

ABSTRACT
The causative agent of Legionnaires’ disease, Legionella pneumophila, replicates in amoebae and macrophages in a distinct membrane-bound compartment, the Legionella-containing vacuole (LCV). LCV formation is governed by the bacterial Icm/Dot type IV secretion system that translocates ~300 different “effector” proteins into host cells. Some of the translocated effectors anchor to the LCV membrane via phosphoinositide (PI) lipids. Here, we use the soil amoeba Dictyostelium discoideum, producing fluorescent PI probes, to analyze the LCV PI dynamics by live-cell imaging. Upon uptake of wild-type or Icm/Dot-deficient L. pneumophila, PtdIns(3,4,5)P3 transiently accumulated for an average of 40 s on early phagosomes, which acquired PtdIns(3)P within 1 min after uptake. Whereas phagosomes containing ΔicmT mutant bacteria remained decorated with PtdIns(3)P, more than 80% of wild-type LCVs gradually lost this PI within 2 h. The process was accompanied by a major rearrangement of PtdIns(3)P-positive membranes condensing to the cell center. PtdIns(4)P transiently localized to early phagosomes harboring wild-type or ΔicmT L. pneumophila and was cleared within minutes after uptake. During the following 2 h, PtdIns(4)P steadily accumulated only on wild-type LCVs, which maintained a discrete PtdIns(4)P identity spatially separated from calnexin-positive endoplasmic reticulum (ER) for at least 8 h. The separation of PtdIns(4)P-positive and ER membranes was even more pronounced for LCVs harboring ΔsidC-sdcA mutant bacteria defective for ER recruitment, without affecting initial bacterial replication in the pathogen vacuole. These findings elucidate the temporal and spatial dynamics of PI lipids implicated in LCV formation and provide insight into host cell membrane and effector protein interactions.
IMPORTANCE
The environmental bacterium Legionella pneumophila is the causative agent of Legionnaires’ pneumonia. The bacteria form in free-living amoebae and mammalian immune cells a replication-permissive compartment, the Legionella-containing vacuole (LCV). To subvert host cell processes, the bacteria secrete the amazing number of ~300 different proteins into host cells. Some of these proteins bind phosphoinositide (PI) lipids to decorate the LCV. PI lipids are crucial factors involved in host cell membrane dynamics and LCV formation. Using Dictyostelium amoebae producing one or two distinct fluorescent probes, we elucidated the dynamic LCV PI pattern in high temporal and spatial resolution. Notably, the endocytic PI lipid PtdIns(3)P was slowly cleared from LCVs, thus incapacitating the host cell’s digestive machinery, while PtdIns(4)P gradually accumulated on the LCV, enabling critical interactions with host organelles. The LCV PI pattern underlies the spatiotemporal configuration of bacterial effector proteins and therefore represents a crucial aspect of LCV formation.
INTRODUCTION
Intracellular pathogenic bacteria intimately interact with their eukaryotic host cells, modulate cellular physiology, and subvert signal transduction as well as vesicle trafficking pathways. The causative agent of Legionnaires’ disease, Legionella pneumophila, is a facultative intracellular bacterium that creates inside phagocytic host cells a distinct replication-permissive compartment termed the Legionella-containing vacuole (LCV) (1, 2). LCVs avoid fusion with lysosomes, yet the pathogen vacuole extensively communicates with the endosomal, secretory, and retrograde vesicle trafficking pathways, as well as with the endoplasmic reticulum (ER) (3–5). The mechanisms of phagocyte resistance, LCV formation, and intracellular replication are largely conserved among protozoan and mammalian phagocytes, and therefore, amoebae are valid model systems to study the cellular virulence of L. pneumophila (6). Over the last decade, the social soil amoeba Dictyostelium discoideum proved to be a particularly versatile model to analyze Legionella-phagocyte interactions (7–11).
The pivotal virulence factor of L. pneumophila is the Icm/Dot type IV secretion system (T4SS), which translocates ~300 so-called effector proteins into eukaryotic cells (4, 12, 13), where some of them target host components implicated in membrane dynamics. These include the small GTPases Arf1 (14), Rab1 (15–17), and Ran (18); the vacuolar H+-ATPase (19); the autophagy machinery (20); the retromer complex (21); and phosphoinositide (PI) lipids (22).
PI lipids are crucial regulators of eukaryotic signal transduction and vesicle trafficking (23, 24). Distinct PIs together with small GTPases define the organelle identity and the trafficking processes between different subcellular compartments (25). PIs are low-abundance phospholipids, composed of diacylglycerol (DAG) and d-myo-inositol 1-phosphate, i.e., phosphatidylinositol (PtdIns). The carbohydrate moiety of PI compounds can be phosphorylated and/or dephosphorylated at the positions 3′, 4′, and/or 5′ by specific PI kinases or PI phosphatases, respectively (26, 27).
The PI lipid pattern undergoes fast and complex temporal and spatial changes, which establish and maintain compartmentalization and membrane flux within the cell. Different PI lipids can be readily interconverted, such that the product of a PI-metabolizing enzyme serves as a substrate for another enzyme. The PI PtdIns(3,4,5)P3 transiently accumulates on the plasma membrane during signal transduction or phagocytosis. PtdIns(3)P is enriched on macropinosomes, phagosomes, early endosomes, and multivesicular bodies, where it is converted into PtdIns(3,5)P2. PtdIns(4)P is predominantly formed on the Golgi apparatus and secretory vesicles (23–25), as well as at the ER (28), but in addition to PtdIns(4,5)P2 also localizes to the plasma membrane. PI-interacting proteins bind their targets via distinct modules, such as PH domains [PtdIns(3,4,5)P3] or FYVE domains [PtdIns(3)P] (29). The fusion of these domains to fluorescent probes created useful tools for the analysis of cellular PI dynamics (30).
Most, if not all, intracellular bacteria target PI metabolism to ensure their survival and replication (31–34). L. pneumophila subverts the host’s PI lipids in manifold and elaborate ways (22). Several of the ~300 Icm/Dot substrates anchor to the LCV membrane by interacting with distinct PIs. PtdIns(4)P is specifically bound by the ER interactor SidC (35, 36) and with high affinity by the Rab1 guanine-nucleotide exchange factor (GEF)/AMPylase SidM (also called DrrA) (4, 5, 17, 37, 38). SidC interacts with PtdIns(4)P through a unique 20-kDa C-terminal domain termed P4CSidC, which as a green fluorescent protein (GFP)-fusion protein has been used to determine the PtdIns(4)P pattern in D. discoideum (36). Moreover, endogenous SidC exclusively and uniformly decorates LCVs (39), a feature that has been exploited to purify intact pathogen vacuoles by immunoaffinity separation (40). PtdIns(3)P is bound by the L. pneumophila retromer interactor RidL (21), the glycosyltransferase SetA (41), and the Rab activator/interactor LidA [which less strongly also binds PtdIns(4)P] (37, 42), as well as by the virulence factor LpnE (43).
L. pneumophila might modulate the LCV PI pattern directly by (Icm/Dot-) translocated PI phosphatase or kinase effector proteins or indirectly through bacterial factors recruiting host PI kinases or phosphatases (22, 44). Recently, two distinct, Icm/Dot-translocated PI phosphatases have been characterized. SidF was identified by screening L. pneumophila effectors for the “CX 5R” motif (45), a characteristic of a PI phosphatase family (46). Upon translocation, SidF localizes through two predicted transmembrane domains to the LCV membrane. Its atomic resolution structure revealed that PtdIns(3,4)P2 is bound in a positively charged groove. SidF is a PI 3-phosphatase that hydrolyzes in vitro phagosomal/endosomal PtdIns(3,4)P2 [and also PtdIns(3,4,5)P3], eventually yielding PtdIns(4)P, to which the above-mentioned effectors can anchor. The “CX 5R” motif is also present in SidP, a PI 3-phosphatase that hydrolyzes in vitro PtdIns(3)P as well as PtdIns(3,5)P2 and thus contributes to evasion of the endocytic pathway by the LCV (47). The primary sequence of SidP is not homologous to SidF, yet the high-resolution enzyme structure revealed that the catalytic domain is surprisingly similar with the exception of a lysine residue, the lack of which might account for the different substrate spectra of the two PI phosphatases.
LpnE might represent an L. pneumophila protein that recruits a host PI-metabolizing enzyme, since the virulence factor binds the PI 5-phosphatase OCRL1 (oculocerebrorenal syndrome of Lowe 1) implicated in retrograde endosome–trans-Golgi trafficking (43). OCRL1 and its Dictyostelium homologue Dd5P4 (D. discoideum 5-phosphatase 4) hydrolyze PtdIns(4,5)P2 and PtdIns(3,4,5)P3 to yield PtdIns(4)P and PtdIns(3,4)P2, respectively (48, 49). Dd5P4 restricts intracellular growth of L. pneumophila, localizes to the LCV via a 132-amino-acid N-terminal Legionella vacuole association (LVA) domain, and is catalytically active on LCVs, thus increasing the PtdIns(4)P available for binding of SidC or SidM (43).
L. pneumophila might also determine the LCV PI pattern by activating small host GTPases by bacterial guanine-nucleotide exchange factors (GEFs), which subsequently leads to the recruitment of host PI phosphatases or kinases (22, 44). Thus, the Arf1 GEF RalF (14) and the Rab1 GEF SidM (15, 16) could modulate the LCV PI pattern through the activation of the small host GTPases and the recruitment of the PI 4-kinase (PI4K) IIIβ (50) or the PI 5-phosphatase OCRL1 (51, 52), respectively. Yet, as outlined above, L. pneumophila does not (exclusively) control the decoration of LCVs with PIs by the indirect recruitment of host PI kinases or phosphatases through GEFs (22, 44). Finally, Icm/Dot-dependent uptake of L. pneumophila appears to require PI 3-kinases (PI3Ks), suggesting a PI3K-dependent macropinocytic process, rather than a phagocytic mechanism (35, 53, 54). For the sake of simplicity, the uptake of wild-type or Icm/Dot-deficient L. pneumophila will be referred to as “phagocytosis.”
