

ABSTRACT
Bacteria living on the aerial parts of plants (the phyllosphere) are globally abundant and ecologically significant communities and can have significant effects on their plant hosts. Despite their importance, little is known about the ecological processes that drive phyllosphere dynamics. Here, we describe the development of phyllosphere bacterial communities over time on the model plant Arabidopsis thaliana in a controlled greenhouse environment. We used a large number of replicate plants to identify repeatable dynamics in phyllosphere community assembly and reconstructed assembly history by measuring the composition of the airborne community immigrating to plant leaves. We used more than 260,000 sequences from the v5v6 hypervariable region of the 16S rRNA gene to characterize bacterial community structure on 32 plant and 21 air samples over 73 days. We observed strong, reproducible successional dynamics: phyllosphere communities initially mirrored airborne communities and subsequently converged to a distinct community composition. While the presence or absence of particular taxa in the phyllosphere was conserved across replicates, suggesting strong selection for community composition, the relative abundance of these taxa was highly variable and related to the spatial association of individual plants. Our results suggest that stochastic events in early colonization, coupled with dispersal limitation, generated alternate trajectories of bacterial community assembly within the context of deterministic selection for community membership.
IMPORTANCE
Commensal bacteria associated with plants help protect their hosts against infection and promote growth. Bacteria associated with plant leaves (the “phyllosphere”) are highly abundant and diverse communities, but we have very limited information about their ecology. Here, we describe the formation of phyllosphere communities on the plant model organism Arabidopsis thaliana. We grew a large number of plants in a greenhouse and measured bacterial diversity in the phyllosphere throughout the Arabidopsis life cycle. We also measured the diversity of airborne microbes landing on leaves. Our findings show that plants develop distinctive phyllosphere bacterial communities drawn from low-abundance air populations, suggesting the plant environment is favorable for particular organisms and not others. However, we also found that the relative abundances of bacteria in the phyllosphere are determined primarily by the physical proximity of individual plants. This suggests that a mixture of selective and random forces shapes phyllosphere communities.
INTRODUCTION
Plants in nature are colonized by a large, diverse array of nonpathogenic microbes (1). Surveys of both root- and leaf-associated microbes demonstrate that these communities are highly abundant (2), are species and ecotype specific (2–4), and can have significant phenotypic impacts on host plants (5). Here, we focus on microbial communities associated with the aerial parts of plants, primarily leaves (the “phyllosphere”) (2). Phyllosphere microbes experience high levels of UV exposure, water stress, large shifts in temperature (6), and heterogeneous nutrient availability (7). Despite this, the global population of phyllosphere microbes is estimated to be ~1026 cells (8), and cell densities in the phyllosphere are typically around 106 to 107 cells/cm2 (6). Phyllosphere bacteria can protect their hosts against pathogen infection (9, 10) and produce plant growth-promoting hormones (11). On a larger scale, phyllosphere communities affect biogeochemical cycling through the breakdown of plant-released methanol (12) and are a major source of bacteria to the atmosphere (13).
Despite the importance and abundance of phyllosphere microbes, little is known about the ecological processes regulating their dynamics. Culture-independent surveys of phyllosphere bacterial diversity in natural communities have identified seasonal variation (14, 15), geographic site (16, 17), and plant species (18) as regulators of community structure. In most of these studies, however, a significant portion of inter- and intraplant variability in phyllosphere composition remains unexplained by environmental structuring factors (17–19). For example, a study of Methylobacterium on natural populations of Medicago and Arabidopsis found that over half the variance in community structure was not explained by site or plant species (17). These results suggest that stochastic processes, including dispersal, ecological drift, and colonization history, may play a significant role in structuring phyllosphere communities.
The intriguing mix of deterministic (niche-based) and stochastic processes operating in phyllosphere communities makes them interesting model systems to address key questions in microbial community assembly (14, 20). The balance between deterministic and stochastic forces in microbial systems is a continuing subject of debate (21, 22). Most studies of bacterial assembly in natural communities find that both sets of processes are important (23–25) but are limited by low replication and the difficulty of controlling assembly in field environments. Laboratory model systems, in contrast, allow high levels of replication and experimental control. They have the potential to generate predictive explanations of community assembly (22).
In this study, we use bacterial communities associated with the phyllosphere of Arabidopsis thaliana to explore the repeatability of community assembly and the importance of deterministic and stochastic processes. The phyllosphere has several advantages as a model. Leaf surfaces are complex natural habitats with extensive microscale variation (26, 27) and diverse substrates for microbial growth (6). We can initialize large numbers of new communities on axenic plants and track community assembly through time using high-resolution sequencing methods. The emergence of new leaves allows us to observe the colonization of new habitats and assembly of the resulting microbial communities. Finally, phyllosphere communities are easily manipulated and replicated in the lab.
Arabidopsis thaliana, the premier model organism in plant biology, is ideal for experimental studies of phyllosphere assembly. The availability of inbred Arabidopsis lines reduces the influence of host-to-host genetic variability on phyllosphere structure. Arabidopsis has a short life cycle and can be grown easily in large quantities (28). Additionally, it is increasingly used as a model to explore plant-microbial community interactions; two recent landmark studies of Arabidopsis rhizosphere microbes discovered an influence of host genotype and developmental stage on microbial community structure (3, 4).
To test the repeatability of phyllosphere community assembly and the influence of deterministic and stochastic forces, we grew many replicate plants of the same genotype in a greenhouse environment (Fig. 1). We measured bacterial community structure in the phyllosphere and the subset of airborne bacteria passively settling near leaves throughout the Arabidopsis life cycle. Replicate phyllosphere communities converged to a shared community composition distinct from air as plants matured, revealing the importance of selective forces. However, we also observed that the relative abundance of these taxa was clearly related to the spatial association of individual plants. Our results suggest a strong influence of assembly history and small-scale dispersal limitation on phyllosphere community structure.
RESULTS
We sampled 32 individual plants and 21 airborne colonizing microbial communities for characterization with tag pyrosequencing of 16S rRNA genes and used quantitative PCR (qPCR) to obtain estimated cell densities in the phyllosphere throughout the Arabidopsis life cycle. Environmental conditions were relatively constant in the greenhouse throughout the experiment (see Fig. S1A in the supplemental material); temperature was held at 20°C, and humidity fluctuated with outside weather. Day length was ~9 h. Plant vegetative biomass continued to increase throughout the experiment (see Fig. S1B), with most plants bolting by day 50. At the end of the experiment, most plants had mature siliques but had not yet entered senescence.
Quantitative PCR.
We obtained 16S rRNA operon copy numbers for 26 of 33 plant samples, representing 10 time points (see Fig. S2); biomass in the remaining samples was too low for accurate quantification. Reaction efficiency was 71.6% (R2 = 0.999), possibly due to high primer degeneracy. While the 783F primer limited chloroplast amplification, it did not prevent 16S rRNA gene amplification from host plant mitochondria. Mitochondrial contamination was minimal in sequenced libraries (<1% in 47/60 datasets, 1 to 5% in 13/60 datasets) and could be discounted in copy number calculations. Pyrosequencing libraries with a high percentage of chloroplast 16S sequences had lower qPCR copy numbers (R2 = 0.3), suggesting that amplification of chloroplast rRNA genes occurred only in the absence of significant bacterial abundance. We estimated operon copy number for each operational taxonomic unit (OTU) using rrndb (29) and corrected read counts for each OTU to generate estimated cell counts in each sample (average rrn copy number/sample = 3.6). We observed a linear correlation between leaf area and rosette dry weight after leaf wash (with trimmed root and stems) across 12 plants (R2 = 0.952). We thus used dry weight data to extrapolate leaf area and deduce bacterial densities on leaves, taking into account both adaxial and abaxial leaf area.
Microbial population density on leaves was relatively constant between days 45 and 67 (1.7 × 104 to 2.7 × 104 cells per cm2), rising to 4.8 × 104 ± 2.9 × 104 cells/cm2 on day 73 (see Fig. S2). Total estimated cell counts per plant rose from 8.0 × 103 ± 0.54 × 103 on day 19 to 2.4 × 106 ± 1.3 × 106 on day 73, an ~300-fold increase. These results suggest that microbial density on Arabidopsis leaves reached a steady-state density of 104 cells/cm2 roughly 25 days following initial colonization. This value is at the lower end of the reported range for phyllosphere microbial densities (30). Because humidity is a strong determinant of cell density in the phyllosphere (31), these lower densities are likely due to the relatively dry conditions in the greenhouse during our experiment.
