

ABSTRACT
CRISPR (clustered regularly interspaced short palindromic repeats)-Cas (CRISPR-associated) systems in bacteria and archaea employ CRISPR RNAs to specifically recognize the complementary DNA of foreign invaders, leading to sequence-specific cleavage or degradation of the target DNA. Recent work has shown that the accidental or intentional targeting of the bacterial genome is cytotoxic and can lead to cell death. Here, we have demonstrated that genome targeting with CRISPR-Cas systems can be employed for the sequence-specific and titratable removal of individual bacterial strains and species. Using the type I-E CRISPR-Cas system in Escherichia coli as a model, we found that this effect could be elicited using native or imported systems and was similarly potent regardless of the genomic location, strand, or transcriptional activity of the target sequence. Furthermore, the specificity of targeting with CRISPR RNAs could readily distinguish between even highly similar strains in pure or mixed cultures. Finally, varying the collection of delivered CRISPR RNAs could quantitatively control the relative number of individual strains within a mixed culture. Critically, the observed selectivity and programmability of bacterial removal would be virtually impossible with traditional antibiotics, bacteriophages, selectable markers, or tailored growth conditions. Once delivery challenges are addressed, we envision that this approach could offer a novel means to quantitatively control the composition of environmental and industrial microbial consortia and may open new avenues for the development of “smart” antibiotics that circumvent multidrug resistance and differentiate between pathogenic and beneficial microorganisms.
IMPORTANCE
Controlling the composition of microbial populations is a critical aspect in medicine, biotechnology, and environmental cycles. While different antimicrobial strategies, such as antibiotics, antimicrobial peptides, and lytic bacteriophages, offer partial solutions, what remains elusive is a generalized and programmable strategy that can distinguish between even closely related microorganisms and that allows for fine control over the composition of a microbial population. This study demonstrates that RNA-directed immune systems in bacteria and archaea called CRISPR-Cas systems can provide such a strategy. These systems can be employed to selectively and quantitatively remove individual bacterial strains based purely on sequence information, creating opportunities in the treatment of multidrug-resistant infections, the control of industrial fermentations, and the study of microbial consortia.
INTRODUCTION
Microorganisms play critical roles in human health and environmental nutrient cycles and are regularly employed in diverse industrial processes. Within each context, a central challenge is controlling the specific composition of a mixed population. A few strategies can remove some microorganisms but not others (Fig. 1): defined growth conditions, conventional antibiotics, and antimicrobial peptides with some strain specificity, lytic bacteriophages, and the expression of antibiotic resistance genes, auxotrophic markers, or toxins under unique expression systems (1). Unfortunately, most of these approaches offer constrained opportunities to selectively remove individual bacterial strains (e.g., antibiotics and antimicrobial peptides) or require detailed knowledge of the genetics, metabolism, and physiology of each constituent of the population (e.g., selective growth conditions). Lytic bacteriophages often offer exquisite specificity (2). However, individual bacteriophages must be isolated against each strain and would require additional screening to determine the degree of specificity. Furthermore, lytic bacteriophages replicate as part of the infection cycle, eventually wiping out the entire target population or breeding resistance. What remains elusive is a generalized and programmable strategy that can distinguish between even closely related microorganisms and that allows for fine control over the composition of a microbial population. We proposed that CRISPR-Cas systems could provide such a strategy.
FIG 1 .
Selective removal of individual bacterial strains. Approaches are needed that can selectively remove individual constituents (green) but not others within a diverse microbial population.
CRISPR-Cas systems are RNA-directed adaptive immune systems in many bacteria and most archaea that recognize nucleic acids of invading plasmids and viruses (3, 4). Recognition is directed by CRISPR RNAs that are processed from transcribed arrays of alternating target-specific “spacer” sequences and identical “repeat” sequences. The spacer region of each CRISPR RNA base pairs with complementary nucleic acids, driving cleavage or degradation by the Cas proteins within minutes of invasion (5–7).
Three types of CRISPR-Cas systems, which vary in their specific target and mechanism of action, have been defined. Type I systems cleave and degrade DNA, type II systems cleave DNA, and type III systems cleave DNA or RNA (8). Type I and II systems require two principal factors to effectively target DNA: (i) complementarity between the CRISPR RNA spacer and the target “protospacer” sequence and (ii) a protospacer-adjacent motif (PAM) specific to each CRISPR-Cas system flanking the protospacer (9–11). Effective targeting can occur even for multiple mismatches between the CRISPR RNA and the protospacer, although mismatches within the “seed” region flanking the PAM are more disruptive (9, 12). Similar factors are required for DNA-targeting by type III systems, where these systems evaluate base pairing between the target sequence and the region flanking the protospacer (13).
While these factors help safeguard against accidental targeting of genomic sequences, they provide a simple set of design rules to achieve DNA targeting. This has primarily been exploited with type II systems for genome editing, whereby cleavage is followed by DNA repair through nonhomologous end joining (NHEJ) or homologous recombination (11, 14, 15). However, within microorganisms with poor or absent NHEJ, genome targeting can be lethal. For instance, natural systems that acquired genome-targeting spacers appear to possess inactive systems or mutated target loci (16), potentially explaining the evolution of Pelobacter carbinolicus harboring a type I-F CRISPR-Cas system (17). In the industrial bacterium Streptococcus thermophilus, cultures under bacteriophage attack rarely integrate genome-targeting spacers and, in such an event, rapidly disappear from the population (18). In the hyperthermophilic archaeon Sulfolobus solfataricus harboring a type III-A system, infecting cells with viral particles encoding a genome-targeting spacer slowed the growth of the culture under selecting conditions and led to recombination between the virally encoded spacer and the endogenous CRISPR array (19). In the bacterium Escherichia coli expressing a type I-E or II-A system in trans, transformation of a plasmid with spacers targeting endogenous genes or a lysogenized bacteriophage led to extremely low recovery of viable transformants (11, 20–23). Similar results were obtained in the bacterial pathogen Streptococcus pneumoniae expressing a type II system in trans, wherein viable transformants contained mutations or deletions that inactivated CRISPR-Cas-mediated targeting (11, 24). Finally, in the bacterial phytopathogen Pectobacterium atrosepticum harboring a native type I-F system, induction of genome-targeting spacers from a tightly regulated plasmid was cytotoxic within a few hours of induction and led to extensive deletions in the target loci (25). This extensive evidence presents an opportunity for the sequence-specific removal of microorganisms by reprogramming CRISPR-Cas systems.
