


Abstract
Translation of the hepatitis C virus (HCV) RNA is mediated by the interaction of ribosomes and cellular proteins with an internal ribosome entry site (IRES) located within the 5′-untranslated region (5′-UTR). We have investigated whether small RNA molecules corresponding to the different stem–loop (SL) domains of the HCV IRES, when introduced in trans, can bind to the cellular proteins and antagonize their binding to the viral IRES, thereby inhibiting HCV IRES-mediated translation. We have found that a RNA molecule corresponding to SL III could efficiently inhibit HCV IRES-mediated translation in a dose-dependent manner without affecting cap-dependent translation. The SL III RNA was found to bind to most of the cellular proteins which interacted with the HCV 5′-UTR. A smaller RNA corresponding to SL e+f of domain III also strongly and selectively inhibited HCV IRES-mediated translation. This RNA molecule interacted with the ribosomal S5 protein and prevented the recruitment of the 40S ribosomal subunit. This study reveals valuable insights into the role of the SL structures of the HCV IRES in mediating ribosome entry. Finally, these results provide a basis for developing anti-HCV therapy using small RNA molecules mimicking the SL structures of the 5′-UTR to specifically block viral RNA translation.
INTRODUCTION
Hepatitis C virus (HCV) is a major human pathogen with an estimated 170 million chronic carriers throughout the world, many of whom are at a significant risk for developing liver cirrhosis and hepatocellular carcinoma (1). Current therapeutic strategies using interferon-α, either alone or in combination with ribavirin, have poor efficacy (2). Moreover, an important lesson obtained from the therapy of other viral infections is that multiple drug targets are required to prevent the emergence of drug-resistant varieties of the virus (3).
HCV is a positive stranded RNA virus and translation of the viral genomic RNA is an early obligatory step of the infection process. Translation initiation of the uncapped viral RNA takes place through the internal ribosome entry site (IRES) located in the 5′-untranslated region (5′-UTR) (4). Translation initiation from the IRES is mediated by a number of cellular trans-acting factors like the La autoantigen (5) and polypyrimidine tract-binding protein (PTB) (6), together with some canonical eukaryotic initiation factors (eIFs), like eIF3 (7), eIF2 and eIF2B (8). It has been suggested that the IRES acts as a structural scaffold with specifically placed recognition sites for recruiting the translation machinery (9). As this mechanism of translation initiation is distinct from the cellular cap-dependent mechanism, it is an attractive target for antiviral therapeutics with high target specificity and low host cytotoxicity (10).
The concept of using RNA molecules as therapeutic agents has aroused increasing interest in the past decade. Antisense RNAs and trans-cleaving ribozymes have been studied as potential inhibitors of HCV translation (11,12). Recently, RNA interference using small interfering RNAs has proved to be highly efficient in inhibiting the replication of a number of viruses, including HCV (3).
Another strategy for blocking the replication of RNA viruses has been to overexpress small and structured viral RNA elements in target cells. These RNAs bind to viral regulatory proteins and prevent their binding to corresponding sequences in the viral RNA, thus inhibiting viral gene expression. Overexpression of human immunodeficiency virus (HIV) trans-activation response region (TAR) and Rev response element (RRE) RNAs in CD4+ T cells prevented the binding of viral Tat and Rev proteins to the viral RNA and made the cells resistant to HIV replication (13,14). Also, a 60 nt RNA termed IRNA, isolated from Saccharomyces cerevisae, has been shown to block IRES-mediated translation of poliovirus and HCV by sequestering cellular trans-acting factors which interact with the viral IRESs (15,16). La and PTB-specific SELEX RNA have also been shown to inhibit HCV IRES-mediated translation (17,18).
In this study we demonstrate a novel approach to inhibit HCV IRES-mediated translation using small RNA molecules mimicking the structure of the defined stem–loop (SL) domains of the HCV IRES. We show that a small RNA molecule corresponding to domain III of the HCV IRES and a smaller molecule corresponding to the SL III e+f subdomain binds to specific cellular proteins interacting with the HCV IRES and strongly inhibit HCV IRES-mediated translation without inhibiting cap-dependent translation.
MATERIALS AND METHODS
Cells and plasmids
HeLa S3 and Huh7 cells were grown in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum. IRES domains II (nucleotides 18–123), III excluding III e and f (nucleotides 121–290) and IV including 42 nt from the coding region (nucleotides 291–383) were PCR amplified from the type 1b HCV 5′-UTR (plasmid pCV, a gift from Dr A. Nomoto, Tokyo University, Japan) and cloned in the vector pCDNA3 (Invitrogen) to generate the plasmids pCD-SL II, SL III and SL IV, respectively. The bicistronic vector pRL-HCV1b having a Rluc (Renilla luciferase)–HCV 5′-UTR–Fluc (firefly luciferase) construct under control of the T7 promoter was a gift from Dr Richard Elliot (Glasgow University, UK). The plasmid pRL-CMV (Promega, Madison, WI) had a Rluc gene under control of the CMV promoter. pCD-HCV-Fluc was constructed by cloning the HCV 5′-UTR (nucleotides 18–383) upstream of the Fluc gene in pCDNA3. The encephalomyocarditis virus (EMCV) bicistronic plasmid was constructed by inserting the EMCV functional IRES, PCR amplified from the vector pCITE (Novagen, Darmstadt, Germany), between Fluc and green fluorescent protein (GFP) genes cloned in the vector pCDNA3. The HCV subgenomic replicon (BB7) was a gift from Apath Inc. (St Louis, MO).
In vitro transcription
The plasmid pRL-HCV1b was linearized downstream of Fluc and transcribed using T7 RNA polymerase in the presence of RNA Cap Analog (Invitrogen) to generate the bicistronic capped RNA. The plasmids pCD-SL II, SLIII and SL IV were linearized with EcoRI and transcribed by in vitro run-off transcription reactions under standard conditions using reagents from Promega. 32P-labeled HCV 5′-UTR RNA and the SL RNAs were transcribed from respective plasmids using T7 RNA polymerase and [α-32P]UTP (Perkin Elmer Life Sciences, Boston, MA).