Verification of sterility.
To accurately compare airborne microbial immigrants with the phyllosphere community, it was important to ensure that contamination was not introduced at any point in the sampling and extraction process. To this end, we implemented rigorous sterility controls throughout the experiment. Initially, we verified the sterility of soil and seedlings germinated from sterile seeds on phytoagar plates. Pyrosequencing of the 16S rRNA gene v4v6 region from seedlings extracted prior to planting produced >99% organellar sequence (chloroplast and mitochondrial 16S rRNA genes). Soil sterility was verified through extraction of soil samples prior to planting and amplification with universal bacterial 16S rRNA gene primers; visualization of reactions on a Bioanalyzer high-sensitivity DNA chip confirmed the absence of amplifiable DNA. Controls from each sampling and extraction event were also amplified with universal bacterial primers and visualized on the Bioanalyzer. Controls from each sampling and extraction event were also amplified with 16S primers, and no contamination was observed in any sample.
Tag sequencing and diversity analysis.
Of the 505,082 raw 454 GS-FLX reads obtained using primers targeting the v4v6 hypervariable region, 346,866 passed our quality control pipeline and were assigned a taxonomic rank using the GAST process (32). We excluded sequences not affiliated with bacteria (43,536 reads), including those derived from chloroplasts or mitochondria (0 to 65%, mostly chloroplast). The remaining sequences, trimmed to the v5v6 region, were clustered into OTUs at 97% similarity using a usearch6-based clustering algorithm (33). This resulted in an abundance matrix of 1,758 OTUs containing 266,043 reads, including singleton and doubleton OTUs. A large number of OTUs were unique to either air (1,003 OTUs) or plants (435 OTUs), while 320 OTUs were shared by both sample types, suggesting that increased sampling depth is required to capture rare OTUs in air that colonize plants. A prior comparison of contemporaneous air and phyllosphere communities using denaturing gradient gel electrophoresis (DGGE), a lower-resolution method, also found that few taxa (2/28 bands) were shared between air and leaves (19).
We calculated within (alpha)-sample diversity on the full observation matrix using both parametric (CatchAll) and nonparametric (Chao1) richness estimators (34). Initial plant community richness varied between 79 and 114 at day 19 and increased 6- to 7-fold by day 60, depending on estimation method (see Fig. S3). The estimated richness of the airborne colonizing community does not show a time-related evolution and oscillated between 131 and 314 OTUs. Between (beta)-sample diversity was computed using QIIME (35) and Vegan (36) software after abundance normalization to the minimum sample depth (896 sequences) and removal of singleton and doubleton OTUs. The resulting abundance matrix contained 261,637 sequences and 397 OTUs over 53 samples, with an average of 4,936 reads per sample. Subsampling and removal of singleton/doubleton OTUs did not affect beta diversity results (data not shown).
Establishment of a “mature” phyllosphere community.
We analyzed plant and airborne colonizing community structure based on OTU presence/absence, using principal coordinate analysis (PCoA) on pairwise unweighted beta diversity metrics (Jaccard [Fig. 2A] and uwUniFrac [see Fig. S4A]). Airborne colonizing communities formed a distinct cluster (Fig. 2A) separate from plant communities and did not show any time-dependent change. Phyllosphere communities initially resembled the airborne colonizing community (day 19) but then departed from the air cluster, converging toward a mature phyllosphere cluster containing all communities from plants at day 60 and later. The composition of day 60+ communities was more phylogenetically homogenous than airborne immigrating communities (see Fig. S4A). In addition, the Jaccard distance between replicate plant communities decreased with time (Fig. 2B), indicating a convergence of phyllosphere communities in OTU membership. Permanova analysis on Jaccard distance matrices indicated a significant effect of both origin (air/plant, P < 0.001) and sampling day (P < 0.001) on community similarity. Sampling day was highly significant when considering plant communities alone (P = 0.005).
The dominant members of mature phyllosphere communities (see Table S1A in the supplemental material) were Acinetobacter (29%), Variovorax (12%), Pseudomonas (11%), unidentified Sphingobacteriaceae (7%), Rhodococcus, Ochrobactrum, and Chryseobacterium (4%). The most abundant taxa in air included Alicyclobacillus (23%), Ralstonia (26%), Acinetobacter (9%), Methylobacterium (5%), and Pseudomonas (3%) (see Fig. S5). We identified some important differences between the phyllosphere community structures of our greenhouse-grown plants and the phyllospheres of field Arabidopsis (2). In particular, we noted an underrepresentation of Methylobacterium and Sphingomonas, which typically dominate in field populations (37–39), and an overrepresentation of Acinetobacter and Pseudomonas taxa in our late-stage communities compared to in field plants.
We identified 51 biomarker OTUs (16 air, 41 plant) that strongly differentiated air and mature plant communities using LEfSe software (40) (see Fig. S6). Sets of air and plant biomarkers are phylogenetically distinctive at the phylum level (see Fig. S6A); air biomarkers are primarily in Firmicutes, while plant biomarkers are associated with Bacteriodetes and Proteobacteria. Biomarker abundance trajectories confirm that they were consistently more abundant in plants or air during the experiment. OTU1 (Alicyclobacillus) dynamics (Fig. 2C) are representative of taxa that were abundant in the pool of colonizing microorganisms but that were rapidly excluded from the phyllosphere microbiota. Conversely, OTU4 (Variovorax) and OTU10 (Rhodococcus) are representative colonists initially rare in air that gradually become dominant members of the mature phyllosphere community (see Fig. 2D and E).
We also used LEfSe to identify OTUs appearing at different stages of community succession. The number of distinct plant biomarkers observed increased as sampling progressed. On each sampling day between day 29 and day 50, less than 5 new biomarker OTUs were identified as statistically significant. On day 60, 19 new biomarker OTUs were identified. These biomarker groups represent distinct stages in ecological succession in the phyllosphere community (see Table S1B). Notably, some OTUs from genera frequently observed in field populations of Arabidopsis (Sphingomonas, Methylobacterium [2]) appeared only later in assembly (day 50+). In contrast, several biomarker OTUs in the order Sphingobacteriales appeared earlier in community development.
We also observed several remarkable cases of host discrimination between closely related OTUs within the same genus. The genus Methylobacterium is one of the most environmentally abundant genera associated with plants, but the factors regulating its distribution are largely unexplained (17). Methylobacterium species are facultative methylotrophs that can grow on a variety of C2, C3, and C4 compounds, and some strains produce plant growth-promoting hormones (11). We identified strong host selection for Methylobacterium OTUs. From a diverse colonizing pool of 13 OTUs present in air communities, a single one (OTU 31) was significantly represented in plants (see Fig. S7). This abundant OTU was identified as a biomarker for plant communities (see Fig. S7; linear discriminant analysis [LDA] score of 3.4), while most others were clearly excluded from Arabidopsis phyllospheres in spite of their abundance in air. Indeed, two other Methylobacterium OTUs were identified as air biomarkers, suggesting they may have originated from other local plant species and could not successfully colonize Arabidopsis.
Community abundance structure.
We examined the structure of phyllosphere communities using PCoA on beta diversity metrics weighted by OTU abundance (Morisita-Horn [Fig. 3A] and wUniFrac [see Fig. S4B in the supplemental material]). The structure of airborne colonizing communities, based on Morisita-Horn distances, remains relatively stable over time, forming a cluster distinct from all but the earliest (day 19) phyllosphere communities. This pattern is thus similar to the one observed using presence/absence metrics. However, plant communities form two groups corresponding to the two trays used for plant growth (Fig. 1). Moreover, this tray effect masks the time-dependent trajectory apparent in Fig. 2. The distinction between trays appears early during community assembly: day 19 plant samples are already distinct, and by day 29, plant samples occupy a central position within their respective groups (Fig. 3). Permanova analysis identified a significant effect of the tray on Morisita-Horn similarity (P < 0.001) but no effect of sampling day (P = 0.5).
FIG 1 .

Experimental design. Plants were placed in four growth trays inside two custom-built shade boxes side by side on a greenhouse bench. Arabidopsis Col-0 plants were placed in trays 1 and 2 (gray). Trays 3 and 4 (crosses) contained Arabidopsis gl-1 plants (data not shown). Trays were rotated at every watering and sampling event by each tray being moved one place to the right. Note that trays 1 and 2 remained in separate shade boxes until day 55 of the experiment. Glass slides coated with CellTak (small black rectangles) were placed in the center of each tray and sampled/replaced simultaneously with sampling of plants. Flats within each tray were rotated back to front and front to back as shown every time trays were rotated.