Here, we investigated the potential of CRISPR-Cas systems for the sequence-specific targeting and selective removal of individual strains of bacteria. Using the E. coli type I system as a model, we found that targeting the E. coli genome led to potent removal of cells as long as the target sequence contained a PAM and was complementary to the spacer. In contrast to targeting of bacteriophages and plasmids, genome targeting accommodated multiple mutations in the seed region. Furthermore, targeting was similarly effective regardless of genomic location, strand, or transcriptional activity. Finally, using genomic sequence information, we could selectively remove closely related bacterial strains whether in pure or mixed cultures. The extent of removal could even be modulated by mixing targeting and nontargeting plasmids. Our findings open the possibility of quantitatively controlling the composition of microbial consortia and selectively treating multidrug-resistant infections, particularly with ongoing advances in the delivery of nucleic acids to microorganisms.
RESULTS
Genome targeting with the type I-E CRISPR-Cas system in E. coli.
We first evaluated the impact of targeting a natural genomic locus with the type I-E CRISPR-Cas system from Escherichia coli K-12, one of the best-characterized CRISPR-Cas systems to date. This system encodes six cas genes in two operons (casABCDE and cas3) required for CRISPR RNA processing and the cleavage and degradation of target DNA (26). Because the casABCDE operon is repressed by H-NS in E. coli K-12 under normal growth conditions (27), we used a previously developed system consisting of two plasmids (pCasA-E and pCas3) (see Fig. S1 in the supplemental material) that inducibly express all six cas genes (26). In addition, we generated a third plasmid, encoding an altered version of the endogenous CRISPR1 array in E. coli K-12 that accommodates the sequential insertion of engineered spacer sequences (pCRISPR) (see Fig. S1 and S2). pCRISPR plasmids encoding engineered, genome-targeting spacers were transformed into E. coli K-12 substrain BW25113 cells equipped with inducible expression of the T7 polymerase (BW25113-T7) and harboring the two cas-expressing plasmids (pCasA-E and pCas3) (see Fig. S1). As part of the assay, we measured the transformation efficiency, a proxy for removal of strains in pure cultures, by dividing the number of viable transformants for each genome-targeting CRISPR plasmid by the number of viable transformants for the original pCRISPR plasmid. Lower ratios or transformation efficiencies indicate a greater extent of removal.
We began with a spacer that is complementary to the template strand of the essential ftsA gene, involved in cell division (Fig. 2A). The selected protospacer was immediately downstream of AAG, one of the four PAMs for this CRISPR-Cas system (9). The resulting anti-ftsA (α-ftsA) plasmid exhibited transformation efficiency ~105-fold lower than that of the original pCRISPR plasmid (Fig. 2B), paralleling the transformation efficiency of plasmids encoding prophage-targeting CRISPR RNAs (21, 22). In the absence of the casABCDE operon, the α-ftsA plasmid and the original pCRISPR plasmid yielded similar transformation efficiencies (Fig. 2B), ruling out transformation issues and confirming the role of the casABCDE operon. Forced expression of the chromosomally encoded cas genes through deletion of the hns gene also resulted in a low transformation efficiency (see Fig. S3A in the supplemental material) (27). In total, these results demonstrate the potency of genome targeting using a type I CRISPR-Cas system.
FIG 2 .
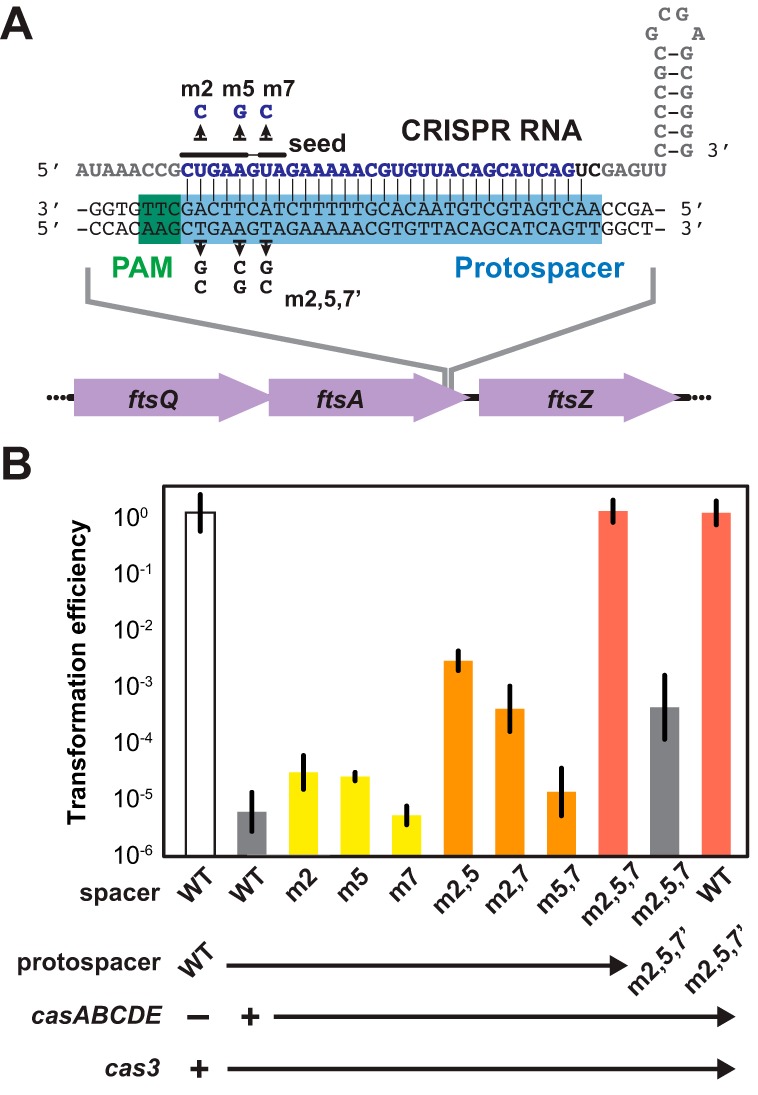
Potent and sequence-specific removal through genome targeting with CRISPR-Cas systems. (A) Design of a CRISPR RNA targeting the ftsA gene in E. coli K-12. The 32-nt spacer sequence is in blue, and the repeat sequence is in gray. The last two nucleotides of the spacer (in black) are fixed to introduce restriction sites used for cloning additional repeat-spacer pairs. Flanking the protospacer (highlighted in blue) is the protospacer-adjacent motif (PAM) (highlighted in green) required for DNA targeting. Point mutations within the established seed region of the spacer (9) and the protospacer tested in panel B are shown. (B) Transformation efficiencies of α-ftsA plasmids containing different mutations in the seed region of the spacer. Single, double, and triple mutations of the spacer sequence are shown in yellow, orange, and red, respectively. The transformations were carried out in BW25113-T7 (wild type [WT]) or BW25113-T7m257′ (m2,5,7′), each harboring two plasmids: pCas3 (+ cas3) and either pCasA-E (+ casABCDE) or pCasA-E′ (− casABCDE). Figure S1 in the supplemental material illustrates the general transformation procedure. Transformation efficiency was calculated as the number of transformants for each tested plasmid divided by the number of transformants for the original pCRISPR plasmid for the same culture. Values represent the geometric means and SEM of data from three independent experiments.