In this study, we analyze the LCV PI dynamics by live-cell imaging in L. pneumophila-infected D. discoideum amoebae producing fluorescent PI probes. Our aim is to define the spatiotemporal PI pattern and to analyze the interactions of L. pneumophila effectors and host factors with these lipids.
RESULTS
Transient accumulation of PtdIns(3,4,5)P3 upon uptake of L. pneumophila.
D. discoideum stably producing GFP-fusion proteins of interest represents an ideal model phagocyte to observe the cellular dynamics of proteins and lipids in real time (55, 56). We used D. discoideum producing the probe PHCRAC-GFP (see Table S1 in the supplemental material), which labels PtdIns(3,4,5)P3 [and PtdIns(3,4)P2], to observe the real-time dynamics of these PI lipids in live cells infected with L. pneumophila. In addition to allowing a defined, time-resolved analysis of the dynamics of host cell components, live-cell imaging avoids common fixation problems, such as the destruction of epitopes and membrane architecture or the production of artifacts.
Live-cell experiments revealed that the PHCRAC-GFP probe labeled dynamic cell membrane protrusions (see Movie S1 in the supplemental material) and accumulated at the entry site of L. pneumophila (Fig. 1A). In amoebae infected with L. pneumophila, PHCRAC-GFP localized to phagocytic/macropinocytic cups and early phagosomes/macropinosomes. The probe was rapidly cleared from these membranes, indicating that PtdIns(3,4,5)P3 transiently accumulated at these sites. Prior infection with L. pneumophila apparently did not affect the transient accumulation of PtdIns(3,4,5)P3 around bacteria subsequently infecting the amoebae. Furthermore, PtdIns(4,5)P2 was excluded from membranes forming a phagosome and reappeared on the plasma membrane after fission of the phagosome (see Fig. S1A and B). Accordingly, L. pneumophila-containing phagosomes as well as maturing LCVs remained devoid of PtdIns(4,5)P2.
FIG 1 .

Transient accumulation of PtdIns(3,4,5)P3 upon uptake of L. pneumophila. (A) Localization of the PtdIns(3,4,5)P3/PtdIns(3,4)P2 probe PHCRAC-GFP in D. discoideum upon contact with L. pneumophila labeled with DsRed (pSW001). The probe (i) lines dynamic cell protrusions and phagocytic pits, (ii) localizes to internalized phagosomes, or (iii) dissociates from pathogen vacuoles. (B) Time-lapse image series for wild-type (WT) or ΔicmT L. pneumophila (pSW001) taken up by D. discoideum producing PHCRAC-GFP as a marker of PtdIns(3,4,5)P3 and PtdIns(3,4)P2. The PI probe accumulates during formation of a phagocytic cup and sealing of the phagosome harboring L. pneumophila (0 s), transiently persists on the internalized phagosome, and finally dissociates from the vacuoles. (C) Quantification of the elapsed time from phagosome formation to clearance of PtdIns(3,4,5)P3 and PtdIns(3,4)P2 from the vacuole. Data were collected for 6 replicates, and 50 events were scored for each data set (mean ± standard deviation). The zero time point was defined by the fusion of the phagosome (infected) or macropinosome (uninfected) membranes, and events were followed until the probe dissociated from the vacuoles. Bars, 5 µm.
To analyze the role of the L. pneumophila Icm/Dot T4SS in PtdIns(3,4,5)P3 dynamics during bacterial uptake, we monitored the infection of D. discoideum producing PHCRAC-GFP with either wild-type or ΔicmT L. pneumophila with a time resolution of seconds (Fig. 1B). Whereas waves of PtdIns(3,4,5)P3 were constantly produced by D. discoideum in the absence of bacteria, the PI lipid accumulated with similar kinetics at every bacterial entry site observed to result in a phagosome. Furthermore, PtdIns(3,4,5)P3 was cleared within an average of approximately 40 s (Fig. 1C), regardless of whether the pathogen vacuole contained wild-type (see Movie S2 in the supplemental material) or ΔicmT (see Movie S3) L. pneumophila. In uninfected D. discoideum, the clearance kinetics of PtdIns(3,4,5)P3 from macropinosomes was also not significantly different (see Movie S1). These results indicate that during uptake of wild-type and Icm/Dot-deficient L. pneumophila, the PtdIns(3,4,5)P3 dynamics are similar and comparable to those of uninfected amoebae. Consequently, the activation of PI 3-kinases and PI-phosphatases during L. pneumophila uptake seems to occur independently of the bacterial Icm/Dot T4SS and other bacterial products.
Rapid acquisition of PtdIns(3)P on Legionella phagosomes.
PtdIns(3)P codefines the endocytic pathway and is formed very early during endocytosis (23, 24). To analyze the dynamics of PtdIns(3)P during L. pneumophila infection, we used a D. discoideum strain producing the probe 2×FYVE-GFP that specifically recognizes PtdIns(3)P. Initial observation of L. pneumophila uptake into D. discoideum producing 2×FYVE-GFP indicated that there was a short elapsed time between phagosome closure and acquisition of PtdIns(3)P. To examine this acquisition more closely, we used the bright-field channel to determine fusion of a phagosome around L. pneumophila and measure the time until the appearance of PtdIns(3)P (Fig. 2A). Wild-type (see Movie S4 in the supplemental material) or ΔicmT mutant (see Movie S5) L. pneumophila could spend several seconds in a phagocytic cup before the projected amoeba lamellipodia fused. In the majority of cases, L. pneumophila was observed to escape the phagocyte prior to phagosome closing, indicating that formation of and entrance into the phagocytic cup were not tantamount to uptake of the bacteria (data not shown).
FIG 2 .
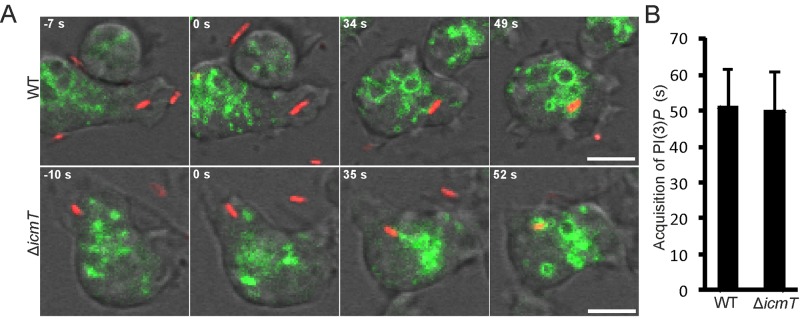
Rapid acquisition of PtdIns(3)P on L. pneumophila phagosomes. (A) Time-lapse images of PtdIns(3)P acquisition by a newly internalized Legionella-containing phagosome (wild-type or ΔicmT strain producing DsRed) in D. discoideum producing 2×FYVE-GFP. The image series shows the formation of a phagocytic cup, fusion of the membranes to seal the phagosome (t = 0 s), the internalized phagosome prior to PtdIns(3)P acquisition, and finally acquisition of PtdIns(3)P by the phagosome. (B) Quantification of PtdIns(3)P acquisition by recently internalized L. pneumophila phagosomes from the time of vacuole formation to the association of the 2×FYVE-GFP probe with the phagosome. Data were collected for 5 replicates, and a total of 40 events were scored for each strain (mean ± standard deviation). Bars, 5 µm.
Upon uptake of L. pneumophila and closure of the phagosome, the compartment was dragged inwards toward the center of the cell by the retracting phagocyte protrusions. After a short period (<30 s), the phagosomes were brought into proximity with cellular PtdIns(3)P-containing membranes and decorated with this PI lipid (see Fig. S1C and Movie S6 in the supplemental material). PtdIns(3)P was acquired by both wild-type- and ΔicmT mutant-containing phagosomes on average 50 s after uptake (Fig. 2B). Taken together, since neither the dynamics of PtdIns(3,4,5)P3 nor those of PtdIns(3)P appear to be affected by the presence of the L. pneumophila Icm/Dot T4SS during the initial minute after contact with the amoebae, the host cell and not the pathogen appears to control these early processes.
Slow loss of PtdIns(3)P on wild-type LCVs and persistence on ΔicmT mutant-containing vacuoles.
Based on the observation that after uptake wild-type and ΔicmT L. pneumophila initially acquire PtdIns(3)P to the same extent, we next followed the fate of PtdIns(3)P for a prolonged time (Fig. 3A). At 30 min postinfection (p.i.), the percentages of PtdIns(3)P-positive vacuoles harboring wild-type and ΔicmT L. pneumophila were virtually the same (approximately 90%) (Fig. 3B). However, during the 2 h following infection more than 80% of wild-type LCVs gradually and slowly lost PtdIns(3)P, with the most drastic change occurring between 30 and 60 min. A significant portion of LCVs (15 to 20%) were still decorated with PtdIns(3)P at 2 h p.i., but all wild-type LCVs eventually lost PtdIns(3)P by 8 h p.i. (see Fig. S1D in the supplemental material). Over the same period of time, the percentage of PtdIns(3)P-positive vacuoles harboring ΔicmT L. pneumophila did not significantly decrease, and most phagosomes containing the mutant bacteria remained decorated with PtdIns(3)P (Fig. 3B). In summary, while the majority of wild-type LCVs slowly and gradually lost PtdIns(3)P in the course of 2 h, most vacuoles harboring ΔicmT L. pneumophila remained decorated with the PI over the same period of time.