FIG 2 .

Mature plant-associated microbial communities have a distinct membership from air communities and show a clear trajectory over time. (A) Principal coordinate plots using the membership-based Jaccard index to measure beta diversity, based on OTUs clustered at 97% similarity. Each dot represents a single community (blue, air; green, plant). Dot size is scaled by sampling day (day 19, small; day 73, large). Arrow indicates the trajectory from early to mature phyllosphere communities. (B) Mean Jaccard distance between replicate plants on each sampling day shows increasing similarity with time. (C) Abundance trajectories over time of the day 60+ air biomarker OTU 1. Blue indicates relative abundance (percentage of total read count) in air, and green indicates relative abundance in plants. Error bars show the standard deviation across triplicates (plants) and duplicates (air). Abundance trajectories over time of day 60+ plant biomarker OTU 4, a day 55 biomarker (D), and OTU 10, a day 50 biomarker (E). Coloring is identical to that described for panel C.
FIG 3 .

Spatially associated plants share similar taxon abundances. (A) Principal coordinates plot using the abundance-based Morisita-Horn index to measure beta diversity, based on OTUs clustered at 97% similarity. Blue, air; green, tray 1 plants; orange, tray 2 plants. Scaling is the same as described for Fig. 1. Arrows indicate trajectories from newly colonized plants to tray-specific communities. (B) Abundance trajectory of the dominant tray 1 biomarker Pseudomonas; error bars indicate the standard deviation of replicate plants. Green bars indicate relative abundance of the OTU in tray 1, orange bars in tray 2, and blue bars in air. (C, D) Abundance profiles for major tray marker taxa identified by LEfSe at successive time points. (C) Abundance of the tray 1 biomarker Rhodococcus. (D) Abundance of the tray 2 biomarker Methylobacterium. (E) Abundance of the dominant tray 2 biomarker Acinetobacter. Note that three plants were sampled at every time point, chosen randomly; possible configurations for sampling between trays 1 and 2 were (2,1), (1,2), (3,0), or (0,3).
LEfSe analysis identified 22 OTUs as biomarkers for either tray 1 or 2, with a prominent role of Pseudomonas in tray 1 and Acinetobacter in tray 2 (Fig. 3). OTUs from both genera were among the strongest tray biomarkers identified. These taxa strongly dominated phyllosphere communities; individual OTUs from these genera appeared at relative frequencies of up to 52% (Pseudomonas, day 50) and 72% (Acinetobacter, day 24). Notably, Pseudomonas and Acinetobacter OTUs were very abundant early in community development (Fig. 3B and E). LEfSe also identified known plant- and soil-associated taxa as tray-specific biomarkers, including Rhodococcus, Methylobacterium, Novosphingobium, and Chryseobacterium (Fig. 3C and D; see also Fig. S8 in the supplemental material), which appear later in the time series.
Ecological differentiation of phyllosphere bacteria at the sub-OTU level.
Interestingly, the strong tray pattern we observed using the Morisita-Horn diversity index almost completely collapsed when the phylogeny-based wUniFrac distance was used (see Fig. S4B). This shows that bacteria accounting for differences between trays are closely related phylotypes. For example, the dominant OTUs driving community abundance patterns belong to the genera Acinetobacter and Pseudomonas, which are both part of the same order (Pseudomonadales).
We hypothesized that these closely related taxa may be ecologically equivalent (41) in phyllosphere communities. We examined whether ecological processes structuring bacterial communities also operate at the sub-OTU level (corresponding to related species, strains, or genotypes of the same species). OTU clustering, while minimizing the effect of sequencing errors on microbial diversity analysis, can obscure biologically significant variations between organisms with highly similar (>97%) 16S rRNA gene sequences. We thus used oligotyping, a powerful new method, to better distinguish true biological variations from sequencing error noise (42). Oligotyping uses Shannon entropy analysis to isolate meaningful positional variation within sets of similar 16S rRNA gene sequences that with standard methods would converge into the same cluster; each unique set of variants is identified as an oligotype.
The 20 most abundant genera, which together accounted for 99 OTUs and 77% of reads in the data set, were individually decomposed by this method, identifying 234 different oligotypes. For six of these genera, oligotype decomposition matched OTU clustering. In all other cases, however, oligotypes demonstrated distinct distributional patterns. As an example, Acinetobacter OTU 0, the most abundant plant-associated taxon overall, could be decomposed into four oligotypes that each exhibited a dramatically different spatial distribution (Fig. 4). In spite of the presence of oligotypes 1 and 2 in airborne immigrating communities and in initial phyllosphere communities, only oligotype 1 successfully colonized the tray 2 phyllosphere. These results show that two Acinetobacter phylotypes (identified as Acinetobacter johnsonii and Acinetobacter junii based on BLAST results) with 98.2% sequence similarity in the 16S rRNA gene had opposite colonization patterns.
FIG 4 .
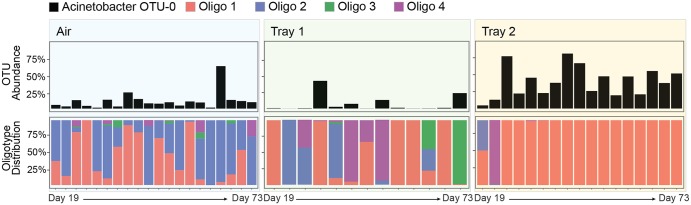
Sub-OTU diversity reveals ecological processes operating at fine taxonomic scale. Within the genus Acinetobacter, sequences belonging to the dominant OTU 0 (top panel, 20% of the data set) could be decomposed into 4 major oligotypes (bottom 4 panels). Each oligotype shows a distinctive distribution indicating fine taxonomic selection within this dominant taxon; oligotype 1 in tray 2 plants, oligotype 2 in air, oligotype 3 in tray 1 plants, and oligotype 4 in both tray 1 and tray 2 plants.
DISCUSSION
In this study, we characterized the development of bacterial communities in the phyllosphere of Arabidopsis thaliana over the plant life cycle. We used a large number of replicate plants grown in a controlled environment to test the repeatability of community assembly, the first such study of phyllosphere assembly to date. Our results demonstrate that the presence or absence of particular taxa in the phyllosphere is under strong deterministic selection (Fig. 2). The membership of phyllosphere communities initially mirrored immigrant airborne microbes but subsequently converged to a phylogenetically distinctive community composition. We observed strong, reproducible successional dynamics in community membership. In contrast, the relative abundance of bacterial taxa in the phyllosphere was highly variable among replicates and was strongly related to the spatial association of individual plants (Fig. 3). Separation by spatial association began very early in community formation and continued throughout the plant life cycle, generating alternate trajectories of community development. These results suggest that stochastic forces play a substantial role in structuring phyllosphere communities.
Stochastic forces in community assembly.
A recent conceptual synthesis (43) identified four main processes in community assembly: selection, drift, speciation, and dispersal. Only one of these forces, selection, predicts a correlation between habitat niche structure and community composition. The other three stochastic processes can operate in conjunction with selection or together to generate the classical neutral model of Hubbell (44). Theory suggests that stochastic variation in colonization order can have a significant impact on community assembly, resulting in high beta diversity among similar sites (45, 46). Dispersal limitation can reinforce the effects of colonization history on beta diversity (46). Implicit in these models is the assumption that niche-based selection operates on sets of functionally equivalent colonizers to generate alternate community structures in similar sites. Stochastic niche theory (47), for example, predicts that primary colonizers rapidly occupy the broadest available niches and preempt further colonization by similar species. Secondary colonizers must be able to fit into the remaining niche space and grow rapidly enough to overcome drift. Under this model, alternate states are easily generated given a sufficient diversity of primary colonizers. Alternatively, stochastic variation in colonization order could lead to priority effects, where the chance arrival of a particular early colonizer alters the habitat in a way that favors the growth of specific secondary colonizers (48).
Our observations suggest that stochastic colonization dynamics and dispersal limitation played a central role in shaping the abundance structure of phyllosphere bacterial populations. The convergence in community membership across replicate plants over time indicates that host-microbe and/or microbe-microbe interactions combined to shape niches favoring the growth of particular taxa. It further suggests that replicate plants have similar niche structures. Given this result, it is difficult to interpret the strong effect of spatial association on bacterial abundance (Fig. 3) as resulting from purely niche-based forces. The two experimental trays were treated identically and frequently rotated, yet plants in one tray had phyllosphere communities dominated by Acinetobacter while communities in the other tray were dominated by Pseudomonas. These two highly abundant taxa appeared very early in community assembly (Fig. 3B and E). They are the strongest, and earliest appearing, tray biomarkers identified by LEfSe (see Fig. S8). Distinct tray community structures were already apparent at 5 days postcolonization (Fig. 3A) and continued until the end of the experiment.