Each experiment yielded a few colonies that presumably escaped genome targeting. These colonies may have escaped through (i) alteration to the target genomic site or (ii) disruption of the expression/activity of the CRISPR RNAs or Cas proteins (16, 28). To initially explore the basis of escape, we inoculated 22 colonies for storage and further analysis. Less than half of the colonies (10/22) exhibited substantial growth after 13 h in liquid culture selecting for all three plasmids, suggesting that the rate of survival is overestimated. We first sequenced the genomic ftsA locus of the 10 viable colonies plus that of 4 additional colonies from a previous experiment. Interestingly, the locus was unaltered in all 14 isolates, unlike previous examples of escape (9, 20). Turning our attention to the expression plasmids, we found that the isolated plasmids conferred resistance to all three antibiotics when transformed into a plasmid-free strain, ruling out integration of the resistance cassette into the E. coli genome. Finally, we sequenced the CRISPR locus on the α-ftsA plasmid, which revealed various deletions that removed the α-ftsA spacer (see Fig. S4 in the supplemental material). In total, alterations to the CRISPR RNAs appear to principally account for surviving colonies, at least within our experimental setup.
We next evaluated the sequence specificity of CRISPR-Cas-mediated removal. We introduced different point mutations into the seed region of the α-ftsA spacer (Fig. 2A), where the seed region for the type I-E CRISPR-Cas system in E. coli was previously identified as the first through fifth, seventh, and eighth nucleotides flanking the PAM (9, 12). Prior work demonstrated that single point mutations within this region of the spacer for type I CRISPR-Cas systems disrupted immunity against bacteriophages (9). However, we found that point mutations to the second (m2), fifth (m5), or seventh (m7) nucleotide of the wild-type (WT) α-ftsA spacer only marginally disrupted removal (Fig. 2B). Pairing point mutations (m2,5; m2,7; m5,7) further disrupted removal, while only the combination of all three point mutations (m2,5,7) fully disrupted removal.
To further probe the specificity of removal, we introduced compensatory mutations within the native ftsA gene (m2,5,7′) (see Fig. S5 in the supplemental material). The matched pairing of the m2,5,7 spacer and the m2,5,7′ strain resulted in a large extent of removal, albeit less than that seen with the pairing of the WT spacer and the WT strain (Fig. 2B). Separately, the mismatched pairing of the WT spacer and the m2,5,7′ strain exhibited negligible removal (Fig. 2B), excluding the possibility of unintended targeting at other genomic loci.
Potent removal by targeting diverse locations throughout the genome.
Programming of CRISPR-Cas systems to remove individual strains would greatly benefit from the ability to readily target any PAM-flanking sequence throughout a genome. Previous examples of genome targeting successfully targeted different genes on both strands of the genome (11, 19, 20, 25). However, a comprehensive and quantitative investigation of genome targeting has not been conducted. Toward this goal, we designed 10 additional spacers that target different protospacers flanked by a PAM throughout the E. coli K-12 genome (Fig. 3A). The corresponding protospacers covered a diverse range of locations, including the positive and negative strands of the genome, template strands and nontemplate strands of genes, and within untranscribed regions. Furthermore, we targeted both essential and nonessential genes because of their relative capacities to tolerate mutations or deletions. In all cases, the extent of removal was statistically similar to that of the original α-ftsA spacer (P values between 0.05 and 0.88) (Fig. 3B), suggesting that removal is based on chromosomal injury rather than on perturbing the natural function of the target locus. Furthermore, in the absence of the casABCDE operon, each plasmid and the original pCRISPR plasmid yielded similar transformation efficiencies (see Fig. S6 in the supplemental material). The PAM was an essential feature, similar to findings of previous studies (11, 20, 25), since targeting a separate site within the ftsA gene lacking a PAM resulted in negligible removal (Fig. 3B). Based on these results, we conclude that potent removal can be achieved by targeting diverse locations throughout the genome as long as a PAM is present. Interestingly, the simultaneous targeting of multiple locations (asd, msbA, ftsA, and nusB) exhibited extents of removal similar to those with targeting of only one of the locations (ftsA) (P = 0.48) (see Fig. S7).
FIG 3 .