Oligonucleotide-driven transcription
Synthetic DNA oligonucleotides corresponding to domain III SL a+c, b, d and e+f structures (sequences: a+c, CGCCTT GGCCACTCATGTGGCCTTAACTCTAAACCCGCACGGGGGCG; b, GGTCCTG CTGGCCCAGGAAAGAACCTA GTTGGGCGAGTTACGGACC; d, ATCGGCTCATCACA ACCCAGCGCTTTCGGAACA; e+f, GGGAGGGCCCTCT CGGTAGAACACCATGACGGACTATCCCACGAACGC TCACGGGGCCCTCC) with T7 promoter sequences at the 5′ end were obtained from Sigma Aldrich (St Louis, MO). The SL III e+f (A297G) oligo had the same sequence as the SL III e+f oligo except for replacement of a T residue by a C at position 37. These oligonucleotides were annealed to T7 RNA polymerase promoter primers and transcribed in vitro using T7 RNA polymerase as described earlier (15). Radioactively labeled RNAs were transcribed using the same templates and [α-32P]UTP.
In vitro translation
In vitro translation was carried out using 1 µg of template RNA in 17 µl of micrococcal nuclease-treated rabbit reticulocyte lysate (RRL) medium (Promega) and either 0.5 µl each of amino acid mixtures minus methionine and minus cysteine or 20 µCi of [35S]methionine (Perkin Elmer). The reaction mixtures were preincubated with in vitro transcribed small RNAs as indicated in Results. After adding template RNA, the reaction mixtures were incubated at 30°C for 90 min and the products were analyzed either using a Dual Luciferase assay system (Promega) in a TD 20/20 Luminometer (Turner Designs, Sunnyvale, CA) or resolved on SDS–12.5% polyacrylamide gels followed by phosphorimaging (Fuji Imaging, Japan).
Purification of S5 ribosomal protein
Escherichia coli JM109 cells were transformed with the plasmid pQE-S5 (a gift from Dr S. Fukushi, Biomedical Laboratories, Japan) expressing the poly(His)-tagged S5 protein. Protein expression in bacterial culture was induced by 0.8 mM IPTG and purified using Ni2+–nitrilotriacetic acid–agarose (Qiagen, Hilden, Germany) under non-denaturing conditions and eluted with 100 mM imidazole.
UV-induced crosslinking of proteins with RNA
The in vitro transcribed 32P-labeled RNAs were incubated with HeLa S10 extract or purified protein in 2× RNA binding buffer and UV-crosslinked as described earlier (19). Unbound RNAs were digested by treatment with 30 µg of RNase A at 37°C for 30 min. The protein–nucleotidyl complexes were electrophoresed on SDS–10% polyacrylamide gels followed by autoradiography.
DNA and RNA transfection
Monolayers (60–70% confluent) of HeLa and Huh7 cells in 35 mm dishes were co-transfected with plasmid DNAs using Tfx 20 reagent (Promega) as indicated in Results. The cells were harvested 48 h after transfection and luciferase activity was assayed. Huh7 cells were co-transfected with in vitro transcribed RNAs using Tfx 20 reagent as indicated in Results. The cells were harvested 16 h after transfection and luciferase activity was assayed. DNA and RNA quantities were normalized using pGEM 3Z DNA (Promega) or an in vitro transcribed RNA corresponding to its polylinker sequence. Huh7 cells were transfected with the BB7 HCV subgenomic replicon RNA followed by retransfection with SL III e+f RNA after 16 h. The cells were harvested 24 h after transfection with SL III e+f and the total RNA was isolated using Tri Reagent (Sigma Aldrich).
RNase protection assay
Equal quantities of total RNA from Huh7 cells co-transfected with the HCV replicon RNA and SL III e+f RNA were alcohol precipitated and resuspended in 30 µl of hybridization buffer (80% deionized formamide, 40 mM PIPES, pH 6.4, 400 mM NaCl and 1 mM EDTA) containing 105 c.p.m. HCV 5′-UTR positive sense RNA probe and incubated at 95°C for 10 min, followed by 55°C for 18 h. An aliquot of 300 µl of RNase digestion buffer (300 mM NaCl, 10 mM Tris–HCl, pH 7.4, 5 mM EDTA, 10 U RNase T1 and 40 µg/ml RNase A) was added to each reaction with incubation at 30°C for 1 h, followed by 20 µl of 10% SDS and 10 µl of 10 mg/ml proteinase K and incubation at 37°C for 30 min. The RNA was alcohol precipitated, resuspended in formamide loading buffer and resolved by 10% urea–PAGE.
Ribosomal assembly assay
32P-labeled HCV 5′-UTR RNA (∼105 c.p.m.) was added to 25 µl of translation reaction containing 17.5 µl RRL, in the presence or absence of a 200-fold excess of SL III e+f RNA. An aliquot of 2 mM 5′-Guanylyl imidophosphate (GMP-PNP) (Sigma Aldrich) was added to the reaction as indicated in the results. The reactions were incubated at 30°C for 15 min, diluted to 150 µl with gradient buffer (20 mM Tris–HCl, pH 7.5, 100 mM KCl, 3 mM MgCl2, 1 mM DTT) and overlaid on a 5–30% linear sucrose gradient. The ribosomal complexes were sedimented by ultracentrifugation for 3 h at 4°C and 30000 r.p.m. Fractions (500 µl) were collected from the bottom of the column and the radioactivity was measured in a liquid scintillation counter.
Statistical analysis
The means ± SD of five independent in vitro translation experiments are represented. Data from the co-transfection experiments are expressed as means ± SD of three independent replicates. The significance of differences between means was tested using Student’s t-test. IC50 values for inhibitory RNAs were determined by non-linear regression analysis of the inhibition curves using SigmaPlot (SPSS Inc., Chicago, IL).