FIG 3 .

Slow loss of PtdIns(3)P on wild-type LCVs and persistence on ΔicmT mutant-containing vacuoles. (A) Time-lapse images showing the clearance of PtdIns(3)P from wild-type LCVs over 120 min and persistence of PtdIns(3)P on vacuoles harboring ΔicmT/pSW001 bacteria in D. discoideum producing 2×FYVE-GFP. (B) Quantification of PtdIns(3)P-positive LCVs following infection (MOI, 20) shows PtdIns(3)P clearance from wild-type but not from ΔicmT mutant LCVs. Data were collected for 4 replicates, and similar numbers of LCVs were scored for wild-type and ΔicmT mutant LCVs for each time point (mean ± standard deviation; 189 to 522 LCVs). Asterisks denote statistical significance of differences between wild-type and ΔicmT values (*, P < 0.005; **, P < 0.001). Bars, 5 µm.
Rearrangement of PtdIns(3)P-positive membranes upon L. pneumophila infection.
Next, we investigated whether the cellular architecture of PtdIns(3)P-positive membranes also underwent changes along with the observed loss of PtdIns(3)P from wild-type LCVs. To this end, we performed live-cell imaging using D. discoideum strains producing tandem 2×FYVE-GFP and either calnexin-mRFPmars or P4CSidC-mRFPmars to simultaneously label PtdIns(3)P and the ER or PtdIns(4)P, respectively.
In uninfected D. discoideum, the probe 2×FYVE-GFP labeled small clustered (endocytic) vesicles throughout the cell, as well as larger vacuoles (likely macropinosomes). Amoebae infected with the L. pneumophila ΔicmT strain were virtually indistinguishable from uninfected cells in terms of their vesicle structure (Fig. 3A). Overall, the avirulent bacteria did not cause any observable rearrangements of the host cell membrane organization. In contrast, in D. discoideum infected with wild-type L. pneumophila, the membrane architecture was notably different. PtdIns(3)P-positive membranes were condensed, and the large characteristic PtdIns(3)P-labeled endosomes/macropinosomes were no longer visible. PtdIns(3)P was removed from the replication-permissive wild-type LCV labeled by the ER marker calnexin-mRFPmars in the course of a 2-h infection (Fig. 4A; also Fig. 3). Thus, the rearrangement of PtdIns(3)P-positive compartments coincided with and followed the clearance of the endocytic PtdIns(3)P from LCVs.
FIG 4 .

Rearrangement of PtdIns(3)P-positive membranes upon L. pneumophila infection. (A) Separation of the replication-permissive, calnexin-positive (ER) compartment and the endocytic marker PtdIns(3)P in D. discoideum producing tandem calnexin-mRFPmars and 2×FYVE-GFP, infected with wild-type L. pneumophila producing GFP (pNT28). PtdIns(3)P remains distributed and vesicular in amoebae infected with ΔicmT bacteria. Images were taken 2 h p.i. Asterisks indicate bacteria of interest. (B) LCVs harboring wild-type L. pneumophila/pSW001 at 2 h p.i. in D. discoideum producing tandem P4CSidC-mRFPmars and 2×FYVE-GFP. The LCV is separate from the condensed mass of PtdIns(3)P in the center of the cell. The characteristic large endosomes/macropinosomes present in uninfected surrounding amoebae (arrows) are not visible in cells infected with wild-type bacteria (MOI, 5). (C) The integrity of large endosomes/macropinosomes was assessed and quantified from the distribution of the 2×FYVE-GFP probe and bright-field images for D. discoideum infected for 2 h with wild-type or ΔicmT L. pneumophila or uninfected amoebae producing tandem P4CSidC-mRFPmars and 2×FYVE-GFP as shown in panel B. Asterisks denote statistical significance compared to wild-type L. pneumophila (**, P < 0.001). Bars, 5 µm.
We also analyzed the distribution of PI lipids in D. discoideum using 2×FYVE-GFP and P4CSidC-mRFPmars as probes for PtdIns(3)P and the LCV marker PtdIns(4)P, respectively. Similar to the findings described above, we observed that 2 h p.i. with wild-type L. pneumophila, PtdIns(4)P-positive LCVs were entirely separated from condensed PtdIns(3)P-positive membranes (Fig. 4B). At the same time, the characteristic PtdIns(3)P-positive endosomes/macropinosomes were clearly visible in uninfected neighboring cells. Quantification of the presence of these endosomes revealed that more than 90% of uninfected or ΔicmT mutant-infected amoebae contained these structures, whereas only 10% or less of the amoebae infected with wild-type L. pneumophila retained them at 2 h p.i. (Fig. 4C). Taken together, the infection of D. discoideum with wild-type L. pneumophila triggers a major remodeling of PtdIns(3)P-positive membranes and the disintegration of the endosomal/macropinocytic network, as well as a complete separation of the endocytic PtdIns(3)P-positive compartments from the replication-permissive PtdIns(4)P-positive LCVs.
Biphasic localization of PtdIns(4)P on wild-type LCVs.
PtdIns(4)P has been previously established as an LCV lipid marker by using fixed cells and a single time point (35). Using live-cell imaging and D. discoideum amoebae stably producing the PtdIns(4)P-specific probe GFP-P4CSidC, we set out to characterize with a high temporal resolution the dynamics of PtdIns(4)P within the amoebae (see Movie S7 in the supplemental material). PtdIns(4)P is a plasma membrane marker in D. discoideum (36) and transiently localizes to phagocytic/macropinocytic membranes of uninfected amoebae for approximately 1 min after vacuole formation until dissociation (Fig. 5A; see Movie S7). Accordingly, PtdIns(4)P was present on early phagosomes containing either L. pneumophila wild-type or ΔicmT mutant bacteria (Fig. 5B). However, the pool of PtdIns(4)P initially present on Legionella vacuoles was rapidly lost, disappearing within the first minutes after uptake of the bacteria. At 5 min p.i., GFP-P4CSidC localized to the plasma membrane and some internal vesicles of amoebae infected with either L. pneumophila wild-type or ΔicmT mutant bacteria but no longer to bacterium-containing phagosomes. These results suggest that the portion of PtdIns(4)P localizing to phagosomes immediately after closure of the membrane is plasma membrane derived and not controlled by the bacterial Icm/Dot T4SS.
FIG 5 .

Biphasic localization of PtdIns(4)P on wild-type LCVs. (A) In uninfected D. discoideum producing GFP-P4CSidC, plasma membrane-derived PtdIns(4)P is initially present on an internalized macropinosome. (B) Upon contact and uptake of L. pneumophila wild-type or ΔicmT bacteria producing DsRed (pSW001), PtdIns(4)P is transiently acquired as labeled by GFP-P4CSidC. (C) Time-resolved accumulation of PtdIns(4)P by L. pneumophila wild-type but not ΔicmT (pSW001) bacteria over the first 120 min of infection (MOI, 10). (D) Quantification of PtdIns(4)P accumulation. In the course of a 120-min infection, wild-type LCVs accumulate PtdIns(4)P to a high level, while ΔicmT mutant-containing vacuoles do not. Data were collected for 3 replicates, and a total of approximately 200 vacuoles were scored per strain for each time point (mean ± standard deviation). Asterisks denote statistical significance between wild-type and ΔicmT bacterial values (**, P < 0.001). Bars, 5 µm.
Next, we analyzed the acquisition of PtdIns(4)P on Legionella vacuoles after the initial removal from phagosomes. At these later time points, PtdIns(4)P stably accumulated on wild-type LCVs (Fig. 5C). At 30 min p.i., more than 40% of LCVs were decorated with PtdIns(4)P on vacuole membranes tightly surrounding the bacteria (Fig. 5D). The number of PtdIns(4)P-positive LCVs increased 1 h or 2 h p.i. to more than 80% or 90%, respectively, and the vacuoles adopted the typical spherical shape. In parallel, the ratio of GFP-P4CSidC signal intensity on LCVs to that on the plasma membrane progressively and prominently increased, and at 2 h p.i., PtdIns(4)P seemed to preferentially localize to the pathogen vacuole (Fig. 5C). PtdIns(4)P remained on the LCV membrane for a prolonged time (18 h p.i. and beyond) and actually was present up to the exit of the bacteria from the infected cells. At these late time points, the LCV membrane was again tightly wrapped around several replicating bacteria (see Fig. S2A and B in the supplemental material). In stark contrast, PtdIns(4)P accumulation was not detectable on vacuoles containing ΔicmT mutant bacteria throughout the 2 h of infection. In summary, PtdIns(4)P localizes to LCVs in a biphasic manner. After an initial transient presence on early phagosomes presumably controlled by the host cell, PtdIns(4)P steadily and stably accumulates on LCVs in a second phase, which is specific for wild-type L. pneumophila and controlled by the Icm/Dot T4SS.
Progressive coating of PtdIns(4)P-positive LCVs with ER membranes.
In the course of their maturation, LCVs attach to and eventually fuse with the ER (57–59). PtdIns(4)P (Fig. 5D) and calnexin (36) are recruited to LCVs with similar kinetics, yielding within 2 h p.i. approximately 90% of LCVs decorated with these lipid and protein markers. In previous studies using fixed samples, the probes GFP-P4CSidC and calnexin-GFP were found to stain “tight” as well as “spacious” (spherical) LCVs in the course of an infection (36). Since fixation of a biological sample might create artifacts, reduce spatial resolution, and disrupt membranes, we sought to use live-cell microscopy and two fluorescent probes in parallel to analyze in high resolution the process of ER attachment to LCVs. To this end, we constructed a D. discoideum strain producing in tandem the PtdIns(4)P probe P4CSidC-mRFPmars and the ER probe calnexin-GFP (Fig. 6).