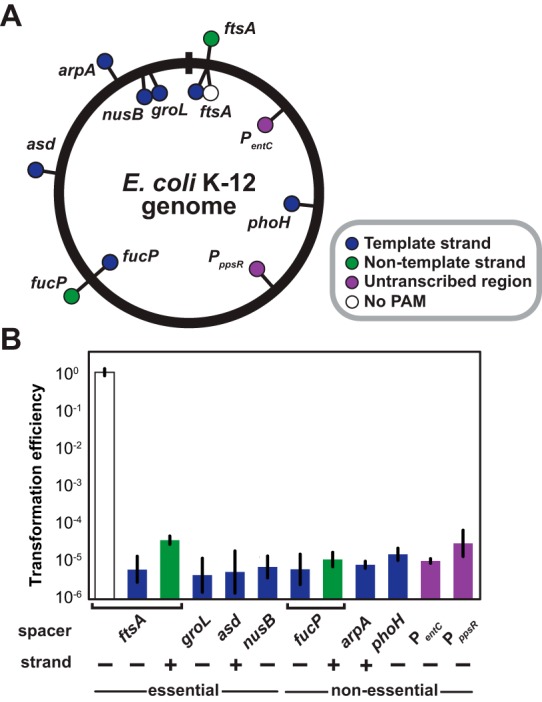
Similar efficiencies when targeting diverse locations throughout the genome. (A) Protospacer locations in the E. coli K-12 genome. Dots inside and outside the circle reflect spacers designed to base pair with the negative (−) or positive (+) strand of the chromosome, respectively. Dots also reflect protospacers flanked by a non-PAM (white), on the template strand (blue), or on the nontemplate strand (green) of coding regions or in nontranscribed regions (purple). (B) Transformation efficiencies for pCRISPR plasmids encoding spacers targeting the sites shown in panel A in BW25113-T7 harboring pCas3 and pCasA-E. See the legend for Fig. 2B for an explanation of the transformation efficiency. Values represent the geometric means and SEM of data from three independent experiments.
To explore the broad potential of our approach through native CRISPR-Cas systems outside of E. coli, we explored the impact of genome targeting in the Gram-positive bacterium Streptococcus thermophilus. In particular, we assessed genome targeting through the two native type II CRISPR-Cas systems (CRISPR1 and CRISPR3) previously shown to be active under normal growth conditions (4, 29). The transformation efficiencies of plasmids encoding CRISPR1 and CRISPR3 RNAs targeting the lacZ gene were near the limit of detection (~103-fold lower than that of the empty plasmid) (see Fig. S3B and Table S1 in the supplemental material). Therefore, potent removal can be achieved through different native CRISPR-Cas systems.
Sequence-specific removal of individual strains.
The flexibility and sequence specificity of genome targeting open the intriguing possibility of using CRISPR-Cas systems to specifically remove individual microbial species and strains. To begin exploring this possibility, we focused on two substrains of E. coli: E. coli K-12 (BW25113-T7) and E. coli B [BL21(DE3)] (Fig. 4A). Because the genomes of these strains bear more than 99% sequence homology and almost all cellular processes are identical (30), selectively removing one of the strains would be extremely difficult with antimicrobial agents or under defined growth conditions. However, the distinguishing sequences afford ample opportunities to selectively target either strain with programmed CRISPR-Cas systems. Using in silico genomic analyses, we identified one PAM-flanking sequence unique to E. coli K-12 (within the fucP gene, involved in the transport of l-fucose), one PAM-flanking sequence unique to E. coli B (within the ogr gene, located within the P2 prophage), and one PAM-flanking sequence shared by both strains (within the groL gene, involved in protein folding). We subsequently designed CRISPR spacers that recognize PAM-adjacent protospacers in each gene and measured removal in pure cultures harboring pCasA-E and pCas3 (see Fig. S1 in the supplemental material). As expected, targeting fucP removed only the K-12 strain, targeting ogr removed only the B strain, and targeting groL removed both strains (Fig. 4B).
FIG 4 .
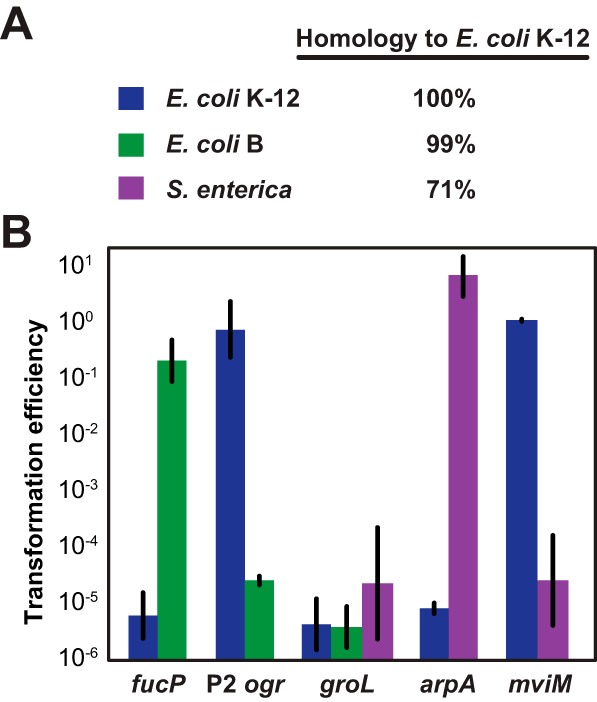
Sequence-specific removal of individual bacterial strains in pure cultures. (A) Estimated homologies between the genomes of E. coli K-12 BW25113-T7 (blue), E. coli B BL21(DE3) (green), and S. enterica SB300A#1 (purple), a derivative of strain LT2 (58). The reported homologies are based on prior comparisons between E. coli K-12 and E. coli B (30) and between E. coli K-12 and S. enterica LT2 (31). (B) Transformation efficiencies for pCRISPR plasmids encoding spacers targeting a PAM-flanking protospacer within the specified gene. The assay was conducted following the scheme depicted in Fig. S1 in the supplemental material for all three strains. See panel A for an explanation of the coloring scheme depicting each transformed strain. See the legend for Fig. 2B for an explanation of the transformation efficiency. Values represent the geometric means and SEM of data from three independent experiments.