RESULTS
Specific inhibition of HCV IRES-mediated translation by RNA corresponding to SL III of the HCV 5′-UTR
RNAs corresponding to the three major domains of the HCV 5′-UTR: SL II, SL III (excluding SL III e+f) and SL IV (including SL III e+f) (Fig. 1A) were transcribed in vitro. 100- and 200-fold molar excesses of these RNAs were exogenously added to in vitro translation reactions of the Rluc–HCV–Fluc bicistronic RNA and the reporter gene products were radiolabeled (Fig. 1B). The three RNA molecules differentially inhibited HCV IRES-mediated translation of Fluc and cap-dependent translation of Rluc in a dose-dependent manner. SL II moderately inhibited HCV IRES-mediated and cap-dependent translation whereas SL IV was found to strongly inhibit both. On the other hand, SL III RNA was found to strongly inhibit HCV IRES-mediated translation with a marginal effect on cap-dependent translation (Fig. 1B). Similar results were obtained when the luciferase activity from non-radiolabeled reporter gene products was assayed (Fig. 1C). SL III RNA caused a maximum 80% reduction in Fluc activity whereas there was 73 and 57% inhibition at corresponding doses of SL IV and SL II, respectively (Fig. 1C). IC50 values for SL III, IV and II were determined to be, respectively, 35-, 60- and 152-fold excesses of the template RNA. There was no significant inhibition of Rluc activity by either SL III or SL II, whereas SL IV caused 68% inhibition at 200-fold excess concentration (Fig. 1D). These observations indicate both an efficient and selective inhibition of HCV IRES-mediated translation by SL III which was significantly higher than that caused by SL II or SL IV RNA (P < 0.01). SL IV caused a strong inhibition of both HCV IRES-mediated translation and cap-dependent translation whereas the effect of SL II on either was not pronounced.
Figure 1.

Specific dose-dependent inhibition of HCV IRES-mediated translation in vitro by HCV SL III RNA. (A) Proposed secondary structure of the HCV IRES RNA spanning nucleotides 40–372 of the 5′-UTR of the viral RNA (adapted from 41). The domains that were PCR amplified and cloned to generate small RNAs are delineated. (B) 100- and 200-fold molar excesses of in vitro transcribed SL II, III and IV RNAs were added to in vitro translation reactions of HCV bicistronic RNA. An aliquot of 5 µl of the translation reactions was resolved by SDS–12.5% PAGE and exposed for phosphoimaging. The Fluc and Rluc protein products are indicated by arrows. (C) The percent Fluc activity, representing the efficiency of HCV IRES-mediated translation from a HCV bicistronic template, in the presence of six increasing concentrations of SL II, III and IV RNAs was plotted. The Fluc activity at each concentration is represented as a percentage of the control reaction (expressed as 100%). The data were fitted to a non-linear regression curve to determine IC50 values. (D) The percent Rluc activity, representing the efficiency of cap-dependent translation from the same set of experiments, was plotted. The Rluc activity at each concentration is represented as a percentage of the control reaction. The translation efficiency was not reduced to below 50% by either SL III or SL II.
SL III RNA binds to a majority of cellular proteins which interact with the HCV 5′-UTR
The inhibitory effect of SL III RNA was hypothesized to be due to its ability to bind to cellular proteins that interact with the HCV IRES. We investigated the protein binding profiles of SL II, III and IV RNAs with HeLa cytoplasmic extract as HeLa cells support efficient HCV IRES activity and compared it with that of the full-length HCV 5′-UTR. SL II, III and IV were found to have distinct protein-binding profiles and SL III was found to bind to a majority of cellular proteins which interacted with the HCV 5′-UTR (Fig. 2A). Competition experiments using excess unlabeled RNAs demonstrated that SL III and IV RNAs competed out the interaction of specific proteins with the HCV 5′-UTR (data not shown). These observations showed that SL III RNA could bind to the majority of proteins that interacted with the HCV IRES and could therefore antagonize their interaction with the full-length 5′-UTR.
Figure 2.

Interaction of HCV 5′-UTR SL RNAs with HeLa cytoplasmic proteins and effect of SL III on HCV IRES-mediated translation in vivo. (A) 32P-labeled RNAs corresponding to the full-length HCV 5′-UTR and SL II, III and IV domains were UV-crosslinked to HeLa S10 cytoplasmic extract, digested with RNase A and resolved by SDS–10% PAGE. M represents 14C-labeled protein molecular weight markers. (B) Three-way co-transfections were performed in HeLa cells using pRL-CMV, pCD-HCV5′-UTR-Fluc and two concentrations of pCD-SL III and pCD-SL II DNAs. DNA quantity per dish was normalized by transfecting pGEM-3Z DNA. The black bars represent Fluc activity (HCV IRES-mediated translation) whereas the gray bars represent Rluc activity (cap-dependent translation). (C) The same experiment was repeated in the Huh7 cell line. Combined data from three independent experiments in each cell line are shown. Luciferase activity in control reactions is expressed as 100%. Values which significantly differ from controls (P < 0.01) are indicated by asterisks.
Specific inhibition of HCV IRES-mediated translation in vivo by SL III RNA
To investigate the inhibitory activity of the SL III RNA in vivo, three-way co-transfections were performed in HeLa cells using pRL-CMV, pCD-HCV-Fluc and two different concentrations of pCD-SL III and pCD-SL II DNAs. SL II, which had not significantly inhibited IRES-mediated or cap-dependent translation in vitro, was utilized as the negative control. SL III significantly reduced (P < 0.01) HCV IRES activity from the control at both concentrations (74 and 85% inhibition, respectively) whereas there was no significant effect on cap-dependent translation (Fig. 2B). SL II did not significantly inhibit either cap-dependent or IRES-mediated translation compared to the control. The co-transfections were repeated in Huh7 cells, a human hepatocellular carcinoma cell line supporting efficient HCV replication (20). There was a significant reduction (P < 0.01) in HCV IRES-mediated translation from the control by SL III although the extent of inhibition was less (37 and 68% inhibition at the two concentrations) than that observed in HeLa cells using similar quantities of transfected DNA (Fig. 2C). These data demonstrate that SL III RNA could selectively inhibit HCV IRES-mediated translation in both non-liver-derived (HeLa) and liver-derived (Huh7) cells.
