to consider the effects of S oxidation to sulfenate, and to evaluate the three binding modes of SO to Cu: pseudoside-on (10), O-bound (11), or S-bound (12). Note that 10, 11, and 12 are large model analogs of 1, 8, and 9, respectively, in the small models. The relevant geometric parameters for 10, 11, and 12 are included in SI Appendix, Table S4. Their structures are given in SI Appendix, Fig. S8. In the large models, both 10 and 11 are found to have Cu(II)–SO− ground states. For 12, the sulfenate group reduces the Cu (as observed in the small model analog). In the large models, 10 and 11 are isoenergetic within <0.5 kcal/mol, whereas 12 is ∼6 kcal/mol higher in energy. We therefore focus on 10 and 11. The TDDFT-predicted UV-vis spectra for 10 and 11 are given in SI Appendix, Fig. S9C. From these calculations, the spectrum of 10 (∼430 nm; ∼23,250 cm−1) best represents the experimental UV-vis data of the air-sensitive species and is consistent with the calculated spectrum of 1. The TDDFT-calculated UV-vis spectrum of 11 exhibits an intense CT transition with a energy lower (∼620 nm; 16,130 cm−1) than that of the L-M121G–H2O model (∼560 nm; ∼17,850 cm−1). This is opposite the trend observed experimentally.
The DFT-calculated vibrational energies and isotope shifts of 10 and 11 are given in SI Appendix, Table S6. Here, 10 gives the best agreement with the experimental rR data on the air-sensitive species. Specifically, the calculated Cu–O vibrational frequency is estimated to be too high in energy for 11 (11: 387, 463, and 477 cm−1; 10: 448 and 457 cm−1). The aggregate 16O/18O shift of the S–O vibration is also larger in 11 relative to 10 (34 vs. 28 cm−1, respectively). Note that the S–O vibration in 11 is mixed approximately equally into two modes, so the average value is used here. Thus, 10 (and its small analog 1), a pseudoside-on Cu(II)–SO− species, best reproduces the experimental absorption and rR data for the air-sensitive species formed in the reaction of Cu(I)–M121G azurin and H2O2.
It is interesting to note which second sphere interactions allow for the stabilization of a Cu(II)–SO− ground state. We systematically removed the second sphere interactions and carried out additional geometry optimizations. The geometric parameters and Mulliken spin densities are given in SI Appendix, Table S7. Reduction of Cu occurs upon removing both the negatively oriented dipole near Cu and the H bonds to the sulfenate (SI Appendix, Fig. S8). Therefore, the secondary protein environment plays an important role in tuning the relative redox couples of the Cu and the sulfenate, allowing for the stabilization of a Cu(II)–SO− species.
The ground-state wave functions [β-Lowest Unoccupied Molecular Orbitals (LUMOs)] of both L-M121G–H2O and 10 are compared in Fig. 5. The calculated Cu(d) and S(p) characters (from Mulliken population analyses) of L-M121G–H2O associated with the Cu(II)–S(thiolate) π-bond are 56% and 25%, respectively. For 10, binding SO− in a pseudoside-on fashion decreases the Cu(d) character to 48%, whereas the SO− character increases to 35%. Thus, the Cu–SO− unit in 10 is more covalent than the Cu–S(Cys) bond in L-M121G–H2O. This is due to the strong σ-overlap associated with two bonding interactions between the SO− π*-donor and the unoccupied 3d(x2−y2) orbital in the side-on structure (Fig. 5, Right). Furthermore, the intense ∼430-nm CT band of 10 is calculated to be a transition between the bonding and antibonding pair of the β-LUMO. Thus, the increased energy of this transition in 10 relative to the Sp(π) → Cu(d(x2−y2)) (∼550 nm) CT transition of L-M121G–H2O reflects the increase in the bonding–antibonding interaction of the Cu–SO− unit compared with the Cu–S(Cys) bond.
Fig. 5.

Comparison of the ground-state wave functions (β-LUMOs) of L-M121G-H2O (Left) and 10 (Right). Results of Mulliken population analyses are indicated.