One potential application of programmable removal with CRISPR-Cas systems is targeting pathogenic bacteria while sparing commensal bacteria. Toward this goal, we focused on E. coli K-12 (BW25113-T7), a derivative of commensal E. coli that naturally inhabits the human digestive tract, and on Salmonella enterica (SB300A#1, a derivative of LT2), a major food pathogen. Both species are Gram-negative enterobacteria, and they share ~71% sequence homology (Fig. 4A) (31). Using genomic analyses, we designed CRISPR spacers targeting a PAM-flanking sequence unique to E. coli (within the arpA gene, involved in the regulation of acetyl-coenzyme A [CoA] biosynthesis), a PAM-flanking sequence unique to S. enterica (within the mviM gene, encoding a putative virulence factor), and a shared PAM-flanking sequence (within the groL gene). The resulting plasmids were transformed into pure cultures harboring pCasA-E and pCas3 (see Fig. S1 in the supplemental material). As expected, targeting arpA removed only E. coli, targeting mviM removed only S. enterica, and targeting groL removed both strains (Fig. 4B).
Selective and titratable removal of individual strains in mixed cultures.
We next proceeded from pure cultures to mixed cultures in order to evaluate the selective removal of target strains. We repeated the transformation experiments with E. coli B [BL21(DE3)] and E. coli K-12 (Bw25113-T7), except that both strains were cocultured and plated on agar with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) and isopropyl-β-d-thiogalactopyranoside (IPTG). Under these plating conditions, BL21(DE3) yields blue colonies, whereas BW25113-T7 yields white colonies (see Fig. S8 in the supplemental material). Similar to the experiments with pure cultures, targeting the PAM-flanking sequence within the ogr gene selectively removed BL21(DE3), targeting the PAM-flanking sequence within the fucP gene selectively removed Bw25113-T7, and targeting with the original pCRISPR plasmid maintained both strains (Fig. 5A). Furthermore, in the absence of the casABCDE operon, the two strains were maintained in similar ratios regardless of the transformed plasmid (Fig. 5A). These results demonstrate that CRISPR-Cas systems can be employed for the selective removal of bacterial strains in mixed cultures.
FIG 5 .

Selective and titratable removal of individual bacterial strains in mixed cultures. (A) Selective removal of BW25113-T7 and BL21(DE3) in mixed cultures. Equal numbers of BW25113-T7 and BL21(DE3) cells harboring pCas3 and either pCasA-E (+ casABCDE) or pCasA-E′ (− casABCDE) were mixed prior to preparation for electroporation. Mixed cultures electroporated with the indicated pCRISPR plasmid were plated on LB agar containing IPTG, X-Gal, l-arabinose, IPTG, and antibiotics. Only BL21(DE3) possesses the lacZ gene, which yields blue colonies (see Fig. S8 in the supplemental material). Plates are representative of three independent experiments. (B) Titratable removal of BL21(DE3) in mixed cultures. Transformations were conducted similarly to the method described for panel A except that cells were transformed with different ratios of the E. coli B-targeting plasmid (pCRISPR-ogr) and the nontargeting plasmid (pCRISPR) for a total of 100 ng. The ratio of blue to white colonies was normalized to the ratio of blue to white colonies for the same culture transformed with the pCRISPR plasmid. Values represent the geometric means and SEM of data from three independent experiments.
The above mixed-culture experiments utilized single plasmids to either remove or maintain individual strains. We hypothesized that transforming combinations of targeting and nontargeting plasmids could remove a portion of targeted cells, conferring control over the composition of the population. To test this hypothesis, we transformed different amounts of the pCRISPR plasmid and the BL21(DE3)-targeting plasmid (total of 100 ng) and then quantitated the ratio of blue and white colonies. Remarkably, the fraction of the BL21(DE3)-targeting plasmid strongly correlated with the selective removal of BL21(DE3) (Fig. 5B). The almost-perfect linear correlation (R2 = 1.00) further suggests that almost all transformed cells received a single plasmid that either removed or sustained E. coli B. We thus conclude that CRISPR-Cas systems can be reprogrammed to quantitatively modulate the composition of a mixed population.
DISCUSSION
We demonstrated the sequence-specific removal of individual strains using CRISPR-Cas systems. While the extent of removal was extremely high (>99.999% for the α-ftsA plasmid [Fig. 2B]), a fraction of the transformed cells consistently survived targeting. Sequencing survivors revealed consistent loss of the genome-targeting spacer in the transformed pCRISPR plasmid (see Fig. S4 in the supplemental material), likely through recombination between the identical repeats. This insight is consistent with findings of a recent study showing loss or inactivation of CRISPR elements under evolutionary pressure (28). This insight also suggests one potential countermeasure against surviving colonies: reducing the number of repeats within the CRISPR array. The array could even be reduced to a single repeat-spacer, paralleling the engineering of single-guide RNAs for use with Cas9 (32). Other potential strategies include expressing multiple CRISPR-Cas systems or eliminating CRISPR-encoding plasmids that underwent recombination. Targeting multiple sites at one time did not appear to be an effective strategy (see Fig. S7), perceivably due to rearrangement of the CRISPR-encoding plasmid (see Fig. S4).
We also demonstrated that potent removal could be achieved using type I and type II CRISPR-Cas systems. An interesting distinction between these systems is that type I systems cleave and degrade DNA through the action of a 3′-to-5′ exonuclease, whereas type II systems only cleave DNA (33). The additional effect of DNA degradation by type I systems may further improve the potency of genome targeting by preventing DNA repair, although a direct comparison between type I and type II systems would be needed to directly evaluate this potential contribution.
We found that multiple mismatches within the seed region were required to fully disrupt targeting by the type I-E system from E. coli (Fig. 2). This may explain the absence of mutations within the protospacer of surviving colonies (see Fig. S4 in the supplemental material). Separately, the number of required mismatches contrasts with the single mismatches that disrupted immunity to the M13 bacteriophage (9). This discrepancy is intriguing considering that the same Cas-encoding plasmids were used in these studies. One possibility is that the seed region can accommodate different numbers of mismatches when targeting genomic DNA or when targeting invader DNA. Such differences may help explain emerging reports of elevated off-target effects associated with genome editing (34–37).