Inhibition of HCV IRES-mediated translation by an RNA corresponding to SL III e+f
As we observed the maximum translation inhibitory effect with RNA molecules corresponding to domain III of the HCV IRES, we tried to generate smaller RNAs corresponding to its specific subdomains that would retain the ability to inhibit HCV IRES-mediated translation. For this purpose, small RNAs corresponding to the SL III a+c, b, d and e+f subdomains (Fig. 3A) were generated by oligonucleotide-driven transcription. Increasing concentrations of these RNAs were exogenously added to in vitro translation reactions of the HCV bicistronic RNA. Interestingly, SL III e+f RNA caused a very strong inhibition (89% reduction from control) of HCV IRES-mediated translation (Fig. 3B) with no significant effect on cap-dependent translation (Fig. 3C). SL III b caused 57% inhibition of HCV IRES-mediated translation but this was accompanied by a 53% decrease in cap-dependent translation (Fig. 3B and C). SL III a+c and III d RNAs showed minor reductions in the efficiency of HCV IRES-mediated translation. The IC50 of SL III e+f RNA was calculated to be a 15-fold molar excess of template RNA. These observations demonstrated that the small RNA corresponding to the SL III e+f subdomain could strongly inhibit HCV IRES-mediated translation in vitro without significantly inhibiting cap-dependent translation.
Figure 3.

Specific inhibition of HCV IRES-mediated translation in vitro by HCV 5′-UTR SL III e+f RNA. (A) Proposed secondary structure of HCV IRES (internal ribosome entry site) domain III (nucleotides 121–315), delineating the SL structures which were generated by oligonucleotide- driven transcription. (B) The percent Fluc activity, representing the efficiency of HCV IRES-mediated translation from a HCV bicistronic template, in the presence of five increasing concentrations of SL III a+c, b, d and e+f RNAs was plotted. Luciferase activity in control reactions is expressed as 100%. The data for SL III e+f was fitted to a non-linear regression curve to determine the IC50 value. (C) The percent Rluc activity representing the efficiency of cap-dependent translation from the same set of experiments was plotted. The reporter gene activity at each concentration is represented as a percentage of the control reaction.
Effect of SL III e+f RNA on HCV IRES-mediated translation and replication in vivo
As SL III e+f RNA showed a strong inhibition of HCV IRES-mediated translation in vitro, we next investigated its effect on HCV translation in vivo. Huh7 cells were co-transfected with in vitro transcribed HCV bicistronic RNA together with two concentrations of SL III e+f RNA. SL III d, which had not shown a significant effect, was used as the negative control. SL III e+f RNA significantly inhibited (P < 0.01) HCV IRES-mediated translation in a dose-dependent manner (45 and 85% inhibition at the two concentrations) with no significant inhibition of cap-dependent translation (Fig. 4A). Addition of SL III d RNA did not cause any inhibition of either IRES-mediated or cap-dependent translation (Fig. 4A).
Figure 4.

Effect of SL III e+f RNA on HCV IRES-mediated translation and replication in vivo. (A) Huh7 cells were co-transfected with 6 µg of in vitro transcribed capped HCV bicistronic RNA (schematically represented) and two concentrations (6 and 12 µg) of either SL III e+f RNA or SL III d RNA. The RNA quantities in each dish were normalized by adding appropriate amounts of an in vitro transcribed RNA corresponding to the polylinker sequence of the pGEM 3Z plasmid. The black bars represent Fluc activity (HCV IRES-mediated translation) whereas the gray bars represent Rluc activity (cap-dependent translation). Luciferase activity in control reactions is expressed as 100%. Values which significantly differ from controls (P < 0.01) are indicated by asterisks. (B) Huh7 cells were either mock transfected (lane 1) or transfected with 8 µg of BB7 HCV replicon RNA (schematically represented). Twelve hours post-transfection the cells were retransfected with two concentrations (6 and 12 µg) of SL III e+f RNA (lanes 3 and 4). The cells were harvested 24 h after retransfection, total RNA was isolated and a RNase protection assay was performed using a [32P]UTP-labeled HCV 5′-UTR positive sense probe. The RNAs were resolved by 10% urea–PAGE. Lane M shows the migration of a radiolabeled 100 bp DNA ladder. (C) 100- and 200-fold molar excesses of in vitro transcribed SL III e+f RNA was added to in vitro translation reactions of Fluc-EMCV-GFP bicistronic RNA in RRL. An aliquot of 10 µl of the translation reactions was resolved by SDS–12.5% PAGE and exposed for phosphorimaging. The 35S-labeled Fluc and GFP protein products are indicated by arrows.
As SL III e+f RNA inhibited HCV IRES-mediated translation in Huh7 cells, we also tried to determine its effect, if any, on HCV replication from a HCV subgenomic replicon. Therefore, the BB7 HCV replicon (21) RNA was transfected into Huh7 cells followed by transfection with two concentrations of SL III e+f RNA corresponding to the amounts used for inhibition of translation from the bicistronic RNA. In a control experiment in which the SL III RNA was co-transfected with the replicon RNA into Huh7 cells, a RNase protection assay using a HCV 5′-UTR antisense probe demonstrated the presence of both the input replicon RNA and the SL III RNA 48 h after transfection (data not shown). A RNase protection assay using a HCV 5′-UTR positive sense RNA probe to assay for RNA replication demonstrated only a marginal reduction in HCV replication by SL III e+f RNA at the concentrations of RNA used (Fig. 4B). This suggested that the inhibitory activity of SL III e+f RNA was specific to HCV IRES-mediated translation rather than replication of the HCV RNA. This also suggested that there was possibly no effect of SL III e+f RNA on EMCV IRES activity, as protein synthesis from the HCV replicon was under the translational control of the EMCV IRES. In order to investigate this possibility, 100- and 200-fold molar excesses of SL III e+f RNA were added to in vitro translation reactions of an EMCV bicistronic RNA, but no effect on EMCV-IRES-mediated translation of the GFP reporter gene was observed (Fig. 4C). This also suggested that the SL III e+f RNA specifically inhibited HCV IRES-mediated and not the picornaviral IRES-mediated translation.