In addition, in going from Cu(II)–M121G azurin to the air-sensitive complex, the g|| value decreases from 2.303 to 2.169, and the A|| value increases in magnitude from ∼20 × 10−4 cm−1 to 139 × 10−4 cm−1. Note that A|| is negative. The deviation of the g|| value from 2.0023 is inversely proportional to energy of the d(xy) → d(x2−y2) ligand field transition. The TDDFT-calculated energies of this transition in L-M121G–H2O and 10 are ∼12,400 and 16,900 cm−1, respectively, consistent with the larger g|| in the former. However, the energy difference between these two species accounts for only ∼60% of the difference in the g|| values. This indicates that the Cu character in the ground state of 10 must be lower than in L-M121G–H2O, which is consistent with the Mulliken population analyses (Fig. 5) and spin densities (SI Appendix, Table S4). Finally, we note that the increase in the magnitude of A|| has two contributions. First, Cu(II)–M121G azurin has a rhombic EPR signal, which is generally associated with dz2/4s mixing. This mixing will reduce the magnitude of A|| in the reference [Cu(II)–M121G azurin]. Second, the decrease in g|| between Cu(II)–M121G azurin and the air-sensitive species decreases the orbital dipolar contributions to the hyperfine, which is positive and will also contribute to the larger |A|||.
Conclusions
In summary, we have shown that anaerobic addition of H2O2 to Cu(I)–M121G azurin resulted in the formation of a side-on Cu(II)–sulfenate species in azurin, as supported by UV-vis, EPR, and rR spectroscopies as well as mass spectrometry and electronic structure calculations correlated to the spectroscopic data. The observation of a Cu(II)–sulfenate species is made possible mainly due to stabilization of the species by noncovalent secondary sphere interactions. The stabilization and characterization of a copper–sulfenate center in proteins opens an avenue for the detection and exploration of other similarly reactive species in metallobiochemistry.
Materials and Methods
Experimental details about protein purification, holo-protein preparation, crystallization, capture and detection of sulfenate with dimedone, and trypsin digest are described in the SI Appendix. Analysis of EPR, rR spectroscopy, ESI-MS and HPLC MS/MS are available in SI Appendix, Figs. S2–S6. Parameters and results of DFT calculation are listed in SI Appendix, Tables S4–S7.
Supplementary Material
Acknowledgments
We thank Drs. Peter M. Yau and Brian S. Imai from the Protein Science Facility in the Biotechnology Center at the University of Illinois at Urbana–Champaign for performing trypsin digest and HPLC MS/MS analysis of the protein. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US DOE under Contract DE-AC02-06CH11357. Use of the life science collaborative access team (LS-CAT) Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). This work is based on work supported by the National Science Foundation under Awards CHE 1058959 (to Y.L.) and CHE 0948211 (to E.I.S.). R.G.H. acknowledges a Gerhard Casper Stanford Graduate Fellowship and the Achievement Rewards for College Scientists (ARCS) Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structures factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4MFH and 4AZU).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1316483111/-/DCSupplemental.