FIG 6 .

Progressive coating of PtdIns(4)P-positive LCVs with ER membranes. (A) D. discoideum strains producing tandem calnexin-GFP and P4CSidC-mRFPmars were infected with L. pneumophila wild type (MOI, 5) producing GFP (pNT28). The image series shows a loose layer of ER wrapping around a PtdIns(4)P-labeled LCV (1 h), an ER network in tight association with the LCV (2 h), and replicating L. pneumophila in a discrete PtdIns(4)P vacuole surrounded by membranous ER (8 h). (B) D. discoideum strains producing tandem calnexin-GFP and P4CSidC-mRFPmars were infected with L. pneumophila ΔsidC-sdcA/pNT28, a strain defective for ER acquisition by LCVs. The image series shows an LCV positive for PtdIns(4)P in the absence of ER association (1 h), partial or loose association of the ER network with LCVs (2 h), and replicating L. pneumophila in a discrete PtdIns(4)P-positive vacuole, with which the membranous ER is ultimately in tight association (8 h). Arrows and arrowheads indicate LCVs incompletely surrounded by calnexin-GFP-labeled ER or gaps between the LCV and surrounding ER network, respectively. The inset histograms in the merged images (8 h) indicate that the maximum fluorescence intensities of the GFP and mRFPmars signals around the LCV do not directly overlap. Bars, 5 µm.
The PtdIns(4)P probe P4CSidC-mRFPmars labeled spherical LCVs that were formed after 1 h and up to 8 h p.i. (Fig. 6A). At 1 h p.i., wild-type LCVs labeled by a concise ring of PtdIns(4)P were loosely surrounded by ER membranes. Thus, the acquisition of PtdIns(4)P precedes and is independent of ER attachment. The ratio of P4CSidC-mRFPmars signal intensity on LCVs versus the plasma membrane progressively increased, and at 2 h or 8 h p.i., PtdIns(4)P seemed to preferentially localize to the pathogen vacuole. These results correspond well to observations using GFP-P4CSidC (Fig. 5C).
Interestingly, however, under live-cell microscopy conditions, the probe calnexin-GFP revealed that for at least 8 h p.i. regions of the membranous ER tightly surrounded a discrete PtdIns(4)P-positive LCV membrane (Fig. 6A). Up until this point, visual observation and histogram plot data indicated that the immediate LCV membrane and the ER layers were separate resolvable entities. As a control for the accuracy of the resolved fluorescence intensity plots, we used D. discoideum producing tandem calnexin-GFP and Arf1-mRFPmars, two host cell factors which are recruited to the LCV. On LCVs produced in these amoebae, the two probes showed colocalization, confirming that the results are not an artifact of chromatic aberration (see Fig. S2C in the supplemental material). Together, these results are in agreement with the notion that early on during LCV maturation a PtdIns(4)P-positive membrane is formed, which serves as an anchoring platform for PtdIns(4)P-binding L. pneumophila effector proteins, promoting later steps of pathogen vacuole formation.
To test the hypothesis that the formation of a PtdIns(4)P-positive membrane and binding of a bacterial effector precede the recruitment of ER, we analyzed an L. pneumophila ΔsidC-sdcA mutant strain, which is defective for ER recruitment (36). The pathogen vacuoles harboring the ΔsidC-sdcA mutant strain shows the same dynamics of PtdIns(4)P (see Fig. S2D in the supplemental material) and PtdIns(3)P (data not shown) as do wild-type LCVs. Upon infection of D. discoideum producing P4CSidC-mRFPmars and calnexin-GFP with the L. pneumophila ΔsidC-sdcA strain, PtdIns(4)P-positive LCVs were formed, as occurred with wild-type bacteria. However, the recruitment of calnexin-GFP to PtdIns(4)P-positive LCVs was severely impaired, yielding obvious PtdIns(4)P-positive LCVs without indication of ER interaction (Fig. 6B). LCVs harboring the L. pneumophila ΔsidC-sdcA strain acquired calnexin-GFP-positive membranes later and to a lesser extent, as previously quantified (36). At 2 h p.i., LCVs containing mutant bacteria had recruited ER, but the interaction was still partial and loose. Moreover, on LCVs completely surrounded by the ER membrane, large gaps could be seen between the two membranes. Finally, at later time points (8 h p.i.), LCVs harboring wild-type and ΔsidC-sdcA L. pneumophila were similar, with the PtdIns(4)P-rich LCV in tight association with, yet still separated from, the surrounding ER. In summary, these findings indicate that the formation of a PtdIns(4)P-positive LCV, binding of bacterial effectors, and further interactions with cell organelles are sequential steps during LCV maturation, where the LCV remains a distinct compartment with a discrete PtdIns(4)P identity.
DISCUSSION
We analyzed the spatiotemporal LCV PI pattern to gain a better understanding of the dynamics and subcellular localization of these lipids during LCV formation and their interactions with L. pneumophila effectors and host factors. Using a real-time method and combining PI and/or host-protein probes, we were able to resolve the dynamic LCV PI pattern and shed light on the sequential assembly and dynamic maintenance of the pathogen vacuole (Fig. 7).
FIG 7 .

Schematic of PI dynamics during L. pneumophila infection in D. discoideum. L. pneumophila enters D. discoideum upon formation of a phagocytic cup rich in PtdIns(3,4,5)P3 and PtdIns(4)P. The phagosome fuses and is internalized, where PtdIns(3,4,5)P3 and PtdIns(3,4)P2, along with PtdIns(4)P, persist for less than 60 s on average. By 60 s, the phagosome has acquired a rich coat of PtdIns(3)P. From this point, the lumen of the phagosome shrinks and wild-type L. pneumophila accumulates PtdIns(4)P on the “tight” LCV. Over the course of 2 h, the concentration of PtdIns(4)P increases, and the LCV lumen expands. PtdIns(3)P is slowly lost, condensed, and excluded from the maturing LCV. The now “spacious” LCV maintains a discrete pool of PtdIns(4)P separate from the surrounding ER, which it acquired 30 to 60 min after uptake. After 8 h of infection, the bacteria have undergone a few rounds of replication. The PtdIns(4)P pattern on the LCV membrane persists and can still be discerned as separate from the surrounding ER network. Regarding the avirulent ΔicmT mutant, the PtdIns(3)P-rich phagosome shrinks and remains tightly associated with the bacterium. The endocytic marker PtdIns(3)P persists beyond 2 h, and the phagosome never acquires PtdIns(4)P. By 8 h postuptake, degradation of ΔicmT mutant bacteria has occurred.
On a time scale resolved to seconds, we demonstrated that upon uptake of L. pneumophila, PtdIns(3,4,5)P3 and PtdIns(4)P are transiently acquired by nascent phagosomes from the plasma membrane and cleared independently of the Icm/Dot T4SS within less than a minute (Fig. 1 and 5). The ensuing acquisition of PtdIns(3)P was also Icm/Dot independent, but from this point on, we observed major Icm/Dot-mediated changes: LCVs harboring wild-type L. pneumophila gradually acquired PtdIns(4)P while slowly shedding PtdIns(3)P, whereas ΔicmT mutant-containing vacuoles never acquired PtdIns(4)P and remained positive for PtdIns(3)P (Fig. 3 and 5).
PtdIns(4)P stably accumulated on LCVs harboring wild-type L. pneumophila during 2 h and up to 8 h after infection (Fig. 5 and 6). The PI was not cleared later but continued to tightly and intensely label the LCV after multiple rounds of replication at 18 h p.i. and likely persisted until bacterial exit from infected cells (see Fig. S2 in the supplemental material). PtdIns(4)P serves as an anchor for several secreted L. pneumophila effectors. The ER interactor SidC and the Rab1 GEF/AMPylase SidM specifically bind to PtdIns(4)P (35, 37). The stable accumulation of PtdIns(4)P on LCV membranes is in agreement with the finding that L. pneumophila effectors bind to this PI lipid throughout the course of infection. SidM was detected on LCV membranes up to 4 h p.i. (60), and SidC was routinely detected at 1 to 2 h p.i. but is likely present on LCVs also much beyond this time point. This notion is supported by the fact that intact LCVs can be purified by immunoaffinity separation using an anti-SidC antibody at 6 h and even at 14 h p.i. with the same efficiency as that at earlier time points (61).
The kinetics of PtdIns(4)P acquisition (Fig. 5D) and calnexin recruitment (36) are similar, yielding within 1 h and 2 h p.i. approximately 80% and 90% of LCVs which are decorated with these lipid and protein markers, respectively. However, a closer inspection revealed that the acquisition of PtdIns(4)P preceded and was independent of ER attachment (Fig. 6). LCVs labeled by a concise ring of PtdIns(4)P were initially loosely surrounded by ER membranes, which wrapped more tightly around the pathogen vacuole (but did not fuse for at least 8 h), as the infection proceeded. Furthermore, PtdIns(4)P-positive LCVs harboring ΔsidC-sdcA mutant bacteria defective for ER recruitment (36) were surrounded by ER membranes only later and less tightly. These findings are in agreement with the notion that LCVs are formed by a sequential acquisition of PtdIns(4)P, the PtdIns(4)P-binding effector SidC and ER membranes. The SidC-dependent progressive coating of PtdIns(4)P-positive LCVs with ER membranes might lead to the eventual fusion of the pathogen compartment with the ER (57–59).