Binding of cellular proteins to the small RNAs corresponding to SL III subdomains: specific interaction of the S5 ribosomal protein with SL III e+f RNA
As SL III e+f RNA was found to strongly inhibit HCV IRES-mediated translation in vitro and in vivo, we investigated the protein-binding profile of the RNA to correlate it with the translation inhibitory activity. The SL III subdomain RNAs demonstrated differential binding to specific proteins that interacted with the HCV domain III RNA (Fig. 5A). A 25 kDa protein was found to interact strongly with SL III e+f RNA but did not interact with the other RNAs. This protein may correspond to the S5 ribosomal protein, as a single mutation in SL III e (A297G) of the HCV 5′-UTR has been shown to abrogate binding of this protein (22). In order to investigate this possibility, bacterially expressed S5 ribosomal protein was used in a UV-crosslinking assay. SL III e+f RNA strongly interacted with the purified protein whereas SL III d, which had not shown any interaction with p25 from HeLa cells, failed to do so (Fig. 5B). The full-length HCV 5′-UTR also interacted with the S5 protein, but to a lesser extent than SL III e+f (Fig. 5B). Therefore, the small RNAs corresponding to the subdomains of domain III could bind to a number of cellular proteins which interacted with the HCV IRES and SL III e+f uniquely interacted with the S5 ribosomal protein. An in vitro transcribed mutant SL III e+f RNA harboring the A297G mutation in SL III e showed a drastic reduction in binding to the p25 protein from HeLa cytoplasmic extract (Fig. 6A) and also purified S5 protein (Fig. 6B). 100- and 200-fold molar excesses of this mutant SL III e+f RNA also failed to inhibit HCV IRES-mediated translation in vitro (Fig. 6C). These observations suggested that the SL III e+f RNA inhibited HCV IRES-mediated translation specifically by binding to S5 ribosomal protein.
Figure 5.

Binding of domain III stem–loop RNAs to HeLa cellular proteins and interaction of SL III e+f RNA with the S5 ribosomal protein. (A) 32P-labeled RNAs corresponding to the HCV SL III a+c, b, d and e+f subdomains were UV-crosslinked to HeLa S10 cytoplasmic extract, digested with RNase A and resolved by SDS–10% PAGE. The position of p25 bound to SL III e+f RNA is indicated by an arrow. (B) Purified recombinant S5 ribosomal protein was UV-crosslinked to HCV SL III e+f, SL III d and full-length HCV 5′-UTR RNA. The nucleoprotein complexes were resolved by SDS–12% PAGE and the position of S5 protein (p25) is indicated.
Figure 6.
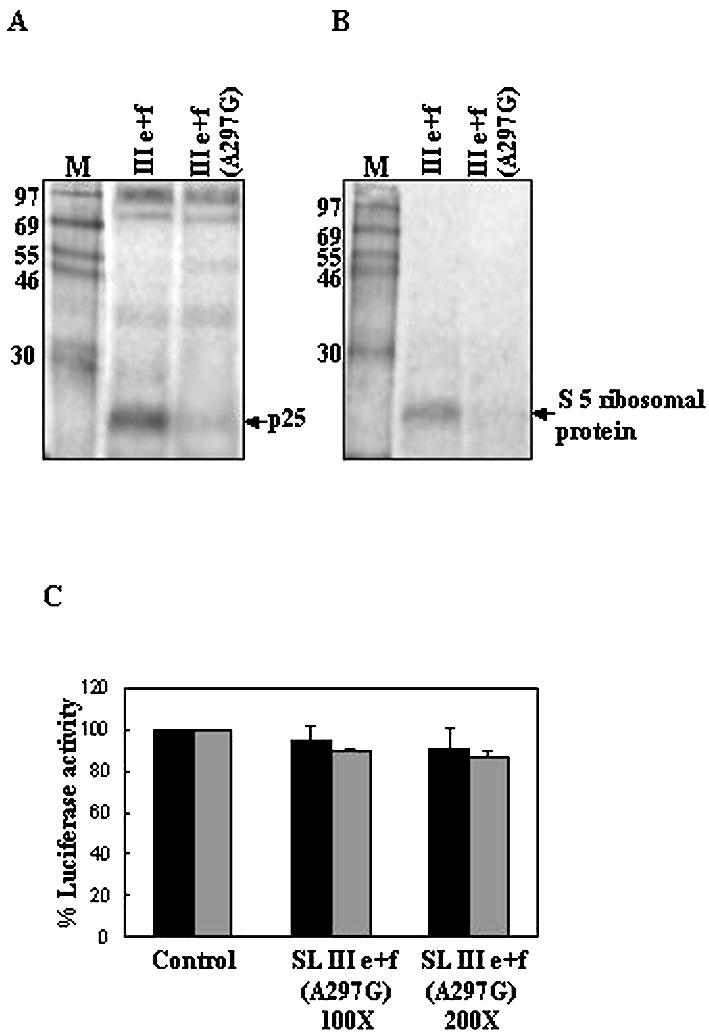
SL III e+f (A297G) RNA fails to bind to S5 ribosomal protein and does not inhibit HCV IRES-mediated translation. (A) 32P-labeled RNAs corresponding to SL III e+f and SL III e+f (A297G) were UV-crosslinked to HeLa S10 extract and digested with RNase A. The nucleoprotein complexes were resolved by SDS–15% PAGE and the position of p25 is indicated. (B) The same RNAs were UV-crosslinked to purified S5 ribosomal protein and the nucleoprotein complexes were resolved by SDS–15% PAGE. (C) 100- and 200-fold molar excesses of in vitro transcribed SL III e+f (A297G) RNA was added to in vitro translation reactions of HCV bicistronic RNA and luciferase activity was assayed. The black bars represent Fluc activity (HCV IRES-mediated translation) whereas the gray bars represent Rluc activity (cap-dependent translation). Luciferase activity in control reactions is expressed as 100%. Combined data from three independent experiments are represented.