The most parsimonious explanation is that random initial colonization events led to the early dominance of each taxon in a particular tray, which is supported by the divergence of tray abundance structures immediately after colonization (day 19; Fig. 3). Populations developing from these early events were maintained on the initial leaves and then propagated to newly formed leaves. Given that trays were separated by roughly 1 m, and plants within a tray were separated by at most 40 cm, it is plausible that dispersal limitation reinforced alternate community trajectories throughout the experiment. This hypothesis is supported by the tray-specific distribution of several late-appearing colonizers, including OTUs from known plant- and soil-associated genera (Rhodococcus, Methylobacterium, Novosphingobium, and Chryseobacterium; Fig. 3C and D). These observations also fit an important prediction of the stochastic niche model (47): initial colonizers have access to more resources and are more likely to develop dominant populations compared to competitors with smaller niche utilization capabilities.
The alternative assembly trajectories in trays 1 and 2 may represent an interesting case of ecological drift between spatially separated communities (22). It shows remarkable similarity to other cases of stochastic community assembly, such as the dependence of mouse gut microbiome composition on housing cage (49). Dispersal limitation as a key factor in phyllosphere assembly is also consistent with field surveys identifying a large effect of geographic site on phyllosphere structure (17). An alternate possibility, which does not require the assumption of dispersal limitation, is that Pseudomonas and Acinetobacter each altered the leaf surface habitat in such a way to favor the growth of particular sets of secondary taxa.
Functional equivalence in microbial populations.
Our observation of alternate phyllosphere assembly trajectories in spatially separated plants raises the obvious question of whether these states are functionally equivalent. Empirical evidence from other microbial systems is mixed. A combination of inoculum source and housing cage led to differences in mouse gut microbiome function (49), and stochastic colonization coupled with biotic interactions led to differences in function across artificial microbial reactors (50). On the other hand, there are many examples of variation in bacterial community structure across similar sites (e.g., multiple wastewater reactors) coupled with conservation of function (23, 51).
The functional equivalence of cooccurring taxa is central to the neutral theory of community assembly (52). Hubbell (53) argued that sets of functionally equivalent species can evolve within the niches that are most prevalent over evolutionary time. Therefore, the dynamics within each niche are neutral, but selection has occurred to set the boundaries of each species set. The necessary condition for this to occur is the absence of factors that promote competitive exclusion between functionally similar species. Other theoretical work suggests that this pattern of sets of functionally similar species (“emergent neutrality”) can appear via several different possible pathways (54, 55) and is more likely to occur in species-rich communities (56).
While our experiment did not directly test for functional equivalence, some of our findings are suggestive. The dominant early colonizing tray biomarker taxa, Acinetobacter and Pseudomonas, arguably fill similar niche spaces in the leaf environment as primary colonizers. Acinetobacter and Pseudomonas have on average 6 and 5 ribosomal operons per genome (29), respectively, making them classic r-strategist taxa capable of rapid growth in response to nutrient availability (57). Additionally, species within both genera are frequently found in association with plants and have growth-promoting properties (58, 59). However, our analyses also identified examples of ecological selection between closely related taxa. Two oligotypes of Acinetobacter, which have 98.2% similar 16S rRNA gene sequences, exhibited distinct distributional patterns; while both were present in the airborne immigrating community, only one was able to establish on tray 2 plants (Fig. 4). We also found that when multiple OTUs of a particular genus were present in airborne immigrants, often only one successfully established on plants (e.g., Methylobacterium; see Fig. S7 in the supplemental material). Testing the hypothesis of functional equivalence between alternate states will require additional replicated experiments, as well as direct assays of community function (for example, through shotgun metagenomics) and community-level competition experiments.
Conclusion.
Through detailed characterization of Arabidopsis-associated leaf microbiota and airborne colonizing microbes over the plant life cycle, we identified key ecological forces driving microbiome assembly. On the one hand, convergence in microbiome membership as plants mature indicates that plants exert a strong selective force on the identity of colonizing microbes (who can colonize). However, variation in the abundance—as opposed to the presence—of dominant taxa is strongly related to spatial associations between plants. This variance is best explained by stochasticity in initial colonization events and subsequent limited dispersal, as predicted by different community assembly models (neutral or stochastic niche). Further experimentation with controlled community assembly is required to assess the repeatability and robustness of these coupled niche and stochastic dynamics.
Our results demonstrating an interaction of niche and stochastic effects are suggestive, but it will be necessary to replicate our experiment in different controlled environments (research greenhouses or hoophouses) and with a larger number of plants to test their generality with respect to microbial colonization of the phyllosphere. Our findings of two alternate community structures also raise the question of how many community trajectories are possible in the phyllosphere environment and whether they differ in relative fitness. Again, similar experiments with a larger number of replicate plants will be needed to address these questions. Additionally, the conspicuous role of dispersal patterns suggested by our results needs to be directly tested and quantified. We anticipate that more complex experimental designs using various dispersal rates between phyllosphere metacommunities will bring valuable insights to this problem.
Our results have implications for the design of experiments aiming at testing the effects of particular treatments or environmental variables on phyllosphere community structure. We show that both stochastic events and dispersal limitation can account for significant beta diversity between spatially separated replicate communities. Large pools of replicates are necessary to account for this inherent stochasticity, and randomization of control and tested individual plants is necessary to avoid confounding ecological drift among pooled individuals with experimental treatments.
Overall, this study provides a novel ecological framework for studies of microbiome assembly and points to the importance of highly replicated, controlled longitudinal studies of microbial community development.
MATERIALS AND METHODS
Experimental design.
We initiated a 73-day time series experiment using 72 microbe-free Arabidopsis thaliana Col-0 plants planted in sterile soil in a greenhouse (Fig. 1). Every 4 to 6 days, three plants were destructively sampled for phyllosphere community analysis. In addition, we used glass slides coated with adhesive protein (CellTak) in order to sample the airborne community. The slides were located among the plants at the level of plant leaves. The slides were intended to act as passive traps for microbes arriving on the leaf surface during a particular time interval rather than be representative of the total airborne microbiota. They allow us to determine, to some extent, the composition of the immigrant community in the absence of the dynamic microbe-microbe and microbe-host interactions shaping leaf surface communities.
Plant germination and growth.
Seeds of Arabidopsis thaliana were surface sterilized, germinated on standard phytoagar plates, and transferred after 14 days of growth to sterile soil in a Conviron greenhouse (Falmouth Technology Park, Falmouth, MA). The light regime was roughly 9 h of light and 15 h of dark. Sterile soil was obtained by saturating dry soil (Lehle Seeds, Round Rocks, TX) with water in an autoclave bag. After 48 h at room temperature, the wet soil was autoclaved twice for 45 min at 24-h intervals (60). Autoclaved soil was tested for the presence of amplifiable bacterial 16S rRNA genes. Prior to being planted, seedlings were sampled from phytoagar plates and tested for sterility with 16S rRNA gene PCR. Upon planting in soil, each plant was transferred to a separate well of a 6-well insert. Inserts were placed in plastic trays under shade boxes constructed with wire screens to reduce light levels to those standard for Arabidopsis growth (28). The first sampling occurred on day 19, 5 days after transfer from plates to soil.
Environmental conditions in the shade boxes were monitored using HOBO sensors (temperature/RH [relative humidity] and light as PAR [photosynthetically active radiation]) and a MicroStation data logger (Onset Computer, Bourne, MA). The greenhouse temperature set point was 20°C, and humidity was not controlled. Plants were watered every 3 days and fertilized with 200 ppm 20/10/20 fertilizer 1× per week starting at day 27. Plant and tray positions were randomized at each watering, fertilization, and sampling event. Plants were randomized within each tray, and trays were moved between the two shade boxes, but plants were not moved from tray to tray (Fig. 1).
Air and phyllosphere sampling.