PtdIns(4)P is a canonical Golgi PI lipid (23–25) but also detectable in the ER (28) and might be present in ER-derived vesicles. The subcellular distribution of PtdIns(4)P is in agreement with the notion that LCVs intercept and fuse with ER-derived vesicles in a Rab1-dependent manner to form a replication-permissive compartment (62, 63). However, in a complex and only partially understood manner, LCVs appear to communicate with and modulate not only the secretory vesicle trafficking pathway but also the endocytic (19, 40), retrograde (21, 36), and autophagy (20, 64, 65) pathways.
PtdIns(4)P-positive LCVs that were enwrapped by the ER but obviously had not yet fused with the organelle frequently contained several replicating L. pneumophila bacteria (Fig. 6). Thus, the PtdIns(4)P-positive LCV already represents a replication-permissive compartment, and the concept that the ER is a main source of nutrients for vacuolar L. pneumophila is likely incomplete (57, 59). In addition to the ER, L. pneumophila seems to acquire nutrients from the cytoplasm. This notion is supported by the finding that the LCV membrane harbors many host amino acid transporters (66) and that intracellular L. pneumophila metabolizes carbohydrates such as glucose (67), which are preferentially found in the host cytoplasm.
Given the importance of PtdIns(4)P as a defining LCV component, the PI lipid might not be produced from a unique source on the pathogen vacuole. Perhaps, PtdIns(4)P is produced on the LCV membrane by a host enzyme such as the PI 5-phosphatase OCRL1 or the PI 4-kinase PI4KIIIβ, which localizes to LCVs (43) or regulates the amount of the PtdIns(4)P-binding effector SidC (37), respectively. Additionally, the activity of the PI 3-phosphatase SidF was proposed to lead to an enrichment of PtdIns(4)P on LCVs (45). The Icm/Dot substrate SidF localizes to the LCV membrane and hydrolyzes specifically the D3 phosphate group of the phagosomal/endosomal PIs PtdIns(3,4)P2 and PtdIns(3,4,5)P3. This might either directly or through the activity of the PtdIns(4,5)P2-specific PI 5-phosphatase OCRL1 yield PtdIns(4)P. However, we observed that the rapid clearance of PtdIns(3,4,5)P3 and PtdIns(3,4)P2 after uptake of L. pneumophila proceeds independently of the Icm/Dot T4SS (Fig. 1). Therefore, it is currently unclear how SidF contributes to the production of PtdIns(4)P in the context of an L. pneumophila infection.
Phagosomes containing either wild-type or ΔicmT L. pneumophila acquired PtdIns(3)P on average 50 s after uptake (Fig. 2). While phagosomes containing Icm/Dot-deficient L. pneumophila remained decorated with PtdIns(3)P beyond 2 h postinfection, wild-type LCVs gradually cleared this PI lipid. Yet, PtdIns(3)P disappeared from wild-type LCVs rather slowly and inefficiently: at 2 h p.i., approximately 20% of the LCVs were still decorated with this PI (Fig. 3). L. pneumophila exploits PtdIns(3)P as a membrane anchor for the Icm/Dot substrates RidL (21) and SetA (41), which decorate LCVs, as well as for the virulence factor LpnE (43). Given the slow removal kinetics, PtdIns(3)P might serve as a membrane anchor for effector proteins for quite some time after the uptake of the bacteria.
PtdIns(3)P promotes the maturation of phagosomes/macropinosomes along the endocytic pathway, and the PI lipid is produced on early endosomes through phosphorylation of PtdIns by class III PI3Ks (68). Thus, the removal of PtdIns(3)P from LCV membranes contributes to the evasion of the pathogen vacuole from the bactericidal endocytic pathway. Only around 20% of wild-type LCVs are positive for PtdIns(3)P after 2 h of infection, and this PI is never observed on LCVs with replicating bacteria (see Fig. S1D in the supplemental material). PtdIns(3)P might be removed from LCVs by the PI 3-phosphatase SidP, which in vitro hydrolyzes PtdIns(3)P as well as PtdIns(3,5)P2, yielding PtdIns and PtdIns(5)P, respectively (47). In agreement with the notion that L. pneumophila interferes with endocytic maturation rather inefficiently and at a late stage, the bacteria produce at least one effector, SidK, which targets the late endosomal marker vacuolar H+-ATPase, thus preventing acidification of the pathogen vacuole (19).
Upon infection of D. discoideum with wild-type L. pneumophila, not only is PtdIns(3)P lost from LCVs but the bulk of PtdIns(3)P-positive membranes condenses and segregates in the cell center (Fig. 4). Thus, PtdIns(3)P-positive cellular compartments are physically removed from PtdIns(4)P-rich LCVs, which likely supports the evasion of the pathogen vacuole from the bactericidal endocytic pathway. Moreover, the architecture of the endosomal/macropinosomal network is destroyed. This rearrangement likely represents a further disabling of the host cell’s digestive machinery and thus facilitates the evasion of L. pneumophila from degradation.
Only seconds after closure, phagosomes containing wild-type L. pneumophila or Icm/Dot-deficient ΔicmT mutant bacteria transiently acquired PtdIns(3,4,5)P3 to the same extent and for the same period of time (Fig. 1). This result suggests that type I PI3Ks, which produce PtdIns(3,4,5)P3, are activated similarly upon infection of D. discoideum with wild-type or ΔicmT L. pneumophila. Whereas type I PI3K appears to be activated by L. pneumophila independently of a functional Icm/Dot T4SS, the requirement of PI3Ks for the uptake of either wild-type or Icm/Dot-deficient L. pneumophila is a matter of debate. Type I PI3Ks can be inhibited by the structurally unrelated inhibitors wortmannin and LY294002 (69, 70). The uptake of wild-type L. pneumophila by nonpermissive murine J774A.1 macrophages or D. discoideum was partially inhibited by wortmannin or LY294002 (54, 71). In contrast, we and other groups previously found that the uptake of L. pneumophila by replication-permissive human U937 macrophages or D. discoideum occurs in a wortmannin-insensitive and PI3K-independent manner, while the uptake of an Icm/Dot mutant was strongly reduced by PI3K inhibitors (35, 72, 73). Perhaps, the conflicting results on the role of PI3Ks during L. pneumophila uptake reflect the different (permissive or nonpermissive) phagocytes and bacterial strains used. In any case, the activation of PI3Ks during uptake of L. pneumophila indicates a macropinocytic process, rather than a phagocytic mechanism (35, 54).
Taken together, the remodeling of the LCV PI pattern has profound effects on the pathogen vacuole host proteome and the array of PI-bound bacterial effectors on LCVs. In addition to changes in the proteome and lipidome, LCVs also undergo a morphological transition from tight to spacious vacuoles, where the membrane detaches from the bacteria and stays connected only at the bacterial poles (35, 55, 74). During this time, ER membrane deposition occurs on the LCV, which retains its own separate identity, while membranes of the endocytic machinery are excluded. The temporal and spatial analysis of PI dynamics in L. pneumophila-infected D. discoideum presented here paves the way for a detailed genetic analysis of pathogen and host factors playing a role in shaping the LCV PI pattern and LCV formation.
MATERIALS AND METHODS
Culture and transformation of amoebae and bacteria.
D. discoideum axenic strains (see Table S1 in the supplemental material) were cultured in HL5 medium (ForMedium, Norfolk, United Kingdom) at 23°C in 75 cm2 culture flasks. The amoebae were cultured to subconfluence, at which point 1 × 104 to 2 × 104 cells/ml were seeded to a new flask. D. discoideum strains were transfected by electroporation (36), followed by repetition of the procedure for tandem fluorescent strains to introduce vector constructs resistant to blasticidin S (Bls). Double fluorescent cell lines were cultured under selection of 20 µg/ml Geneticin (G418) and 10 µg/ml Bls (Sigma).
L. pneumophila strains (see Table S1 in the supplemental material) were grown for 2 days on charcoal yeast extract (CYE) agar plates, buffered with N-(2-acetamido)-2-aminoethanesulfonic acid (ACES). Liquid cultures were inoculated in ACES yeast extract (AYE) medium at an optical density at 600 nm (OD600) of 0.1 and grown at 37°C for 14 to 16 h to an OD600 of 3.0 to 3.4. Cultures were checked for fitness (namely, motility and homogeneity of rods) with an inverted light microscope (40× objective).
Molecular biology.
The plasmid pRM006 (see Table S1 in the supplemental material) was constructed as follows: the full 2×FYVE tandem domain was amplified by PCR using peGFP-2×FYVE as a template (kind gift from H. Stenmark), cut with SacI/XhoI, and cloned into pSW102. To construct pWS021, the D. discoideum cnxA gene was amplified by PCR using pCaln-GFP as a template and primers containing a HindIII or a BamHI restriction site, respectively, and cloned into vector pBsrH encoding C-terminal mRFPmars. pWS022 was constructed in the same way, using the P4CSidC domain of SidC instead and vector pSU04 as a template. All PCR products were sequenced.
Real-time imaging of L. pneumophila infection of D. discoideum.
D. discoideum amoebae were harvested from approximately 70% confluent cultures. HL5 medium was removed, and cultures were washed with 5 ml LoFlo medium (ForMedium) and resuspended in fresh LoFlo medium. Cells were seeded (300 µl) at a density of 2 × 105 to 4 × 105/ml in 8-well µ-slides (Ibidi, Martinsried, Germany). Cells were allowed to adhere for 1 h, after which LoFlo medium was replaced. The microscope stage thermostat was set to regulate between 22°C and 25°C. Samples were viewed with a Leica TCS SP5 confocal microscope (HCX PL APO CS, objective 63×/1.4 to 0.60 oil; Leica Microsystems, Mannheim, Germany). For select applications, samples were observed with a Nikon Eclipse TE300 microscope with the PerkinElmer UltraVIEW spinning disk system and a Hamamatsu Orca ER camera (PerkinElmer, Cambridge, United Kingdom). A Nikon 100×/1.40 Plan Apo oil objective was used in combination with filters 488–10BP/525–50BP and 568–10BP/607–45BP. Data evaluation was carried out with Velocity 6.0.1 (PerkinElmer).