SL III e+f RNA prevented the assembly of ribosomal complexes on the HCV IRES
Binding of the 25 kDa S5 ribosomal protein has been suggested to be crucial for efficient translation mediated by the HCV IRES (23). As SL III e+f RNA was found to interact strongly with the S5 protein, we investigated its effect on ribosome recruitment by the HCV IRES. For this purpose, ribosomal assembly reactions containing radiolabeled HCV 5′-UTR were incubated with a 200-fold molar excess of SL III e+f RNA and analyzed by sucrose density gradient ultracentrifugation. In the absence of SL III e+f RNA, HCV IRES showed the formation of both 48S and 80S ribosomal complexes (Fig. 7A). However, in the presence of SL III e+f RNA, formation of both of these complexes was significantly reduced (Fig. 7A), suggesting that SL III e+f prevented the assembly of ribosomal complexes on the HCV IRES. To further elucidate the role of SL III e+f RNA, the ribosomal assembly reactions were incubated with GMP-PNP, which inhibits translation initiation at the 48S stage by preventing the release of eIF2. Addition of GMP-PNP abolished the 80S peak in the control reaction, demonstrating that only the 48S complex was being assembled (Fig. 7B). In the presence of SL III e+f the 48S complex formation was also abrogated, suggesting that SL III e+f prevented binding of the 40S ribosomal subunit to the HCV IRES (Fig. 7B). This supported the earlier observation that SL III e+f interacted with the S5 ribosomal protein, a component of the 40S subunit. Taken together, these observations suggest that the SL III e+f RNA inhibited HCV IRES-mediated translation by interacting with a component of the 40S ribosomal subunit and thereby prevented ribosomal assembly on the HCV IRES.
Figure 7.

SL III e+f prevents 40s ribosomal subunit recruitment by the HCV IRES. (A) Sucrose gradient sedimentation profiles of [32P]UTP-labeled HCV 5′-UTR RNA incubated in RRL in the absence and presence of a 200-fold excess of unlabeled SL III e+f RNA. (B) Sedimentation profile of radiolabeled HCV 5′-UTR RNA in the presence of 2 mM GMP-PNP in the presence or absence of a 200-fold excess of unlabeled SL III e+f RNA. The filled circles represent the control reaction profile and the open circles show the profile in the presence of SL III e+f. Both profiles show the counts per minute as a percentage of the total counts added to the reaction (∼105 c.p.m.) against the fraction number of the gradient. The fractions were collected from the bottom upwards. The 80S and 48S ribosomal peaks are indicated.
DISCUSSION
The process of IRES-mediated translation is an attractive target for antiviral drug design (24). Selective inhibition of the HCV IRES-mediated mechanism by the SL III e+f RNA of the HCV 5′-UTR has a potential to be used as a therapeutic strategy with many associated advantages. Firstly, as the interactions between host cellular proteins and a highly conserved region of the viral RNA is targeted, the chance of generation of viral escape mutants is very low. Approaches like siRNA treatment have demonstrated the rapid emergence of escape mutants in poliovirus (25). Although the rate of HCV replication is not as high as that of poliovirus (3), any sequence-specific antiviral molecule would exert a selection pressure for the generation of escape variants, unlike a strategy targeting host protein–viral RNA interactions. Moreover, the activity of the HCV IRES being highly structure dependent, only mutational events that can alter the IRES structure would allow the virus to circumvent inhibition by this approach. Secondly, the RNA molecule being a part of the viral genome, if administered prophylactically to patients harboring the viral RNA it would not be expected to give rise to non-specific immune responses as in the case of antisense RNAs (26). Thirdly, as binding of the cellular proteins is probably dependent on the RNA structure, stabler derivatives and small molecule structural analogs of the RNA could be utilized. We found that the amounts of SL III e+f RNA required to inhibit HCV IRES-mediated translation was more (IC50 ≈ 0.6 µM) than the amount of antisense RNA targeted against SL III d (IC50 < 10 nM) required for a similar inhibition (27). However, the absolute values of RNA required to inhibit translation from an HCV IRES-containing construct by the two approaches cannot be compared as the amount of competitor SL RNA required to inhibit translation is dependent on the concentration of template RNA used as well as the levels of trans-acting factors present that interact with the IRES RNA.
The major domains of the HCV IRES that have been shown to interact with cellular factors and the 40S ribosomal subunit are SL III and SL IV (28,29). In our study, among the major domains of the HCV IRES, SL III RNA strongly inhibited HCV IRES-mediated translation and could efficiently bind to most proteins that interacted with the HCV 5′-UTR. The SL III e+f RNA, which inhibited HCV IRES-mediated translation even more efficiently, interacted with ribosomal protein S5 (30). Binding of this protein to the HCV IRES has been suggested to be crucial for the efficient translation of HCV RNA (23). A single mutation in SL III e (A297G), which abrogated binding of this protein, has been found to abolish HCV IRES activity (22,31). In our study, a SL III e+f RNA containing this mutation failed to bind the S5 protein and was also unable to inhibit HCV IRES-mediated translation. However, direct binding of the 40S ribosomal subunit to the HCV IRES involves SL III d, SL III e+f, the pseudoknot region and SL IV (32). Our observations suggest that although SL III e+f might not sequester the 40S ribosomal subunit independently, it could prevent its interaction with the HCV IRES by interacting with one of its component proteins. The 40S subunit S5 protein has not been found to interact with eukaryotic mRNAs either in the 48S preinitiation complex or in polysomes (33). This possibly explains why SL III e+f RNA did not inhibit cap-dependent translation. However, the prokaryotic homolog of S5 protein, S7, has been shown to be present near the E site of the bacterial 30S ribosome subunit and interacts with tRNA and mRNA (30,34). Therefore, the HCV IRES, which has been suggested to have a prokaryotic-like mode of interaction with the 40S ribosomal subunit (32), may interact with S5 protein, although eukaryotic mRNAs and picornaviral IRESs (35) have not been reported to interact with this protein. The SL III RNA lacking SL III e+f also failed to significantly inhibit cap-dependent translation, probably due to its inability to bind efficiently to the 40S ribosomal subunit. This suggests that only RNA that can constitute the intact ribosome-binding site of the HCV IRES is able to recruit 40S subunits. Our strategy of using individual SLs from this region prevented the sequestration of ribosomes and the consequent inhibition of cap-dependent translation. On the other hand, SL IV RNA inhibited both IRES-mediated and cap-dependent translation, probably because it contained SL III e+f and the pseudoknot region together with domain IV, which generated the core ribosome-binding site (36). SL III b inhibited both IRES-mediated and cap-dependent translation, probably because of its strong interaction with eIF3 and PTB, which interacts with this region of the HCV IRES (37,38). However, the larger SL III RNA did not inhibit cap-dependent translation probably because its different secondary structure conferred a lower affinity for binding to these proteins. SL II RNA did not inhibit HCV translation by interacting with the 40S subunit, which is consistent with earlier reports that a domain II-deleted IRES construct could bind the 40S subunit with nearly wild-type affinity (37).