References
- 1.Giles NM, Giles GI, Jacob C. Multiple roles of cysteine in biocatalysis. Biochem Biophys Res Commun. 2003;300(1):1–4. doi: 10.1016/s0006-291x(02)02770-5. [DOI] [PubMed] [Google Scholar]
- 2.Claiborne A, et al. Protein-sulfenic acids: Diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38(47):15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 3.Rehder DS, Borges CR. Cysteine sulfenic acid as an intermediate in disulfide bond formation and nonenzymatic protein folding. Biochemistry. 2010;49(35):7748–7755. doi: 10.1021/bi1008694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poole LB. Curr Protoc Toxicol. 2003. Formation and Functions of Protein Sulfenic Acids. 10.1002/0471140856.tx1701s18. [DOI] [PubMed] [Google Scholar]
- 5.Paulsen CE, Carroll KS. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem Rev. 2013;113(7):4633–4679. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng X, et al. Proteome-wide quantification and characterization of oxidation-sensitive cysteines in pathogenic bacteria. Cell Host Microbe. 2013;13(3):358–370. doi: 10.1016/j.chom.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arakawa T, et al. Structural basis for catalytic activation of thiocyanate hydrolase involving metal-ligated cysteine modification. J Am Chem Soc. 2009;131(41):14838–14843. doi: 10.1021/ja903979s. [DOI] [PubMed] [Google Scholar]
- 8.Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J Biol Chem. 2001;276(44):41279–41287. doi: 10.1074/jbc.M106958200. [DOI] [PubMed] [Google Scholar]
- 9.Michalek RD, et al. The requirement of reversible cysteine sulfenic acid formation for T cell activation and function. J Immunol. 2007;179(10):6456–6467. doi: 10.4049/jimmunol.179.10.6456. [DOI] [PubMed] [Google Scholar]
- 10.Abate C, Patel L, Rauscher FJ, 3rd, Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science. 1990;249(4973):1157–1161. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- 11.Aslund F, Zheng M, Beckwith J, Storz G. Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc Natl Acad Sci USA. 1999;96(11):6161–6165. doi: 10.1073/pnas.96.11.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauer CE, Elsen S, Bird TH. Mechanisms for redox control of gene expression. Annu Rev Microbiol. 1999;53(1):495–523. doi: 10.1146/annurev.micro.53.1.495. [DOI] [PubMed] [Google Scholar]
- 13.Poor CB, Chen PR, Duguid E, Rice PA, He C. Crystal structures of the reduced, sulfenic acid, and mixed disulfide forms of SarZ, a redox active global regulator in Staphylococcus aureus. J Biol Chem. 2009;284(35):23517–23524. doi: 10.1074/jbc.M109.015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ge W, et al. Isopenicillin N synthase mediates thiolate oxidation to sulfenate in a depsipeptide substrate analogue: Implications for oxygen binding and a link to nitrile hydratase? J Am Chem Soc. 2008;130(31):10096–10102. doi: 10.1021/ja8005397. [DOI] [PubMed] [Google Scholar]
- 15.Padmakumar R, Oriel P. Bioconversion of acrylonitrile to acrylamide using a thermostable nitrile hydratase. Appl Biochem Biotechnol. 1999;77-79(1):671–679. doi: 10.1385/abab:79:1-3:671. [DOI] [PubMed] [Google Scholar]
- 16.Murakami T, et al. Post-translational modification is essential for catalytic activity of nitrile hydratase. Protein Sci. 2000;9(5):1024–1030. doi: 10.1110/ps.9.5.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Claiborne A, Mallett TC, Yeh JI, Luba J, Parsonage D. Structural, redox, and mechanistic parameters for cysteine-sulfenic acid function in catalysis and regulation. Adv Protein Chem. 2001;58:215–276. doi: 10.1016/s0065-3233(01)58006-7. [DOI] [PubMed] [Google Scholar]
- 18.Giles NM, et al. Metal and redox modulation of cysteine protein function. Chem Biol. 2003;10(8):677–693. doi: 10.1016/s1074-5521(03)00174-1. [DOI] [PubMed] [Google Scholar]