We captured airborne microbes representing the colonizing community using sterilized standard microscope slides coated with the biological adhesive CellTak (BD Biosciences) at a density of 3.5 µg/cm2. We constructed small platforms to hold slides at the level of plant leaves. Slides were left in place during each sampling interval (~5 days), collected, and replaced at the next sampling event. In preliminary experiments, fluorescent microscopy using 4′,6-diamidino-2-phenylindole (DAPI) staining indicated the presence of intact cells on slides exposed to air for ~1-week periods (data not shown). Slides were incubated with trypsin to remove CellTak and detach microbes; the wash solution was collected on sterile 0.22-μm-pore-size filters, and DNA was extracted using the Biostic bacteremia DNA extraction kit (MoBio Labs). Due to the low biomass present in air, we used rigorous sterile technique and extensive negative controls to exclude contaminant microbes (see Results).
Three whole Col-0 plants, randomly selected, were sampled at each of 11 time points. Plants were randomly drawn from both trays, resulting in an unbalanced number of plants from each tray at different time points. Roots and flowering stems were trimmed off, and then rosettes were placed into a solution of 0.2% Silwet in TE (10 mM Tris, 1 mM EDTA, pH 7) (37, 61) and sonicated in a bath sonicator for 10 min. Rosettes were removed, dried at 70°C overnight, and subsequently weighed. Wash solutions were prefiltered through 5-μm-pore-size filters and then collected on 0.22-μm-pore-size sterile filters. DNA was extracted from filters with Biostic bacteremia kits. Plant leaf area was calculated based on dissected plant photos using ImageJ software (62). Plant dry weight was measured on a high-precision scale after overnight drying of the plants at 70°C. It was not possible to obtain wet weights due to the addition of Silwet/buffer to plants in the wash procedure. One of the day 60 plants and all day 45 CellTak slide replicates were omitted after manipulation problems (breakage of the filtration membrane).
Sterility tests.
During each sampling event, we collected four negative controls to detect possible contamination during plant and air sampling. First, we filtered sterile Silwet wash solution through our Swinnex apparatus onto a 0.22-μm-pore-size filter. Second, we sampled 500 µl of Silwet wash solution alone. Third, we filtered 30 ml of the trypsin solution used in CellTak slide extraction onto a 0.22-μm-pore-size filter. Finally, prior to each sampling event, we placed a sterile CellTak slide inside a 50-ml Eppendorf tube within the laminar flow hood. This control slide was transported to the greenhouse, placed on the bench near exposed slides, and subsequently analyzed. We also included a blank control filter in every batch of DNA extractions, as well as standard PCR-negative controls. All filters were extracted with MoBio bacteremia DNA kits, and the resulting DNA was amplified with standard v4v6 primers. The lack of amplification signals from DNA extracted from sterilized soil using a Bioanalyzer high-sensitivity DNA chip (detection limit of 5 pg/µl, manufacturer’s specs) was interpreted as sufficient evidence for soil sterility. Because sterilized soil exposed to air for 5 days showed a strong PCR product using the same assay, we conclude that our initial results are not due to inhibition of PCR amplification by elements from soil coextracted with DNA.
qPCR.
A SYBR green quantitative PCR (qPCR) assay was developed for quantification of bacterial 16S rRNA gene copy number among rosette leaf washes using a modified16S rRNA gene primer that limits chloroplast 16S amplification coupled with the universal primer 1046R (63). A 10-fold serially diluted, 5-point standard curve (range, 3 × 103 to 3 × 107) was generated with plasmids containing 16S rRNA gene 783F/1046R inserts from 9 microbial species of various GC content. The standard curve, environmental samples, and NTCs (no template controls) were run in triplicate on a StepOnePlus real-time PCR system (Life Technologies). All rosette samples were run on the same qPCR plate to avoid variation between assays due to variable efficiency. Representative sequences for each operational taxonomic unit were submitted to rrndb (29) to obtain a copy number correction factor to translate qPCR counts into estimated cell number.
Amplification and 454 sequencing of the v4v6 region of 16S rRNA genes.
The v4v6 variable region of 16S rRNA genes was amplified in triplicate with 454 fusion primers containing adapters and bar codes with bacterial primer sequences 518F (5′ CCAGCAGCYGCGGTAAN 3′) and 1046R (5′ CGACRRCCATGCANCACCT 3′) and sequenced on a 454 GS-Ti instrument at the MBL as previously described (64). Individual plant samples were separately amplified, but CellTak slides from each shade box were pooled prior to amplification due to low biomass.
Bioinformatic analyses.
Processing and filtering of v4v6 pyrosequencing reads were carried out using a standard MBL pipeline (32, 64). Reads were trimmed to the v5v6 region using a conserved anchor sequence due to low quality at the v4 end, and all subsequent analyses were performed on these data set. Sequences were clustered using a standard Usearch6 pipeline into 97% similarity OTUs (33). We used the Catchall software (34) for both parametric and nonparametric richness estimation. We then used QIIME (35) and Vegan (36) software for beta diversity analysis based on a corrected abundance matrix: (i) OTUs containing 1 or 2 reads were discarded in order to further reduce the number of spurious OTUs generated by errors introduced during PCR and sequencing, and (ii) observation counts were subsampled to the number of reads present in the smallest library (895 reads) for calculation of beta diversity indices. We used LEfSE software (40) to identify biomarker OTUs. We used oligotyping (42) to identify significant sub-OTU-level variation in each of the 20 most abundant genera and analyzed the abundance pattern of oligotypes across sample sets.
Nucleotide sequence accession number.
All 16S rRNA gene sequences described in this study have been deposited in the VAMPS archive (http://vamps.mbl.edu) with the accession number “SLS_PHY_Bv6v4.”
SUPPLEMENTAL MATERIAL
Supplemental Materials and Methods. Download
(A) Environmental conditions in the greenhouse during the time series, as measured with PAR and combined temperature/humidity sensors and logged on a HOBO MicroStation logger (Onset Computer). Instantaneous measurements were averaged across a 5-day sampling interval. (a) Temperature (°C). The spike in temperature from 28 October 2011 to 31 October 2011 (days 21 to 24) reflects a partial failure of the greenhouse air conditioning unit. (b) Daytime light levels; for each day, the maximum light level was recorded. The minimum value for the interval therefore indicates a day where maximum light levels were low, usually due to cloud cover. (c) Relative humidity (%). Blue, maximum reading during interval; orange, minimum reading during interval; red, average across interval. (B) Plant dry weight; error bars are the standard deviations from three replicates. Plants were dried overnight at 70°C prior to being weighed to remove Silwet/buffer remaining from the leaf wash. Day 19 data are missing. Download
Microbial population density estimated with qPCR. Estimated cell counts were obtained by applying rrn copy number correction factors to raw qPCR counts (see the text). Top, mean estimated cell count per plant; bottom, estimated microbial population density (cells/cm2) on leaves was calculated by dividing corrected cell counts by total leaf area (see the text). Error bars show the standard deviation of combined technical and biological replicates; day 19 error bar indicates only technical error (only one day 19 sample could be amplified). The spike in estimated cell counts on day 34 is unexplained but may indicate recovery from a temperature spike in the greenhouse due to air conditioning failure. Leaf area data are not available for samples prior to day 34. Download
OTU richness in airborne colonizing and leaf communities. Evolution of OTU richness in airborne and plant leaf microbial communities as a function of time. Both observed (Sobs) and estimated OTU numbers are shown. Nonparametric Chao1 and parametric estimates of richness were computed with the CatchAll software (34). Note that richness was calculated based on the uncorrected observation matrix containing singletons and doubletons, as these are critical values for (non)parametric estimation. Estimated richness in air communities oscillated between 106 and 319 OTUs. Phyllosphere richness, as estimated with the Chao index, increased from 79 OTUs at the beginning of the time series (day 19) to 474 OTUs at day 66, with much higher estimation from Catchall (840 OTUs). Download