Delivery arguably poses the most immediate challenge to the downstream use of CRISPR-Cas systems for the selective and titratable removal of microorganisms. However, opportunities in nanoparticle development and the engineering of bacteriophages present potential solutions. Nanoparticles have been used to deliver nucleic acids to bacteria (38), but little subsequent work has been done; the delivery of CRISPR RNAs may provide the impetus to further investigate nanoparticles as vehicles of delivery to microorganisms. Separately, bacteriophages have been widely used for heterologous protein expression, gene delivery, and the treatment of bacterial infections (39, 40). Lysogenic bacteriophages or phagemids with broad host ranges would be particularly beneficial for the delivery of CRISPR-Cas-encoding constructs (41–43). While silver nanoparticles and lytic bacteriophages also could be used to remove bacteria (44, 45), they lack the specificity or the programmability offered by genome-targeting CRISPR-Cas systems and cannot be easily dosed to quantitatively control the composition of a microbial consortium.
Once delivery challenges are overcome, we foresee CRISPR-Cas systems being exploited to control bacterial populations in diverse ecological niches and scientific fields. In biotechnology, CRISPR-Cas systems could be used to selectively clear contaminating microorganisms or to quantitatively control the composition of microbial consortia in industrial processes or in environmental samples. In medicine, CRISPR-Cas systems could be used to control the composition of the gut flora or as “smart” antibiotics that circumvent commonly transmitted modes of antibiotic resistance and distinguish between beneficial and pathogenic bacteria. For applications that require the removal of more than one strain, multiple spacers that target shared or unique sequences could be encoded in a single CRISPR array. The arrays could also be combined with a complete set of cas genes to instigate removal of strains lacking functional CRISPR-Cas systems (15, 46). Because of the sequence specificity of targeting, CRISPR-Cas systems could be used to distinguish strains separated by only a few base pairs. The use of CRISPR-Cas systems would require detailed knowledge of the genomic sequences of the bacterial population, although the dwindling cost and increasing speed of high-throughput sequencing along with powerful metagenomics tools would alleviate this challenge. Overall, CRISPR-Cas systems offer a unique opportunity for the selective and titratable removal of microorganisms for industrial and medical purposes, which can be added to the ever-expanding applications of this versatile immune system (11, 20, 23, 47, 48).
MATERIALS AND METHODS
Strains and plasmid construction.
See Table S2 in the supplemental material for a list of all strains used in this work. E. coli K-12 strain BW25113-T7 was generated by transferring araB::T7-RNAp-tetA from IY5163 to BW25113 by P1 transduction. Successful transduction was verified by PCR. BW25113-T7m2,5,7′ (Fig. 2A) was generated using three rounds of oligonucleotide-mediated recombination with ftsA-m257-spacer.recomb and the pKD46 plasmid encoding the λ red recombination genes (49, 50). The oligonucleotide contained two phosphorothioate linkages at each end to improve the recombination efficiency (51, 52). Successful recombinants were verified by PCR and by sequencing.
See Table S2 in the supplemental material for a list of all plasmids used in this work. The origins of replication for the pCas3, pCasA-E, and pCRISPR plasmids used with E. coli and S. enterica belong to different incompatibility groups (26, 53). To generate the pCasA-E plasmid lacking the casABCDE operon (pCasA-E′), pCasA-E was digested with NcoI/NotI, blunt ended using Pfu polymerase, and ligated.
To generate the pCRISPR plasmid, the pBAD18 plasmid (53) was linearized with XbaI and amplified by PCR using primers pBAD18.fwd/pBAD18.rev. A chemically synthesized gBlock (IDT) was then inserted downstream of the PBAD promoter by Gibson assembly (54). The gBlock encoded four repeats and three intervening spacers from the endogenous CRISPR locus in E. coli K-12 MG1655 (see Table S2 in the supplemental material), where the first spacer was modified to include a KpnI restriction site and an XhoI restriction site. These restriction sites allow the sequential insertion of engineered repeat-spacer pairs (see Fig. S2). Each pair was chemically synthesized as two oligonucleotides (IDT), phosphorylated with polynucleotide kinase, annealed, and ligated into the pCRISPR plasmid digested with KpnI and XhoI.
The pBAD18-asd,msbA,ftsA,nusB plasmid was constructed in a manner similar to that for the pCRISPR plasmid, wherein a chemically synthesized gBlock (IDT) was inserted downstream of the PBAD promoter of the linearized pBAD18 plasmid by Gibson assembly (54). The gBlock encoded the first repeat-spacer sequence from the endogenous E. coli CRISPR locus, followed by five repeats and four intervening spacers targeting four different locations in E. coli Bw25113 (asd, msbA, ftsA, and nusB) (see Table S2 in the supplemental material).
To generate pORI28 (55) with engineered spacers, pORI28 and each insert generated through PCR assembly were digested with BamHI and SacI and ligated together. To generate the insert encoding the lacZ1 spacer, template-free PCR was conducted with C1-lacZ1.fwd/C1-lacZ1.rev, followed by using the resulting product in a subsequent PCR with C1-BamHI.fwd/C1C3-SacI.rev. To generate the inserts encoding the lacZ2 and lacZ3 spacers, first the CRISPR3 leader region was amplified by PCR from LMD-9 genomic DNA with C3-leader.fwd/C3-leader.rev. Next, the resulting product was used as the template in a subsequent PCR with C3-leader.fwd/C3-lacZ2.rev or C3-leader.fwd/C3-lacZ3.rev. Finally, each PCR product was used as the template in a final round of PCR with C3-leader.fwd/C1C3-SacI.rev. All oligonucleotides and enzymes were purchased from IDT and NEB, respectively. All cloned plasmids were verified by sequencing.
Growth conditions.
All E. coli and Salmonella strains were cultured in LB medium (10 g/liter tryptone, 5 g/liter yeast extract, and 10 g/liter sodium chloride) at 37°C and 250 rpm with appropriate antibiotics. The same strains were plated on LB agar (LB medium with 1.5% agar) supplemented with appropriate inducers and incubated at 37°C. S. thermophilus LMD-9 was cultured in Elliker broth (Elliker medium [Difco] supplemented with 1% beef extract) and plated on Elliker agar (Elliker broth with 1.5% agar) (56). Both culturing and plating of LMD-9 were conducted at 37°C. Antibiotics were administered at the following final concentrations: 50 µg/ml streptomycin, 50 µg/ml kanamycin, 50 µg/ml ampicillin, 2 µg/ml chloramphenicol, and 2 µg/ml erythromycin. Inducers were administered at the following final concentrations: 0.1 mM IPTG and 0.02% l-arabinose.