The inhibitory effect of the small RNAs was not by RNAi as replication of HCV RNA was not inhibited by SL III e+f RNA. RNAi using siRNA against the HCV 5′-UTR has been demonstrated using the HCV replicon system (39). However, studying the inhibition of translation by the approach used in our study might not be feasible using the replicon system as the HCV replicons reported in the literature are under translational control of the EMCV IRES and the SL III e+f RNA did not inhibit EMCV IRES activity. Therefore, other models, like mice containing chimeric mouse/human livers supporting HCV infection and replication (40), may provide a suitable system to test the efficacy of this strategy. With recent advances in targeted gene delivery, this approach of selectively targeting HCV translation, in conjunction with siRNAs targeting HCV replication, may provide a basis for developing an effective anti-HCV therapy.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr K. Tsukiyama-Kohara and Dr A. Nomoto for the pCV clone, Dr Richard Elliot for the pRL-HCV1b plasmid, Dr Charles M. Rice and Apath Inc. for the BB7 HCV replicon and Dr S. Fukushi for the pQE-S5 plasmid. We gratefully acknowledge our laboratory members for their help and discussion. This work is supported by research grants to S.D. from the Life Sciences Research Board, DRDO and Department of Science and Technology, Government of India.
REFERENCES
- 1.Jenny-Avital E.R. (1998) Hepatitis C. Curr. Opin. Infect. Dis., 11, 293–299. [DOI] [PubMed] [Google Scholar]
- 2.Lindsay K.L. (1997) Therapy of hepatitis C: overview. Hepatology, 26, 715–755. [DOI] [PubMed] [Google Scholar]
- 3.Randall G., Grakoui,A. and Rice,C.M. (2003) Clearance of replicating hepatitis C virus replicon RNAs in cell culture by small interfering RNAs. Proc. Natl Acad. Sci. USA, 100, 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsukiyama-Kohara K., Iizuka,N., Kohara,M. and Nomoto A. (1992) Internal ribosome entry site within hepatitis C virus RNA. J. Virol., 66, 1476–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pudi R., Abhiman,S., Srinivasan,N. and Das,S. (2003) Hepatitis C virus internal ribosome entry site-mediated translation is stimulated by specific interaction of independent regions of human La autoantigen. J. Biol. Chem., 278, 12231–12240. [DOI] [PubMed] [Google Scholar]
- 6.Ali N. and Siddiqui,A. (1995) Interaction of polypyrimidine tract-binding protein with the 5′ noncoding region of the hepatitis C virus RNA genome and its functional requirement in internal initiation of translation. J. Virol., 69, 6367–6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sizova D.V., Kolupaeva,V.G., Pestova,T.V., Shatsky,I.N. and Hellen,C.U.T. (1998) Specific interaction of eukaryotic translation initiation factor 3 with the 5′ nontranslated regions of hepatitis C virus and classical swine fever virus RNAs. J. Virol., 72, 4775–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kruger M., Beger,C., Li,Q.X., Welch,P.J., Tritz,R., Leavitt,M., Barber,J.R. and Wong-Staal,F. (2000) Identification of eIF2Bγ and eIF2γ as cofactors of hepatitis C virus internal ribosome entry site-mediated translation using a functional genomics approach. Proc. Natl Acad. Sci. USA, 97, 8566–8571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kieft J.S., Zhou,K., Jubin,R., Murray,M.G., Lau,J.Y.N and Doudna,J.A. (1999) The hepatitis C virus internal ribosome entry site adopts an ion-dependent tertiary fold. J. Mol. Biol., 292, 513–529. [DOI] [PubMed] [Google Scholar]
- 10.Lott W.B., Takyar,S.S., Tuppen,J., Crawford,D.H.G., Harrison,M., Sloots,T.P. and Gowans,E.J. (2001) Vitamin B12 and hepatitis C: molecular biology and human pathology. Proc. Natl Acad. Sci. USA, 98, 4916–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu C.H. and Wu,G.Y. (1998) Targeted inhibition of hepatitis C virus-directed gene expression in human hepatoma cell lines. Gastroenterology, 114, 1304–1312. [DOI] [PubMed] [Google Scholar]
- 12.Welch P.J., Tritz,R., Yei,S., Leavitt,M., Yu,M. and Barber,J. (1996) A potential therapeutic application of hairpin ribozymes: in vitro and in vivo studies of gene therapy for hepatitis C virus infection. Gene Ther., 3, 994–1001. [PubMed] [Google Scholar]
- 13.Lee T.C., Sullenger,B.A., Gallardo,H.F., Ungers,G.E. and Gilboa,E. (1992) Overexpression of RRE-derived sequences inhibits HIV-1 replication in CEM cells. New Biol., 4, 66–74. [PubMed] [Google Scholar]
- 14.Sullenger B.A., Gallardo,H.F., Ungers,G.E. and Gilboa,E. (1990) Overexpression of TAR sequences renders cells resistant to Human Immunodeficiency Virus replication. Cell, 63, 601–608. [DOI] [PubMed] [Google Scholar]
- 15.Das S., Kenan,D.J., Bocskai,D., Keene,J.D. and Dasgupta,A. (1996) Sequences within a small yeast RNA required for inhibition of internal initiation of translation: interaction with La and other cellular proteins influences its inhibitory activity. J. Virol., 70, 1624–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Das S., Ott,M., Yamane,A., Tsai,W., Gromeier,M., Lahser,F., Gupta,S. and Dasgupta,A. (1998) A small yeast RNA blocks hepatitis C virus internal ribosome entry site (HCV IRES)-mediated translation and inhibits replication of a chimeric poliovirus under translational control of the HCV IRES element. J. Virol., 72, 5638–5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ali N., Pruijn,G.J.M, Kenan,D.J., Keene,J.D. and Siddiqui,A. (2000) Human La antigen is required for the hepatitis C virus internal ribosome entry site-mediated translation. J. Biol. Chem., 275, 27531–27540. [DOI] [PubMed] [Google Scholar]