Phylogenetically informed beta diversity analysis using unweighted and weighted UniFrac performed on trimmed v5v6 reads. (A) Unweighted UniFrac PCoA plot with points colored to show sampling day and origin (air/plant). Mature plant communities (green) are closely associated phylogenetically and are distinct from air communities (blue). Dots are scaled by sampling day (day 19, small; day 73, large). (B) Weighted UniFrac PCoA plot with points colored to show origin of plants by tray (green, tray 1; orange, tray 2; blue, air). The separation of tray communities observed in beta diversity PCoA plots using nonphylogenetic abundance metrics (Fig. 3, main text) collapses when phylogeny is considered, due to the close phylogenetic relatedness of highly abundant tray markers (Pseudomonas, tray 1; Acinetobacter, tray 2). Download
Overall taxonomic distribution in air and plant communities. Annotations of all quality trimmed reads generated with the GAST process were pooled for all plant (A) and air (B) communities. The summed counts were used to generate hierarchical Krona (65) charts showing the relative abundances of genus-level taxonomic groupings. Download
(A) LEfSe (40) biomarker analyses of mature plant (day 50+) and air communities identify clusters and higher-level taxonomic groups characteristic of each sample type that are highly statistically significant (P < 0.05). Terminal tips represent individual clusters. Phylogenetic groupings enriched in plant samples are highlighted in green (b, Microbacteriaceae; c, Nocardiaceae; f, Patulibacteraceae; g, Cytophagaceae; h, sphingobacteriaceae; i, Sphingobacteriales; j, Sphingobacteria; K, OTU_142; r, OTU_105; S, Brucellaceae; t, Rhizobiaceae; u, unassigned; y, OTU_95; a0, Comamonadaceae; a3, Xanthomonadaceae; a4, Xanthomonadales; a5, verrucomicrobiaceae; a6, Verrucomicrobiales; a7, verrucomicrobiae). Those in red are enriched in air (a, Corynebacteriaceae; d, Propionibacteriaceae; e, Streptomycetaceae; I, Alicyclobacillaceae; m, Staphylococcoceae; n, Bacillales; o, Streptococcaceae; p, Lactobacillales; q, Bacillus; v, Rhodobacteraceae; w, Rhodobacterales; x, Rickettsiales; z, Burkholderiaceae; A1, Enterobacteriaceae; a2, Enterobacteriales). (B) Biomarkers for air and mature plant communities (day 60+) identified with LEfSe software. Top, LDA scores for air biomarkers (blue); bottom, LDA scores for plant biomarkers (green). Download
Example of OTU partitioning: distribution of OTUs classified as genus Methylobacterium as a function of time in airborne communities (T0; far left), plant-associated communities in tray 1 (T1; middle), and plant-associated communities in tray 2 (T2; right). A diverse population of methylobacterial OTUs is present in air, but most are unable to grow successfully on plant leaves. Download
OTUs and higher-level taxonomic groupings identified by LeFSe as biomarkers distinguishing tray 1 and tray 2 plant-associated communities, including all sampling dates. Bars indicate the value of the LDA score, which is a measure of the strength of the biomarker identification. All biomarkers shown were significant at P values of <0.05. Green, tray 1; orange, tray 2. Download
(A) Average abundance of the top 20 OTUs (percentage of total read counts) in day 60+ mature phyllosphere communities. (B) Plant-specific biomarker OTUs identified by LEfSE (40) that become statistically significant on each sampling day. Abundance matrices were trimmed to identify later-appearing biomarkers. For example, to identify biomarkers appearing on day 55, we removed all plant and air samples prior to day 55 and ran LEfSE analysis on only day 55+ samples. The resulting set of biomarker OTUs from day 55 contained some biomarkers that first appeared earlier and some newly appearing biomarkers. The newly appearing plant-specific biomarkers are shown as “day 55” in the above table (light green). Biomarker groupings identified in this fashion are color-coded by the sampling day at which they were first identified by LEfSE.
ACKNOWLEDGMENTS
We thank M. Sogin, Z. Cardon, and E. López-Peredo for critical discussions and reading of the manuscript, Z. Cardon for use of the MBL greenhouse facility, H. Morrison, J. Vineis, and S. Grim for assistance with sequencing, S. Huse for assistance with bioinformatic analysis, A. Aicher for assistance with greenhouse protocols, and Norm Pace for suggesting CellTak as a substrate for capture of airborne microbes.
Funding was provided by the J. Unger Vetleson Foundation to S.L.S.
Footnotes
Citation Maignien L, DeForce EA, Chafee ME, Eren AM, Simmons SL. 2014. Ecological succession and stochastic variation in the assembly of Arabidopsis thaliana phyllosphere communities. mBio 5(1):e00682-13. doi:10.1128/mBio.00682-13.
REFERENCES
- 1. Bulgarelli D, Schlaeppi K, Spaepen S, Ver Loren van Themaat E, Schulze-Lefert P. 2013. Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 64:807–838. 10.1146/annurev-arplant-050312-120106 [DOI] [PubMed] [Google Scholar]
- 2. Vorholt JA. 2012. Microbial life in the phyllosphere. Nat. Rev. Microbiol. 10:828–840. 10.1038/nrmicro2910 [DOI] [PubMed] [Google Scholar]
- 3. Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, Tremblay J, Engelbrektson A, Kunin V, del Rio TG, Edgar RC, Eickhorst T, Ley RE, Hugenholtz P, Tringe SG, Dangl JL. 2012. Defining the core Arabidopsis thaliana root microbiome. Nature 488:86–90. 10.1038/nature11237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bulgarelli D, Rott M, Schlaeppi K, van Loren van Themaat E, Ahmadinejad N, Assenza F, Rauf P, Huettel B, Reinhardt R, Schmelzer E, Peplies J, Gloeckner FO, Amann R, Eickhorst T, Schulze-Lefert P. 2012. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488:91–95. 10.1038/nature11336 [DOI] [PubMed] [Google Scholar]
- 5. Kim YC, Leveau J, McSpadden Gardener BB, Pierson EA, Pierson LS, III, Ryu CM. 2011. The multifactorial basis for plant health promotion by plant-associated bacteria. Appl. Environ. Microbiol. 77:1548–1555. 10.1128/AEM.01867-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lindow SE, Brandl MT. 2003. Microbiology of the phyllosphere. Appl. Environ. Microbiol. 69:1875–1883. 10.1128/AEM.69.4.1875-1883.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leveau JH, Lindow SE. 2001. Appetite of an epiphyte: quantitative monitoring of bacterial sugar consumption in the phyllosphere. Proc. Natl. Acad. Sci. U. S. A. 98:3446–3453. 10.1073/pnas.061629598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morris CE, Kinkel LL, Lindow SE, Hecht-Poinar EI, Elliott VJ. 2002. Fifty years of phyllosphere microbiology: significant contributions to research in related fields, p 365–375 In Lindow SE, Hecht-Poinar EI, Elliott VJ. (ed), Phyllosphere microbiology. APS Publishing, St. Paul, MN. [Google Scholar]
- 9. Wilson M, Lindow SE. 1994. Coexistence among epiphytic bacterial populations mediated through nutritional resource partitioning. Appl. Environ. Microbiol. 60:4468–4477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Innerebner G, Knief C, Vorholt JA. 2011. Protection of Arabidopsis thaliana against leaf-pathogenic Pseudomonas syringae by Sphingomonas strains in a controlled model system. Appl. Environ. Microbiol. 77:3202–3210. 10.1128/AEM.00133-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lidstrom ME. 2006. Aerobic methylotrophic prokaryotes, p 618–634 Springer, New York, NY. [Google Scholar]
- 12. Galbally IE, Kirstine W. 2002. The production of methanol by flowering plants and the global cycle of methanol. J. Atmos. Chem. 43:195-229-229. 10.1023/A:1020684815474 [DOI] [Google Scholar]
- 13. Lighthart B. 1997. The ecology of bacteria in the alfresco atmosphere. FEMS Microbiol. Ecol. 23:263–274. 10.1016/S0168-6496(97)00036-6 [DOI] [Google Scholar]
- 14. Redford AJ, Fierer N. 2009. Bacterial succession on the leaf surface: a novel system for studying successional dynamics. Microb. Ecol. 58:189–198. 10.1007/s00248-009-9495-y [DOI] [PubMed] [Google Scholar]
- 15. Jackson CR, Denney WC. 2011. Annual and seasonal variation in the phyllosphere bacterial community associated with leaves of the southern magnolia (Magnolia grandiflora). Microb. Ecol. 61:113–122. 10.1007/s00248-010-9742-2 [DOI] [PubMed] [Google Scholar]