Design of native CRISPR RNAs.
An overview of the approach to design and insert spacer sequences into the CRISPR array within the pCRISPR plasmid is shown in Fig. S2 in the supplemental material. The spacers were designed by identifying one of the known PAMs for the type I-E CRISPR-Cas system in E. coli (AAG, GAG, GAG, and ATG). The downstream 32 nucleotides (nt) were then used as the spacer within the engineered repeat-spacer pair. Note that the last two nucleotides of the spacer are fixed as TC because of the adopted cloning strategy (see Fig. S2). However, these nucleotides fall well outside the seed region and therefore are expected to have a negligible effect on targeting.
The spacers for S. thermophilus were designed by identifying a known PAM for CRISPR1 (NNAGAAW) or for CRISPR3 (NGGNG) (10). The sequence of the 31 nt upstream of each PAM was integrated into oligonucleotides that were used to generate a leader region followed by a single repeat-spacer-repeat that was subsequently cloned into pORI28. This construct relies on processing through the native tracrRNA and RNase III.
Transformation assay.
Freezer stocks of E. coli and Salmonella strains harboring pCas3 and pCasA-E (or pCasA-E′) were streaked to isolation on LB agar. Individual colonies were inoculated into 3 ml of LB medium and shaken overnight at 37°C. The cultures then were back diluted into 25 ml of LB medium and grown to an A600 of 0.6 to 0.8, which was measured on a Nanodrop 2000c spectrophotometer (Thermo Scientific). The cells were then pelleted and washed with ice-cold 10% glycerol 2 times before being resuspended in 150 to 350 µl of 10% glycerol. The resuspended cells (50 µl) were transformed with 50 ng of pCRISPR or pCRISPR encoding the indicated spacer, using a MicroPulser electroporator (Bio-Rad), and recovered in 300 µl of SOC medium (Quality Biological) for 1 h (E. coli) or for 2 h (Salmonella). After the recovery period, 200 µl of different dilutions of the cells were plated on LB agar with inducers. The transformation efficiency was calculated by dividing the number of transformants for the tested plasmid by the number of transformants for the original pCRISPR plasmid. To normalize for experimental variability in transformation efficiency, the same batch of cells prepared for electroporation was transformed with each tested plasmid and the original pCRISPR plasmid.
S. thermophilus strain LMD-9 harboring pTRK669 was grown in 50 ml of Elliker broth and prepared for electroporation as described previously, which concentrated the culture 100-fold (57). The resuspended cells (50 µl) were transformed with 1 µg of the pORI28 control plasmid or pORI28 containing the indicated spacer. Transformed cells were recovered in 950 µl of Elliker broth overnight and plated on Elliker agar. Plates were then incubated for 48 h in a Coy anaerobic chamber with a gas mixture of 10% hydrogen, 5% carbon dioxide, and 85% nitrogen before the colonies were counted. The transformation efficiency was calculated by dividing the number of transformants for the tested plasmid by the number of transformants for the pORI28 control plasmid.
The average limit of detection of the killing assay, calculated as 1/(no. of transformants for the control plasmid) was 7 × 10−7 for E. coli, 4 × 10−7 for Salmonella, and 2 × 10−3 for S. thermophilus. The high transformation efficiency for Salmonella was achieved by purifying the pCRISPR plasmids, the pCas3 plasmid, and the pCasA-E plasmid individually from SB300A#1.
Mixed-culture transformation assay.
The transformation assay for mixed cultures resembled that for the pure culture with a few notable differences. Cultures of E. coli K-12 and E. coli B strains harboring pCas3 and pCasA-E (or pCasA-E′) were grown separately to an A600 of ~0.8, and then equal numbers of cells were mixed from the back dilutions prior to preparing the culture for electroporation. An aliquot of the resuspended cell mixture (50 µl) was then transformed with the pCRISPR plasmid, pCRISPR encoding the indicated spacer, or a defined mixture of both plasmids for a total of 100 ng. The transformed cells were recovered in 300 µl of SOC medium for 90 min. After the recovery period, 200 µl of different dilutions of the cells were plated on LB agar with inducers and appropriate antibiotics. The ratio of blue (E. coli B) to white (E. coli K-12) colonies on the sample plate was divided by the same ratio on the pCRISPR plate, yielding the normalized ratio. To normalize for experimental variability in transformation efficiency, the same batches of cell mixtures prepared for electroporation were transformed with each tested plasmid mixture and the pCRISPR control plasmid.
Analysis of escape mutants.
Colonies from the transformation assay with the α-ftsA plasmid (pCB304) were inoculated into 5 ml of LB medium with appropriate antibiotics and inducers. Growth was assessed based on the A600 after 13.5 h of growth. Cultures exhibiting measurable growth (A600 > 0.01) were stored as glycerol stocks. Plasmids were then isolated from each escape mutant, and equal amounts of DNA were resolved by agarose gel electrophoresis. Each isolated set of plasmids was also transformed into E. coli K-12 and plated on LB agar containing one of the three antibiotics. Finally, the plasmid mixture from each escape mutant was sequenced using primers that specifically bind within the PBAD promoter or the double terminator of the α-ftsA plasmid. To analyze the protospacers, approximately 400 bp surrounding the protospacer within the ftsA gene of the escape mutant was PCR amplified and subjected to sequencing.
Statistical analyses.
All P values were calculated using the Student t test, assuming log-normal distributions, two tails, and unequal variances.