To monitor rapid events, the amoebae were brought into focus on the microscope stage. Up to 5 µl of an L. pneumophila culture 10-fold diluted in LoFlo medium was introduced by submerging a pipette tip directly above the objective position. Video capture was initiated immediately as motile L. pneumophila arrived at the focal plane within seconds of addition. The focus was maintained manually, and events were followed for up to a maximum of 15 min before moving to a fresh well to repeat the process.
To monitor slow (long-term) events, a bacterial multiplicity of infection (MOI) between 5 and 20 in 300 µl LoFlo medium was used. LoFlo in the observation dishes was replaced by 300 µl LoFlo containing L. pneumophila (t = 0). Dishes were immediately centrifuged (1,000 × g, 5 min), and the cells were brought into focus on the microscope stage. Three representative images for each strain were captured at defined time points. As images cannot be captured simultaneously, image acquisition was staggered so that capture was half completed when the specific time point was reached.
Statistical methods.
To compare potential Icm/Dot-dependent differences between L. pneumophila strains or uninfected amoebae, the two-sample t test was applied, assuming unequal variance.
SUPPLEMENTAL MATERIAL
Exclusion of PtdIns(4,5)P2 from the L. pneumophila entry site and long-term loss of PtdIns(3)P on LCVs. (A) Formation of a vacuole (macropinosome) by D. discoideum producing the PtdIns(4,5)P2 probe PHPLCδ1-GFP. The probe labels the resting plasma membrane but dissociates at dynamic membrane sites and does not label the internalized macropinosome (asterisk). After macropinosome internalization, the probe relocalizes to the resting plasma membrane. (B) LCV containing wild-type L. pneumophila producing DsRed (pSW001) does not accumulate PtdIns(4,5)P2 (2 h p.i.). (C) Acquisition of PtdIns(3)P by the internalized phagosome, resolved for vesicle structure (see also Movie S6). Still images show the newly internalized wild-type L. pneumophila-containing phagosome, the vacuole coming into proximity with the PtdIns(3)P pool, and acquiring PtdIns(3)P, followed by intensification and persistence of the signal. (D) Replication of L. pneumophila/pSW001 at 8 h p.i. (MOI, 10) in D. discoideum producing 2×FYVE-GFP. The endocytic lipid marker PtdIns(3)P is not visible on any wild-type LCV containing replicating bacteria. There is also a general decrease in the PtdIns(3)P pool. Cells infected with ΔicmT bacteria show normal morphology and PtdIns(3)P labeling of endosomes. At this time, many of the avirulent bacteria have been digested. Bars, 5 µm. Download
Long-term accumulation of PtdIns(4)P and colocalization of Arf1-mRFPmars with calnexin-GFP on LCVs. (A) P4CSidC-mRFPmars produced in D. discoideum infected with wild-type L. pneumophila/pSW001 tightly labels LCVs at 18 h p.i. (B) Three-dimensional representation of the PtdIns(4)P pattern and the LCV structure for D. discoideum producing GFP-P4CSidC infected with wild-type L. pneumophila at 18 h p.i. (C) Arf1-mRFPmars and calnexin-GFP coproduced in D. discoideum colocalize on LCVs containing L. pneumophila wild-type producing GFP (pNT28) at 2 h p.i. The histogram indicates that the fluorescence intensity maxima overlap. This overlap is visualized as a yellow ring around the LCV. (D) Accumulation of PtdIns(4)P by L. pneumophila wild-type and ΔsidC-sdcA bacteria producing DsRed (pSW001) over the first 120 min of infection (MOI, 10). At 5 min (data not shown), 60 min, and 120 min after infection, no differences were observed between strains. Data were collected for 2 replicates, and a total of approximately 100 vacuoles were scored per strain for each time point (mean ± standard deviation). Bars, 5 µm. Download
The PHCRAC-GFP probe labels dynamic membranes and macropinosomes in D. discoideum. Download
Localization of PtdIns(3,4,5)P in D. discoideum producing PHCRAC-GFP infected with the L. pneumophila wild type. Download
Localization of PtdIns(3,4,5)P in D. discoideum producing PHCRAC-GFP infected with the L. pneumophila ΔicmT mutant. Download
Acquisition of PtdIns(3)P in D. discoideum producing 2×FYVE-GFP infected with the L. pneumophila wild type. Download
Acquisition of PtdIns(3)P in D. discoideum producing 2×FYVE-GFP infected with the L. pneumophila ΔicmT mutant. Download
Acquisition of PtdIns(3)P by Legionella-containing phagosomes and vacuole architecture in D. discoideum producing 2×FYVE-GFP. Download
Plasma membrane-derived PtdIns(4)P is rapidly removed from macropinosomes in D. discoideum producing GFP-P4CSidC. Download
Strains and plasmids.
ACKNOWLEDGMENTS
We thank Roger Meier for constructing plasmid pRM006, Annette Müller-Taubenberger (University of Munich) for the mRFPmars vector, and Harald Stenmark (Oslo University Hospital) for peGFP-2×FYVE.
The work in the group of H.H. was funded by the Max von Pettenkofer-Institute, Ludwig-Maximilians University Munich, and the German Research Foundation (DFG; HI 1511/1-1, SFB914, and SPP1580). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Citation Weber S, Wagner M, Hilbi H. 2014. Live-cell imaging of phosphoinositide dynamics and membrane architecture during Legionella infection. mBio 5(1):e00839-13. doi:10.1128/mBio.00839-13.
REFERENCES
- 1. Newton HJ, Ang DK, van Driel IR, Hartland EL. 2010. Molecular pathogenesis of infections caused by Legionella pneumophila. Clin. Microbiol. Rev. 23:274–298. 10.1128/CMR.00052-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hilbi H, Hoffmann C, Harrison CF. 2011. Legionella spp. outdoors: colonization, communication and persistence. Environ. Microbiol. Rep. 3:286–296. 10.1111/j.1758-2229.2011.00247.x [DOI] [PubMed] [Google Scholar]
- 3. Isberg RR, O’Connor TJ, Heidtman M. 2009. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7:13–24. 10.1038/nrmicro1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hubber A, Roy CR. 2010. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26:261–283. 10.1146/annurev-cellbio-100109-104034 [DOI] [PubMed] [Google Scholar]
- 5. Hilbi H, Haas A. 2012. Secretive bacterial pathogens and the secretory pathway. Traffic 13:1187–1197. 10.1111/j.1600-0854.2012.01344.x [DOI] [PubMed] [Google Scholar]
- 6. Hoffmann C, Harrison CF, Hilbi H. 2014. The natural alternative: protozoa as cellular models for Legionella infection. Cell. Microbiol. 16:15–26. 10.1111/cmi.12235 [DOI] [PubMed] [Google Scholar]
- 7. Solomon JM, Isberg RR. 2000. Growth of Legionella pneumophila in Dictyostelium discoideum: a novel system for genetic analysis of host-pathogen interactions. Trends Microbiol. 8:478–480. 10.1016/S0966-842X(00)01852-7 [DOI] [PubMed] [Google Scholar]
- 8. Steinert M, Heuner K. 2005. Dictyostelium as host model for pathogenesis. Cell. Microbiol. 7:307–314. 10.1111/j.1462-5822.2005.00493.x [DOI] [PubMed] [Google Scholar]
- 9. Hilbi H, Weber SS, Ragaz C, Nyfeler Y, Urwyler S. 2007. Environmental predators as models for bacterial pathogenesis. Environ. Microbiol. 9:563–575. 10.1111/j.1462-2920.2007.01238.x [DOI] [PubMed] [Google Scholar]
- 10. Cosson P, Soldati T. 2008. Eat, kill or die: when amoeba meets bacteria. Curr. Opin. Microbiol. 11:271–276. 10.1016/j.mib.2008.05.005 [DOI] [PubMed] [Google Scholar]
- 11. Clarke M. 2010. Recent insights into host-pathogen interactions from Dictyostelium. Cell. Microbiol. 12:283–291. 10.1111/j.1462-5822.2009.01413.x [DOI] [PubMed] [Google Scholar]
- 12. Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, Luo ZQ. 2011. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One 6:e17638. 10.1371/journal.pone.0017638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lifshitz Z, Burstein D, Peeri M, Zusman T, Schwartz K, Shuman HA, Pupko T, Segal G. 2013. Computational modeling and experimental validation of the Legionella and Coxiella virulence-related type-IVB secretion signal. Proc. Natl. Acad. Sci. U. S. A. 110:E707–E715. 10.1073/pnas.1215854110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR. 2002. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 295:679–682. 10.1126/science.1067025 [DOI] [PubMed] [Google Scholar]
- 15. Murata T, Delprato A, Ingmundson A, Toomre DK, Lambright DG, Roy CR. 2006. The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat. Cell Biol. 8:971–977. 10.1038/ncb1463 [DOI] [PubMed] [Google Scholar]