- 16. Finkel OM, Burch AY, Elad T, Huse SM, Lindow SE, Post AF, Belkin S. 2012. Distance-decay relationships partially determine diversity patterns of phyllosphere bacteria on Tamarix trees across the Sonoran desert. Appl. Environ. Microbiol. 78:6187–6193. 10.1128/AEM.00888-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Knief C, Ramette A, Frances L, Alonso-Blanco C, Vorholt JA. 2010. Site and plant species are important determinants of the Methylobacterium community composition in the plant phyllosphere. ISME J. 4:719–728. 10.1038/ismej.2010.9 [DOI] [PubMed] [Google Scholar]
- 18. Redford AJ, Bowers RM, Knight R, Linhart Y, Fierer N. 2010. The ecology of the phyllosphere: geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ. Microbiol. 12:2885–2893. 10.1111/j.1462-2920.2010.02258.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vokou D, Vareli K, Zarali E, Karamanoli K, Constantinidou HI, Monokrousos N, Halley JM, Sainis I. 2012. Exploring biodiversity in the bacterial community of the Mediterranean phyllosphere and its relationship with airborne bacteria. Microb. Ecol. 64:714–724. 10.1007/s00248-012-0053-7 [DOI] [PubMed] [Google Scholar]
- 20. Meyer KM, Leveau JH. 2012. Microbiology of the phyllosphere: a playground for testing ecological concepts. Oecologia 168:621–629. 10.1007/s00442-011-2138-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gonzalez A, King A, Robeson MS, Song S, Shade A, Metcalf JL, Knight R. 2012. Characterizing microbial communities through space and time. Curr. Opin. Biotechnol. 23:431–436. 10.1016/j.copbio.2011.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JB. 2012. Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Microbiol. 10:497–506 [DOI] [PubMed] [Google Scholar]
- 23. Ofiteru ID, Lunn M, Curtis TP, Wells GF, Criddle CS, Francis CA, Sloan WT. 2010. Combined niche and neutral effects in a microbial wastewater treatment community. Proc. Natl. Acad. Sci. U. S. A. 107:15345–15350. 10.1073/pnas.1000604107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Langenheder S, Berga M, Östman O, Székely AJ. 2012. Temporal variation of β-diversity and assembly mechanisms in a bacterial metacommunity. ISME J. 6:1107–1114. 10.1038/ismej.2011.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dumbrell AJ, Nelson M, Helgason T, Dytham C, Fitter AH. 2010. Relative roles of niche and neutral processes in structuring a soil microbial community. ISME J. 4:337–345. 10.1038/ismej.2009.122 [DOI] [PubMed] [Google Scholar]
- 26. Remus-Emsermann MN, Leveau JH. 2010. Linking environmental heterogeneity and reproductive success at single-cell resolution. ISME J. 4:215–222. 10.1038/ismej.2009.110 [DOI] [PubMed] [Google Scholar]
- 27. Mercier J, Lindow SE. 2000. Role of leaf surface sugars in colonization of plants by bacterial epiphytes. Appl. Environ. Microbiol. 66:369–374. 10.1128/AEM.66.1.369-374.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boyes DC, Zayed AMAR, Ascenzi R, McCaskill AJ, Hoffman NE, Davis KR, Görlach J. 2001. Growth stage-based phenotypic analysis of Arabidopsis: a model for high throughput functional genomics in plants. Plant Cell 13:1499–1510. 10.2307/3871382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klappenbach JA, Saxman PR, Cole JR, Schmidt TM. 2001. rrndb: the ribosomal RNA operon copy number database. Nucleic Acids Res. 29:181–184. 10.1093/nar/29.1.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Remus-Emsermann MN, Tecon R, Kowalchuk GA, Leveau JH. 2012. Variation in local carrying capacity and the individual fate of bacterial colonizers in the phyllosphere. ISME J. 6:756–765. 10.1038/ismej.2011.209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beattie GA. 2011. Water relations in the interaction of foliar bacterial pathogens with plants. Annu. Rev. Phytopathol. 49:533–555. 10.1146/annurev-phyto-073009-114436 [DOI] [PubMed] [Google Scholar]
- 32. Huse SM, Dethlefsen L, Huber JA, Mark Welch D, Relman DA, Sogin ML. 2008. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 4: e1000255. 10.1371/journal.pgen.1000255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 34. Bunge J. 2011. Estimating the number of species with catchall. Pac. Symp. Biocomput. 2011:121–130 [DOI] [PubMed] [Google Scholar]
- 35. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Henry M, Stevens H, Wagner H. Vegan: community ecology package. 2011. R package version 2.0-10. [Google Scholar]
- 37. Delmotte N, Knief C, Chaffron S, Innerebner G, Roschitzki B, Schlapbach R, von Mering C, Vorholt JA. 2009. Community proteogenomics reveals insights into the physiology of phyllosphere bacteria. Proc. Natl. Acad. Sci. U. S. A. 106:16428–16433. 10.1073/pnas.0905240106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Knief C, Frances L, Vorholt JA. 2010. Competitiveness of diverse Methylobacterium strains in the phyllosphere of Arabidopsis thaliana and identification of representative models, including M. extorquens PA1. Microb. Ecol. 60:440–452. 10.1007/s00248-010-9725-3 [DOI] [PubMed] [Google Scholar]
- 39. Kniskern JM, Traw MB, Bergelson J. 2007. Salicylic acid and jasmonic acid signaling defense pathways reduce natural bacterial diversity on Arabidopsis thaliana. Mol. Plant Microbe Interact. 20:1512–1522. 10.1094/MPMI-20-12-1512 [DOI] [PubMed] [Google Scholar]
- 40. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. 2011. Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hubbell SP. 2006. Neutral theory and the evolution of ecological equivalence. Ecology 87:1387–1398. 10.1890/0012-9658(2006)87[1387:NTATEO]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- 42. Eren AM, Maignien L, Sul WJ, Murphy LG, Grim SL, Morrison HG, Sogin ML. 2013. Oligotyping: differentiating between closely related microbial taxa using 16S rRNA gene data. Methods Ecol. Evol. 4:1111–1119. 10.1111/2041-210X.12114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vellend M. 2010. Conceptual synthesis in community ecology. Q. Rev. Biol. 85:183–206. 10.1086/652373 [DOI] [PubMed] [Google Scholar]
- 44. Hubbell SP. 2001. The unified neutral theory of biodiversity and biogeography. Monogr. Popul. Biol. 32:375 [Google Scholar]
- 45. Fukami T. 2004. Community assembly along a species pool gradient: implications for multiple-scale patterns of species diversity. Popul. Ecol. 46:137–147. 10.1007/s10144-004-0182-z [DOI] [Google Scholar]
- 46. Chase JM. 2003. Community assembly: when should history matter? Oecologia 136:489–498. 10.1007/s00442-003-1311-7 [DOI] [PubMed] [Google Scholar]
- 47. Tilman D. 2004. Niche tradeoffs, neutrality, and community structure: a stochastic theory of resource competition, invasion, and community assembly. Proc. Natl. Acad. Sci. U. S. A. 101:10854–10861. 10.1073/pnas.0403458101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Odling-Smee J, Erwin DH, Palkovacs EP, Feldman MW, Laland KN. 2013. Niche construction theory: a practical guide for ecologists. Q. Rev. Biol. 88:3–28. 10.1086/669266 [DOI] [PubMed] [Google Scholar]
- 49. McCafferty J, Mühlbauer M, Gharaibeh RZ, Arthur JC, Perez-Chanona E, Sha W, Jobin C, Fodor AA. 2013. Stochastic changes over time and not founder effects drive cage effects in microbial community assembly in a mouse model. ISME J. 7:2116–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou J, Liu W, Deng Y, Jiang YH, Xue K, He Z, Van Nostrand JD, Wu L, Yang Y, Wang A. 2013. Stochastic assembly leads to alternative communities with distinct functions in a bioreactor microbial community. mBio 4(2):e00584-12. 10.1128/mBio.00584-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Microbiome Human, Project Consortium 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hubbell SP. 2001. The unified neutral theory of biodiversity and biogeography. Princeton University Press, Princeton, NJ. [DOI] [PubMed] [Google Scholar]
- 53. Hubbell SP. 2005. Neutral theory in community ecology and the hypothesis of functional equivalence. Funct. Ecol. 19:166–172. 10.1111/j.0269-8463.2005.00965.x [DOI] [Google Scholar]
- 54. Scheffer M, van Nes EH. 2006. Self-organized similarity, the evolutionary emergence of groups of similar species. Proc. Natl. Acad. Sci. USA 103:6230–6235. 10.1073/pnas.0508024103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gravel D, Canham CD, Beaudet M, Messier C. 2006. Reconciling niche and neutrality: the continuum hypothesis. Ecol. Lett. 9:399–409. 10.1111/j.1461-0248.2006.00884.x [DOI] [PubMed] [Google Scholar]
- 56. Holt RD. 2006. Emergent neutrality. Trends Ecol. Evol. 21:531–533. 10.1016/j.tree.2006.08.003 [DOI] [PubMed] [Google Scholar]