- 18.Anwar A., Ali,N., Tanveer,R. and Siddiqui,A. (2000) Demonstration of functional requirement of polypyrimidine tract-binding protein by SELEX RNA during hepatitis C virus internal ribosome entry site-mediated translation initiation. J. Biol. Chem., 275, 34231–34235. [DOI] [PubMed] [Google Scholar]
- 19.Ray P.S. and Das,S. (2002) La autoantigen is required for the internal ribosome entry site-mediated translation of coxsackievirus B3 RNA. Nucleic Acids Res., 30, 4500–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lohmann V., Körner,F., Koch,J.O., Herian,U., Theilmann,L. and Bartenschlager,R. (1999) Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science, 285, 110–113. [DOI] [PubMed] [Google Scholar]
- 21.Blight K.J., Kolykhalov,A.A. and Rice,C.M. (2000) Efficient Initiation of HCV RNA replication in cell culture. Science, 290, 1972–1974. [DOI] [PubMed] [Google Scholar]
- 22.Odreman-Macchioli F., Baralle,F.E. and Buratti,E. (2001) Mutational analysis of the different bulge regions of hepatitis C virus Domain II and their influence on internal ribosome entry site translational ability. J. Biol. Chem., 276, 41648–41655. [DOI] [PubMed] [Google Scholar]
- 23.Fukushi S., Kurihara,C., Ishiyama,N., Hoshino,F.B., Oya,A. and Katayama,K. (1997) The sequence element of the internal ribosome entry site and a 25-kilodalton cellular protein contribute to efficient internal initiation of translation of hepatitis C virus RNA. J. Virol., 71, 1662–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gosert R. and Moradpour,D. (2002) A new twist to translation initiation of the hepatitis C virus. Hepatology, 35, 724–726. [DOI] [PubMed] [Google Scholar]
- 25.Gitlin L., Karelsky,S. and Andino,R. (2002) Short interfering RNA confers intracellular antiviral immunity in human cells. Nature, 418, 430–434. [DOI] [PubMed] [Google Scholar]
- 26.Dove A. (2002) Antisense and sensibility. Nat. Biotechnol., 20, 121–124. [DOI] [PubMed] [Google Scholar]
- 27.Tallet-Lopez B., Aldaz-Carroll,L., Chabas,S., Dausse,E., Staedel,C. and Toulme,J.J. (2003) Antisense oligonucleotides targeted to the domain IIId of the hepatitis C virus IRES compete with 40S ribosomal subunit binding and prevent in vitro translation. Nucleic Acids. Res., 31, 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolupaeva V.G., Pestova,T.V. and Hellen,C.U.T. (2000) An enzymatic footprinting analysis of the interaction of 40S ribosomal subunits with the internal ribosomal entry site of the hepatitis C virus. J. Virol., 74, 6242–6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spahn C.M.T., Kieft,J.S., Grassucci,R.A., Penczek,P.A., Zhou,K., Doudna,J.A. and Frank,J. (2001) Hepatitis C virus IRES RNA-induced change in the conformation of the 40S ribosomal subunit. Science, 291, 1959–1962. [DOI] [PubMed] [Google Scholar]
- 30.Fukushi S., Okada,M., Stahl,J., Kageyama,T., Hoshino,F.B. and Katayama,K. (2001) Ribosomal protein S5 interacts with the internal ribosome entry site of hepatitis C virus. J. Biol. Chem., 276, 20824–20826. [DOI] [PubMed] [Google Scholar]
- 31.Psaridi L., Georgopolou,U., Varaklioti,A. and Mavromara,P. (1999) Mutational analysis of a conserved tetraloop in the 5′ untranslated region of hepatitis C virus identifies a novel RNA element essential for the internal ribosome entry site function. FEBS Lett., 453, 49–53. [DOI] [PubMed] [Google Scholar]
- 32.Pestova T., Shatsky,I.N., Fletcher,S.P., Jackson,R.J. and Hellen,C.U.T. (1992) A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev., 6, 1643–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westermann P. and Nygard,O. (1984) Cross-linking of mRNA to initiation factor eIF-3, 24 kDa cap binding protein and ribosomal proteins S1, S3/3a, S6 and S11 within the 48S pre-initiation complex. Nucleic Acids Res., 12, 8887–8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brodersen D.E., Carter,A.P., Clemons,W.M., Morgan-Warren,R.J., Murphy,F.V., Ogle,J.M., Tarry,M.J., Wimberley,B.T. and Ramakrishnan,V. (2001) Atomic structures of the 30S subunit and its complexes with ligands and antibiotics. Cold Spring Harbor Symp. Quant. Biol., LXVI, 17–32. [DOI] [PubMed] [Google Scholar]
- 35.Fukushi S., Okada,M., Kageyama,T., Hoshino,F.B. and Katayama,K. (1999) Specific interaction of a 25-kilodalton cellular protein, a 40S ribosomal subunit protein, with the internal ribosome entry site of hepatitis C virus genome. Virus Genes, 19, 153–161. [DOI] [PubMed] [Google Scholar]
- 36.Lytle J.R., Wu,L. and Robertson,H.D. (2001) The ribosome binding site of hepatitis C virus mRNA. J. Virol., 75, 7629–7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kieft J.S., Zhou,K., Jubin,R. and Doudna,J.A. (2001) Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA, 7, 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beales L.P., Rowlands,D.J. and Holzenburg,A. (2001) The internal ribosome entry site (IRES) of hepatitis C virus visualized by electron microscopy. RNA, 7, 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seo M.Y., Abrignani,S., Houghton,M. and Han,J.H. (2003) Small interfering RNA-mediated inhibition of hepatitis C virus replication in the human hepatoma cell line Huh-7. J. Virol., 77, 810–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mercer D.F., Schiller,D.E., Elliott,J.F., Douglas,D.N., Hao,C., Rinfret,A., Addison,W.R., Fischer,K.P., Churchill,T.A., Lakey,J.R., Tyrrell,D.L. and Kneteman,K.M. (2001) Hepatitis C virus replication in mice with chimeric human livers. Nat. Med., 7, 927–933. [DOI] [PubMed] [Google Scholar]
- 41.Brown E.A., Zhang,H., Ping,L.H. and Lemon,S.M. (1992) Secondary structure of the 5′ nontranslated regions of hepatitis C virus and pestivirus genomic RNAs. Nucleic Acids Res., 20, 5041–5045. [DOI] [PMC free article] [PubMed] [Google Scholar]