- 19.Kovacs JA. Synthetic analogues of cysteinate-ligated non-heme iron and non-corrinoid cobalt enzymes. Chem Rev. 2004;104(2):825–848. doi: 10.1021/cr020619e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu Y, Berry SM, Pfister TD. Engineering novel metalloproteins: Design of metal-binding sites into native protein scaffolds. Chem Rev. 2001;101(10):3047–3080. doi: 10.1021/cr0000574. [DOI] [PubMed] [Google Scholar]
- 21.Lu Y, Yeung N, Sieracki N, Marshall NM. Design of functional metalloproteins. Nature. 2009;460(7257):855–862. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hay M, Richards JH, Lu Y. Construction and characterization of an azurin analog for the purple copper site in cytochrome c oxidase. Proc Natl Acad Sci USA. 1996;93(1):461–464. doi: 10.1073/pnas.93.1.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lancaster KM, Yokoyama K, Richards JH, Winkler JR, Gray HB. High-potential C112D/M121X (X = M, E, H, L) Pseudomonas aeruginosa azurins. Inorg Chem. 2009;48(4):1278–1280. doi: 10.1021/ic802322e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lancaster KM, DeBeer George S, Yokoyama K, Richards JH, Gray HB. Type-zero copper proteins. Nat Chem. 2009;1(9):711–715. doi: 10.1038/nchem.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berry SM, Mayers JR, Zehm NA. Models of noncoupled dinuclear copper centers in azurin. J Biol Inorg Chem. 2009;14(1):143–149. doi: 10.1007/s00775-008-0432-1. [DOI] [PubMed] [Google Scholar]
- 26.Fraczkiewicz G, Bonander N, Czernuszewicz RS. Metal-ligand interactions in azido, cyano and thiocyanato adducts of Pseudomonas aeruginosa Met121X (X=Gly, Ala, Val or Leu) azurins monitored by resonance Raman spectroscopy. J Raman Spectrosc. 1998;29(10-11):983–995. [Google Scholar]
- 27.Tsai LC, et al. Mutant Met121Ala of Pseudomonas aeruginosa azurin and its azide derivative: Crystal structures and spectral properties. Acta Crystallogr D Biol Crystallogr. 1996;52(Pt 5):950–958. doi: 10.1107/S0907444996004982. [DOI] [PubMed] [Google Scholar]
- 28.Dave BC, Germanas JP, Czernuszewicz RS. The first direct evidence for copper(II)-cysteine vibrations in blue copper proteins: Resonance Raman spectra of 34S-Cys-labeled azurins reveal correlation of copper-sulfur stretching frequency with metal site geometry. J Am Chem Soc. 1993;115(25):12175–12176. [Google Scholar]
- 29.Gray HB, Malmström BG, Williams RJP. Copper coordination in blue proteins. J Biol Inorg Chem. 2000;5(5):551–559. doi: 10.1007/s007750000146. [DOI] [PubMed] [Google Scholar]
- 30.Lu Y. In: Electron Transfer: Cupredoxins. Biocoordination Chemistry, Comprehensive Coordination Chemistry II: From Biology to Nanotechnology. Que JL, Tolman WB, editors. Vol 8. Oxford: Elsevier; 2004. pp. 91–122. [Google Scholar]
- 31.Jones CW, Clark JH. Activation of hydrogen peroxide using inorganic and organic species. In: Jones CW, Clark JH, editors. Applications of Hydrogen Peroxide and Derivatives. Cambridge, UK: Royal Society of Chemistry; 1999. [Google Scholar]
- 32.Allison WS. Formation and reactions of sulfenic acids in proteins. Acc Chem Res. 1976;9(8):293–299. [Google Scholar]
- 33.O'Donnell JS, Schwan AL. Generation, structure and reactions of sulfenic acid anions. J Sulfur Chem. 2004;25(2-3):183–211. [Google Scholar]
- 34.Seo YH, Carroll KS. Quantification of protein sulfenic acid modifications using isotope-coded dimedone and iododimedone. Angew Chem Int Ed Engl. 2011;50(6):1342–1345. doi: 10.1002/anie.201007175. [DOI] [PubMed] [Google Scholar]
- 35.Chen P, Fujisawa K, Solomon EI. Spectroscopic and theoretical studies of mononuclear copper(II) alkyl- and hydroperoxo complexes: Electronic structure contributions to reactivity. J Am Chem Soc. 2000;122(41):10177–10193. [Google Scholar]
- 36.Hadt RG, et al. Spectroscopic and DFT studies of second-sphere variants of the type 1 copper site in azurin: Covalent and nonlocal electrostatic contributions to reduction potentials. J Am Chem Soc. 2012;134(40):16701–16716. doi: 10.1021/ja306438n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.