- 16. Machner MP, Isberg RR. 2006. Targeting of host Rab GTPase function by the intravacuolar pathogen Legionella pneumophila. Dev. Cell 11:47–56. 10.1016/j.devcel.2006.05.013 [DOI] [PubMed] [Google Scholar]
- 17. Itzen A, Goody RS. 2011. Covalent coercion by Legionella pneumophila. Cell Host Microbe 10:89–91. 10.1016/j.chom.2011.08.002 [DOI] [PubMed] [Google Scholar]
- 18. Rothmeier E, Pfaffinger G, Hoffmann C, Harrison CF, Grabmayr H, Repnik U, Hannemann M, Wölke S, Bausch A, Griffiths G, Müller-Taubenberger A, Itzen A, Hilbi H. 2013. Activation of Ran GTPase by a Legionella effector promotes microtubule polymerization, pathogen vacuole motility and infection. PLoS Pathog. 9:e1003598. 10.1371/journal.ppat.1003598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu L, Shen X, Bryan A, Banga S, Swanson MS, Luo ZQ. 2010. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog. 6:e1000822. 10.1371/journal.ppat.1000822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Choy A, Dancourt J, Mugo B, O’Connor TJ, Isberg RR, Melia TJ, Roy CR. 2012. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 338:1072–1076. 10.1126/science.1227026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Finsel I, Ragaz C, Hoffmann C, Harrison CF, Weber S, van Rahden VA, Johannes L, Hilbi H. 2013. The Legionella effector RidL inhibits retrograde trafficking to promote intracellular replication. Cell Host Microbe 14:38–50. 10.1016/j.chom.2013.06.001 [DOI] [PubMed] [Google Scholar]
- 22. Hilbi H, Weber S, Finsel I. 2011. Anchors for effectors: subversion of phosphoinositide lipids by Legionella. Front. Microbiol. 2:91. 10.3389/fmicb.2011.00091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Di Paolo G, De Camilli P. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–657. 10.1038/nature05185 [DOI] [PubMed] [Google Scholar]
- 24. Michell RH. 2008. Inositol derivatives: evolution and functions. Nat. Rev. Mol. Cell Biol. 9:151–161. 10.1038/nrm2334 [DOI] [PubMed] [Google Scholar]
- 25. Behnia R, Munro S. 2005. Organelle identity and the signposts for membrane traffic. Nature 438:597–604. 10.1038/nature04397 [DOI] [PubMed] [Google Scholar]
- 26. De Matteis MA, Godi A. 2004. PI-loting membrane traffic. Nat. Cell Biol. 6:487–492. 10.1038/ncb0604-487 [DOI] [PubMed] [Google Scholar]
- 27. Sasaki T, Takasuga S, Sasaki J, Kofuji S, Eguchi S, Yamazaki M, Suzuki A. 2009. Mammalian phosphoinositide kinases and phosphatases. Prog. Lipid Res. 48:307–343. 10.1016/j.plipres.2009.06.001 [DOI] [PubMed] [Google Scholar]
- 28. Blumental-Perry A, Haney CJ, Weixel KM, Watkins SC, Weisz OA, Aridor M. 2006. Phosphatidylinositol 4-phosphate formation at ER exit sites regulates ER export. Dev. Cell 11:671–682. 10.1016/j.devcel.2006.09.001 [DOI] [PubMed] [Google Scholar]
- 29. Lemmon MA. 2008. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9:99–111. 10.1038/nrm2328 [DOI] [PubMed] [Google Scholar]
- 30. Várnai P, Balla T. 2006. Live cell imaging of phosphoinositide dynamics with fluorescent protein domains. Biochim. Biophys. Acta 1761:957–967. 10.1016/j.bbalip.2006.03.019 [DOI] [PubMed] [Google Scholar]
- 31. Pizarro-Cerdá J, Cossart P. 2004. Subversion of phosphoinositide metabolism by intracellular bacterial pathogens. Nat. Cell Biol. 6:1026–1033. 10.1038/ncb1104-1026 [DOI] [PubMed] [Google Scholar]
- 32. Hilbi H. 2006. Modulation of phosphoinositide metabolism by pathogenic bacteria. Cell. Microbiol. 8:1697–1706. 10.1111/j.1462-5822.2006.00793.x [DOI] [PubMed] [Google Scholar]
- 33. Weber SS, Ragaz C, Hilbi H. 2009. Pathogen trafficking pathways and host phosphoinositide metabolism. Mol. Microbiol. 71:1341–1352. 10.1111/j.1365-2958.2009.06608.x [DOI] [PubMed] [Google Scholar]
- 34. Ham H, Sreelatha A, Orth K. 2011. Manipulation of host membranes by bacterial effectors. Nat. Rev. Microbiol. 9:635–646. 10.1038/nrmicro2602 [DOI] [PubMed] [Google Scholar]
- 35. Weber SS, Ragaz C, Reus K, Nyfeler Y, Hilbi H. 2006. Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS Pathog. 2:e46. 10.1371/journal.ppat.0020046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ragaz C, Pietsch H, Urwyler S, Tiaden A, Weber SS, Hilbi H. 2008. The Legionella pneumophila phosphatidylinositol-4 phosphate-binding type IV substrate SidC recruits endoplasmic reticulum vesicles to a replication-permissive vacuole. Cell. Microbiol. 10:2416–2433. 10.1111/j.1462-5822.2008.01219.x [DOI] [PubMed] [Google Scholar]
- 37. Brombacher E, Urwyler S, Ragaz C, Weber SS, Kami K, Overduin M, Hilbi H. 2009. Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J. Biol. Chem. 284:4846–4856. 10.1074/jbc.M807505200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schoebel S, Blankenfeldt W, Goody RS, Itzen A. 2010. High-affinity binding of phosphatidylinositol 4-phosphate by Legionella pneumophila DrrA. EMBO Rep. 11:598–604. 10.1038/embor.2010.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luo ZQ, Isberg RR. 2004. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc. Natl. Acad. Sci. U. S. A. 101:841–846. 10.1073/pnas.0304916101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Urwyler S, Nyfeler Y, Ragaz C, Lee H, Mueller LN, Aebersold R, Hilbi H. 2009. Proteome analysis of Legionella vacuoles purified by magnetic immunoseparation reveals secretory and endosomal GTPases. Traffic 10:76–87. 10.1111/j.1600-0854.2008.00851.x [DOI] [PubMed] [Google Scholar]
- 41. Jank T, Böhmer KE, Tzivelekidis T, Schwan C, Belyi Y, Aktories K. 2012. Domain organization of Legionella effector SetA. Cell. Microbiol. 14:852–868. 10.1111/j.1462-5822.2012.01761.x [DOI] [PubMed] [Google Scholar]
- 42. Neunuebel MR, Mohammadi S, Jarnik M, Machner MP. 2012. Legionella pneumophila LidA affects nucleotide binding and activity of the host GTPase Rab1. J. Bacteriol. 194:1389–1400. 10.1128/JB.06306-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weber SS, Ragaz C, Hilbi H. 2009. The inositol polyphosphate 5-phosphatase OCRL1 restricts intracellular growth of Legionella, localizes to the replicative vacuole and binds to the bacterial effector LpnE. Cell. Microbiol. 11:442–460. 10.1111/j.1462-5822.2008.01266.x [DOI] [PubMed] [Google Scholar]
- 44. Haneburger I, Hilbi H. 2013. Phosphoinositide lipids and the Legionella pathogen vacuole. Curr. Top. Microbiol. Immunol. 376:155–173. 10.1007/82_2013_341 [DOI] [PubMed] [Google Scholar]
- 45. Hsu F, Zhu W, Brennan L, Tao L, Luo ZQ, Mao Y. 2012. Structural basis for substrate recognition by a unique Legionella phosphoinositide phosphatase. Proc. Natl. Acad. Sci. U. S. A. 109:13567–13572. 10.1073/pnas.1207903109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Norris FA, Wilson MP, Wallis TS, Galyov EE, Majerus PW. 1998. SopB, a protein required for virulence of Salmonella dublin, is an inositol phosphate phosphatase. Proc. Natl. Acad. Sci. U. S. A. 95:14057–14059. 10.1073/pnas.95.24.14057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Toulabi L, Wu X, Cheng Y, Mao Y. 2013. Identification and structural characterization of a Legionella phosphoinositide phosphatase. J. Biol. Chem. 288:24518–24527. 10.1074/jbc.M113.474239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Loovers HM, Kortholt A, de Groote H, Whitty L, Nussbaum RL, van Haastert PJ. 2007. Regulation of phagocytosis in Dictyostelium by the inositol 5-phosphatase OCRL homolog Dd5P4. Traffic 8:618–628. 10.1111/j.1600-0854.2007.00546.x [DOI] [PubMed] [Google Scholar]
- 49. Zhang X, Jefferson AB, Auethavekiat V, Majerus PW. 1995. The protein deficient in Lowe syndrome is a phosphatidylinositol-4,5-bisphosphate 5-phosphatase. Proc. Natl. Acad. Sci. U. S. A. 92:4853–4856. 10.1073/pnas.92.11.4853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Godi A, Pertile P, Meyers R, Marra P, Di Tullio G, Iurisci C, Luini A, Corda D, De Matteis MA. 1999. ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat. Cell Biol. 1:280–287. 10.1038/12993 [DOI] [PubMed] [Google Scholar]
- 51. Lichter-Konecki U, Farber LW, Cronin JS, Suchy SF, Nussbaum RL. 2006. The effect of missense mutations in the RhoGAP-homology domain on ocrl1 function. Mol. Genet. Metab. 89:121–128. 10.1016/j.ymgme.2006.04.005 [DOI] [PubMed] [Google Scholar]
- 52. Hyvola N, Diao A, McKenzie E, Skippen A, Cockcroft S, Lowe M. 2006. Membrane targeting and activation of the Lowe syndrome protein OCRL1 by rab GTPases. EMBO J. 25:3750–3761. 10.1038/sj.emboj.7601274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hilbi H, Segal G, Shuman HA. 2001. Icm/Dot-dependent upregulation of phagocytosis by Legionella pneumophila. Mol. Microbiol. 42:603–617. 10.1046/j.1365-2958.2001.02645.x [DOI] [PubMed] [Google Scholar]