- 57. Klappenbach JA, Dunbar JM, Schmidt TM. 2000. rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 66:1328–1333. 10.1128/AEM.66.4.1328-1333.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Loper JE, Hassan KA, Mavrodi DV, Davis EW, II, Lim CK, Shaffer BT, Elbourne LD, Stockwell VO, Hartney SL, Breakwell K, Henkels MD, Tetu SG, Rangel LI, Kidarsa TA, Wilson NL, van de Mortel JE, Song C, Blumhagen R, Radune D, Hostetler JB, Brinkac LM, Durkin AS, Kluepfel DA, Wechter WP, Anderson AJ, Kim YC, Pierson LS, III, Pierson EA, Lindow SE, Kobayashi DY, Raaijmakers JM, Weller DM, Thomashow LS, Allen AE, Paulsen IT. 2012. Comparative genomics of plant-associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS Genet. 8:e1002784. 10.1371/journal.pgen.1002784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rokhbakhsh-Zamin F, Sachdev D, Kazemi-Pour N, Engineer A, Pardesi KR, Zinjarde S, Dhakephalkar PK, Chopade BA. 2011. Characterization of plant-growth-promoting traits of Acinetobacter species isolated from rhizosphere of Pennisetum glaucum. J. Microbiol. Biotechnol. 21:556–566 [PubMed] [Google Scholar]
- 60. Wolf DC, Dao TH, Scott HD, Lavy TL. 1989. Influence of sterilization methods on selected soil microbiological, physical, and chemical properties. J. Environ. Qual. 18:39. 10.2134/jeq1989.00472425001800010007x [DOI] [Google Scholar]
- 61. Tornero P, Dangl JL. 2001. A high-throughput method for quantifying growth of phytopathogenic bacteria in Arabidopsis thaliana. Plant J. 28:475–481 [DOI] [PubMed] [Google Scholar]
- 62. Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9:671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sakai M, Matsuka A, Komura T, Kanazawa S. 2004. Application of a new PCR primer for terminal restriction fragment length polymorphism analysis of the bacterial communities in plant roots. J. Microbiol. Methods 59:81–89. 10.1016/j.mimet.2004.06.005 [DOI] [PubMed] [Google Scholar]
- 64. Filkins LM, Hampton TH., Gifford AH, Gross MJ, Hogan DA, Sogin ML, Morrison HG, Paster BJ, O'Toole GA. 2012. The prevalence of streptococci and increased polymicrobial diversity associated with cystic fibrosis patient stability. J. Bacteriol. 194:4709–4717. 10.1128/JB.00566-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ondov BD, Bergman NH, Phillippy AM. 2011. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. 10.1186/1471-2105-12-385 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Materials and Methods. Download
(A) Environmental conditions in the greenhouse during the time series, as measured with PAR and combined temperature/humidity sensors and logged on a HOBO MicroStation logger (Onset Computer). Instantaneous measurements were averaged across a 5-day sampling interval. (a) Temperature (°C). The spike in temperature from 28 October 2011 to 31 October 2011 (days 21 to 24) reflects a partial failure of the greenhouse air conditioning unit. (b) Daytime light levels; for each day, the maximum light level was recorded. The minimum value for the interval therefore indicates a day where maximum light levels were low, usually due to cloud cover. (c) Relative humidity (%). Blue, maximum reading during interval; orange, minimum reading during interval; red, average across interval. (B) Plant dry weight; error bars are the standard deviations from three replicates. Plants were dried overnight at 70°C prior to being weighed to remove Silwet/buffer remaining from the leaf wash. Day 19 data are missing. Download
Microbial population density estimated with qPCR. Estimated cell counts were obtained by applying rrn copy number correction factors to raw qPCR counts (see the text). Top, mean estimated cell count per plant; bottom, estimated microbial population density (cells/cm2) on leaves was calculated by dividing corrected cell counts by total leaf area (see the text). Error bars show the standard deviation of combined technical and biological replicates; day 19 error bar indicates only technical error (only one day 19 sample could be amplified). The spike in estimated cell counts on day 34 is unexplained but may indicate recovery from a temperature spike in the greenhouse due to air conditioning failure. Leaf area data are not available for samples prior to day 34. Download
OTU richness in airborne colonizing and leaf communities. Evolution of OTU richness in airborne and plant leaf microbial communities as a function of time. Both observed (Sobs) and estimated OTU numbers are shown. Nonparametric Chao1 and parametric estimates of richness were computed with the CatchAll software (34). Note that richness was calculated based on the uncorrected observation matrix containing singletons and doubletons, as these are critical values for (non)parametric estimation. Estimated richness in air communities oscillated between 106 and 319 OTUs. Phyllosphere richness, as estimated with the Chao index, increased from 79 OTUs at the beginning of the time series (day 19) to 474 OTUs at day 66, with much higher estimation from Catchall (840 OTUs). Download
Phylogenetically informed beta diversity analysis using unweighted and weighted UniFrac performed on trimmed v5v6 reads. (A) Unweighted UniFrac PCoA plot with points colored to show sampling day and origin (air/plant). Mature plant communities (green) are closely associated phylogenetically and are distinct from air communities (blue). Dots are scaled by sampling day (day 19, small; day 73, large). (B) Weighted UniFrac PCoA plot with points colored to show origin of plants by tray (green, tray 1; orange, tray 2; blue, air). The separation of tray communities observed in beta diversity PCoA plots using nonphylogenetic abundance metrics (Fig. 3, main text) collapses when phylogeny is considered, due to the close phylogenetic relatedness of highly abundant tray markers (Pseudomonas, tray 1; Acinetobacter, tray 2). Download
Overall taxonomic distribution in air and plant communities. Annotations of all quality trimmed reads generated with the GAST process were pooled for all plant (A) and air (B) communities. The summed counts were used to generate hierarchical Krona (65) charts showing the relative abundances of genus-level taxonomic groupings. Download
(A) LEfSe (40) biomarker analyses of mature plant (day 50+) and air communities identify clusters and higher-level taxonomic groups characteristic of each sample type that are highly statistically significant (P < 0.05). Terminal tips represent individual clusters. Phylogenetic groupings enriched in plant samples are highlighted in green (b, Microbacteriaceae; c, Nocardiaceae; f, Patulibacteraceae; g, Cytophagaceae; h, sphingobacteriaceae; i, Sphingobacteriales; j, Sphingobacteria; K, OTU_142; r, OTU_105; S, Brucellaceae; t, Rhizobiaceae; u, unassigned; y, OTU_95; a0, Comamonadaceae; a3, Xanthomonadaceae; a4, Xanthomonadales; a5, verrucomicrobiaceae; a6, Verrucomicrobiales; a7, verrucomicrobiae). Those in red are enriched in air (a, Corynebacteriaceae; d, Propionibacteriaceae; e, Streptomycetaceae; I, Alicyclobacillaceae; m, Staphylococcoceae; n, Bacillales; o, Streptococcaceae; p, Lactobacillales; q, Bacillus; v, Rhodobacteraceae; w, Rhodobacterales; x, Rickettsiales; z, Burkholderiaceae; A1, Enterobacteriaceae; a2, Enterobacteriales). (B) Biomarkers for air and mature plant communities (day 60+) identified with LEfSe software. Top, LDA scores for air biomarkers (blue); bottom, LDA scores for plant biomarkers (green). Download
Example of OTU partitioning: distribution of OTUs classified as genus Methylobacterium as a function of time in airborne communities (T0; far left), plant-associated communities in tray 1 (T1; middle), and plant-associated communities in tray 2 (T2; right). A diverse population of methylobacterial OTUs is present in air, but most are unable to grow successfully on plant leaves. Download
OTUs and higher-level taxonomic groupings identified by LeFSe as biomarkers distinguishing tray 1 and tray 2 plant-associated communities, including all sampling dates. Bars indicate the value of the LDA score, which is a measure of the strength of the biomarker identification. All biomarkers shown were significant at P values of <0.05. Green, tray 1; orange, tray 2. Download
(A) Average abundance of the top 20 OTUs (percentage of total read counts) in day 60+ mature phyllosphere communities. (B) Plant-specific biomarker OTUs identified by LEfSE (40) that become statistically significant on each sampling day. Abundance matrices were trimmed to identify later-appearing biomarkers. For example, to identify biomarkers appearing on day 55, we removed all plant and air samples prior to day 55 and ran LEfSE analysis on only day 55+ samples. The resulting set of biomarker OTUs from day 55 contained some biomarkers that first appeared earlier and some newly appearing biomarkers. The newly appearing plant-specific biomarkers are shown as “day 55” in the above table (light green). Biomarker groupings identified in this fashion are color-coded by the sampling day at which they were first identified by LEfSE.