- 54. Peracino B, Balest A, Bozzaro S. 2010. Phosphoinositides differentially regulate bacterial uptake and Nramp1-induced resistance to Legionella infection in Dictyostelium. J. Cell Sci. 123:4039–4051. 10.1242/jcs.072124 [DOI] [PubMed] [Google Scholar]
- 55. Lu H, Clarke M. 2005. Dynamic properties of Legionella-containing phagosomes in Dictyostelium amoebae. Cell. Microbiol. 7:995–1007. 10.1111/j.1462-5822.2005.00528.x [DOI] [PubMed] [Google Scholar]
- 56. Gerisch G, Müller-Taubenberger A. 2003. GFP-fusion proteins as fluorescent reporters to study organelle and cytoskeleton dynamics in chemotaxis and phagocytosis. Methods Enzymol. 361:320–337. 10.1016/S0076-6879(03)61017-7 [DOI] [PubMed] [Google Scholar]
- 57. Swanson MS, Isberg RR. 1995. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect. Immun. 63:3609–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tilney LG, Harb OS, Connelly PS, Robinson CG, Roy CR. 2001. How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J. Cell Sci. 114:4637–4650 [DOI] [PubMed] [Google Scholar]
- 59. Robinson CG, Roy CR. 2006. Attachment and fusion of endoplasmic reticulum with vacuoles containing Legionella pneumophila. Cell. Microbiol. 8:793–805. 10.1111/j.1462-5822.2005.00666.x [DOI] [PubMed] [Google Scholar]
- 60. Ingmundson A, Delprato A, Lambright DG, Roy CR. 2007. Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature 450:365–369. 10.1038/nature06336 [DOI] [PubMed] [Google Scholar]
- 61. Urwyler S, Finsel I, Ragaz C, Hilbi H. 2010. Isolation of Legionella-containing vacuoles by immuno-magnetic separation. Curr. Protoc. Cell Biol. Chapter 3:Unit 3.34. 10.1002/0471143030.cb0334s46 [DOI] [PubMed] [Google Scholar]
- 62. Kagan JC, Roy CR. 2002. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat. Cell Biol. 4:945–954. 10.1038/ncb883 [DOI] [PubMed] [Google Scholar]
- 63. Kagan JC, Stein MP, Pypaert M, Roy CR. 2004. Legionella subvert the functions of Rab1 and Sec22b to create a replicative organelle. J. Exp. Med. 199:1201–1211. 10.1084/jem.20031706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Otto GP, Wu MY, Clarke M, Lu H, Anderson OR, Hilbi H, Shuman HA, Kessin RH. 2004. Macroautophagy is dispensable for intracellular replication of Legionella pneumophila in Dictyostelium discoideum. Mol. Microbiol. 51:63–72. 10.1046/j.1365-2958.2003.03826.x [DOI] [PubMed] [Google Scholar]
- 65. Tung SM, Unal C, Ley A, Peña C, Tunggal B, Noegel AA, Krut O, Steinert M, Eichinger L. 2010. Loss of Dictyostelium ATG9 results in a pleiotropic phenotype affecting growth, development, phagocytosis and clearance and replication of Legionella pneumophila. Cell. Microbiol. 12:765–780. 10.1111/j.1462-5822.2010.01432.x [DOI] [PubMed] [Google Scholar]
- 66. Wieland H, Ullrich S, Lang F, Neumeister B. 2005. Intracellular multiplication of Legionella pneumophila depends on host cell amino acid transporter SLC1A5. Mol. Microbiol. 55:1528–1537. 10.1111/j.1365-2958.2005.04490.x [DOI] [PubMed] [Google Scholar]
- 67. Heuner K, Eisenreich W. 2013. The intracellular metabolism of Legionella by isotopologue profiling. Methods Mol. Biol. 954:163–181. 10.1007/978-1-62703-161-5_8 [DOI] [PubMed] [Google Scholar]
- 68. Steinberg BE, Grinstein S. 2008. Pathogen destruction versus intracellular survival: the role of lipids as phagosomal fate determinants. J. Clin. Invest. 118:2002–2011. 10.1172/JCI35433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Araki N, Johnson MT, Swanson JA. 1996. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J. Cell Biol. 135:1249–1260. 10.1083/jcb.135.5.1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cox D, Tseng CC, Bjekic G, Greenberg S. 1999. A requirement for phosphatidylinositol 3-kinase in pseudopod extension. J. Biol. Chem. 274:1240–1247. 10.1074/jbc.274.3.1240 [DOI] [PubMed] [Google Scholar]
- 71. Tachado SD, Samrakandi MM, Cirillo JD. 2008. Non-opsonic phagocytosis of Legionella pneumophila by macrophages is mediated by phosphatidylinositol 3-kinase. PLoS One 3:e3324. 10.1371/journal.pone.0003324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Khelef N, Shuman HA, Maxfield FR. 2001. Phagocytosis of wild-type Legionella pneumophila occurs through a wortmannin-insensitive pathway. Infect. Immun. 69:5157–5161. 10.1128/IAI.69.8.5157-5161.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Harada T, Tanikawa T, Iwasaki Y, Yamada M, Imai Y, Miyake M. 2012. Phagocytic entry of Legionella pneumophila into macrophages through phosphatidylinositol 3,4,5-trisphosphate-independent pathway. Biol. Pharm. Bull. 35:1460–1468. 10.1248/bpb.b11-00011 [DOI] [PubMed] [Google Scholar]
- 74. Li Z, Solomon JM, Isberg RR. 2005. Dictyostelium discoideum strains lacking the RtoA protein are defective for maturation of the Legionella pneumophila replication vacuole. Cell. Microbiol. 7:431–442. 10.1111/j.1462-5822.2004.00472.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Exclusion of PtdIns(4,5)P2 from the L. pneumophila entry site and long-term loss of PtdIns(3)P on LCVs. (A) Formation of a vacuole (macropinosome) by D. discoideum producing the PtdIns(4,5)P2 probe PHPLCδ1-GFP. The probe labels the resting plasma membrane but dissociates at dynamic membrane sites and does not label the internalized macropinosome (asterisk). After macropinosome internalization, the probe relocalizes to the resting plasma membrane. (B) LCV containing wild-type L. pneumophila producing DsRed (pSW001) does not accumulate PtdIns(4,5)P2 (2 h p.i.). (C) Acquisition of PtdIns(3)P by the internalized phagosome, resolved for vesicle structure (see also Movie S6). Still images show the newly internalized wild-type L. pneumophila-containing phagosome, the vacuole coming into proximity with the PtdIns(3)P pool, and acquiring PtdIns(3)P, followed by intensification and persistence of the signal. (D) Replication of L. pneumophila/pSW001 at 8 h p.i. (MOI, 10) in D. discoideum producing 2×FYVE-GFP. The endocytic lipid marker PtdIns(3)P is not visible on any wild-type LCV containing replicating bacteria. There is also a general decrease in the PtdIns(3)P pool. Cells infected with ΔicmT bacteria show normal morphology and PtdIns(3)P labeling of endosomes. At this time, many of the avirulent bacteria have been digested. Bars, 5 µm. Download
Long-term accumulation of PtdIns(4)P and colocalization of Arf1-mRFPmars with calnexin-GFP on LCVs. (A) P4CSidC-mRFPmars produced in D. discoideum infected with wild-type L. pneumophila/pSW001 tightly labels LCVs at 18 h p.i. (B) Three-dimensional representation of the PtdIns(4)P pattern and the LCV structure for D. discoideum producing GFP-P4CSidC infected with wild-type L. pneumophila at 18 h p.i. (C) Arf1-mRFPmars and calnexin-GFP coproduced in D. discoideum colocalize on LCVs containing L. pneumophila wild-type producing GFP (pNT28) at 2 h p.i. The histogram indicates that the fluorescence intensity maxima overlap. This overlap is visualized as a yellow ring around the LCV. (D) Accumulation of PtdIns(4)P by L. pneumophila wild-type and ΔsidC-sdcA bacteria producing DsRed (pSW001) over the first 120 min of infection (MOI, 10). At 5 min (data not shown), 60 min, and 120 min after infection, no differences were observed between strains. Data were collected for 2 replicates, and a total of approximately 100 vacuoles were scored per strain for each time point (mean ± standard deviation). Bars, 5 µm. Download
The PHCRAC-GFP probe labels dynamic membranes and macropinosomes in D. discoideum. Download
Localization of PtdIns(3,4,5)P in D. discoideum producing PHCRAC-GFP infected with the L. pneumophila wild type. Download
Localization of PtdIns(3,4,5)P in D. discoideum producing PHCRAC-GFP infected with the L. pneumophila ΔicmT mutant. Download
Acquisition of PtdIns(3)P in D. discoideum producing 2×FYVE-GFP infected with the L. pneumophila wild type. Download
Acquisition of PtdIns(3)P in D. discoideum producing 2×FYVE-GFP infected with the L. pneumophila ΔicmT mutant. Download
Acquisition of PtdIns(3)P by Legionella-containing phagosomes and vacuole architecture in D. discoideum producing 2×FYVE-GFP. Download
Plasma membrane-derived PtdIns(4)P is rapidly removed from macropinosomes in D. discoideum producing GFP-P4CSidC. Download
Strains and plasmids.