SUPPLEMENTAL MATERIAL
Quantitating the removal capacity of genome-targeting CRISPR spacers. A genomic sequence is inserted within the native CRISPR array of E. coli K-12 in the pCRISPR plasmid (see Fig. S2) and transformed into BW25113-T7 cells harboring the two cas-expressing plasmids, pCasA-E and pCas3. The transformation efficiency is then assessed on plates containing l-arabinose, IPTG, and antibiotics that select for all three plasmids. Although this scheme uses three plasmids, more compact schemes are possible, such as using type II systems with a single-guide RNA. Download
Design and insertion of spacer sequences into the pCRISPR plasmid. The protospacer is selected as the 32 nt immediately downstream of a protospacer-adjacent motif, or PAM (AAG, AGG, GAG, and ATG for E. coli K-12 MG1655). The sequence and selected spacer within the ftsA gene used in Fig. 2 are shown at the top. The first 30 bp of the protospacer are integrated into two chemically synthesized oligonucleotides. The last two nucleotides of the spacer (outlined in blue) are fixed because they are part of the XhoI overhang. The overhangs of the annealed oligonucleotides allow insertion into the pCRISPR plasmid digested with KpnI and XhoI. Insertion recreates the KpnI and XhoI sites at the 5′ end and destroys the XhoI site (XhoI′) at the 3′ end. Recreating the KpnI and XhoI sites allows the sequential insertion of additional repeat-spacer pairs into the CRISPR array as long as the 30th nt in each spacer is not a C. The base CRISPR array is contained within the gBlock shown in Table S2. Download
Removal of bacteria using native CRISPR-Cas systems. (A) Removal of Escherichia coli through the native type I-E system. Pure cultures of the wild-type strain (WT) (BW25113) or the hns deletion strain (Δhns; JW1225-2) were transformed with the α-ftsA CRISPR plasmid depicted in Fig. 2A. Transformed cells were plated on LB agar supplemented with l-arabinose and ampicillin. See Fig. 2B for an explanation of the transformation efficiency. Values represent the geometric means and SEM of data from three independent experiments. (B) Removal of Streptococcus thermophilus through the native type II systems. The two systems, termed CRISPR1 and CRISPR3, possess distinct repeat sequences, Cas proteins, and PAMs. Strain LMD-9, harboring the pTRK669 plasmid, was transformed with the pORI28 plasmid, encoding a spacer targeting PAM-flanking protospacers within the lacZ gene (STER_1366; protein ID ABJ66539.1). See the legend for Fig. 2B for an explanation of the transformation efficiency; only the original pORI28 plasmid served as the control plasmid. The gray background indicates the limit of detection of the transformation assay. Values represent the arithmetic means and SEM of data from five independent experiments. The arithmetic means and SEM were employed because many of the experiments resulted in zero colonies. Download
Analysis of viable transformants that escaped removal by the α-ftsA CRISPR plasmid. Plasmid DNA was isolated from viable colonies following transformation with the α-ftsA plasmid. The CRISPR locus then was sequenced using upstream and downstream primers, resulting in the depicted truncations. Red lines designate deletions. NR, no read from sequencing starting upstream and downstream of the CRISPR locus, suggestive of the absence of the entire locus. Download
Compensatory mutations within the ftsA coding region. The three mutations (m2, m5, and m7) were selected to minimize changes to the amino acid sequence of FtsA. The first two mutations (m2 and m5) are silent, whereas the third mutation (m7) changes a valine to an alanine. Numbers above each amino acid designate the location within the coding region of ftsA. The short black bar above the wild type (WT) sequence designates the seed region. Download
Transformation efficiencies with an incomplete type I CRISPR-Cas system in E. coli. Transformations paralleled those shown in Fig. 3, except that the cells harbored pCasA-E′ (− casABCDE) and pCas3 (+ cas3). See the legend for Fig. 2B for an explanation of the transformation efficiency. Download
Impact of simultaneously targeting multiple sites on the transformation efficiency in E. coli K-12. A plasmid encoding a synthetic CRISPR array targeting four different endogenous genes (asd, msbA, ftsA, and nusB) was used. See the legend for Fig. 2B for an explanation of the transformation efficiency. Values represent the geometric means and SEM of data from three independent experiments. In comparison to the α-ftsA plasmid (Fig. 2B), targeting the three additional sites did not significantly alter the relative transformation efficiency (P = 0.48; n = 3). Download
E. coli strains BW25113-T7 and BL21(DE3) can be readily distinguished on LB agar with IPTG and X-Gal. The lacZ gene is disrupted in BW25113-T7 and is intact in BL21(DE3). As a result, BW25113-T7 colonies remain white, whereas BL21(DE3) colonies turn blue on LB agar supplemented with IPTG and X-Gal. Download
Protospacer sequences
Strains, plasmids, and oligonucleotides used in this work
ACKNOWLEDGMENTS
We thank G. Seelig for initial discussions, R. M. Kelly for E. coli BL21(DE3), D. M. Downs for Salmonella SB300A#1, and U. Qimron for E. coli IYB5163 and plasmids pWUR397 and pWUR400.
This work was supported by university start-up funds and the Faculty Research and Professional Development program [to C.L.B.] and a graduate fellowship through the North Carolina Agriculture Foundation and through the Dannon Company, Inc. [to K.S.].
Footnotes
Citation Gomaa AA, Klumpe HE, Luo ML, Selle K, Barrangou R, Beisel CL. 2014. Programmable removal of bacterial strains by use of genome-targeting CRISPR-Cas systems. mBio 5(1):e00928-13. doi:10.1128/mBio.00928-13.
REFERENCES
- 1. Parisien A, Allain B, Zhang J, Mandeville R, Lan CQ. 2008. Novel alternatives to antibiotics: bacteriophages, bacterial cell wall hydrolases, and antimicrobial peptides. J. Appl. Microbiol. 104:1–13. 10.1111/j.1365-2672.2007.03498.x [DOI] [PubMed] [Google Scholar]
- 2. Koskella B, Meaden S. 2013. Understanding bacteriophage specificity in natural microbial communities. Viruses 5:806–823. 10.3390/v5030806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sorek R, Lawrence CM, Wiedenheft B. 2013. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu. Rev. Biochem. 82:237–266. 10.1146/annurev-biochem-072911-172315 [DOI] [PubMed] [Google Scholar]
- 4. Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. 10.1126/science.1138140 [DOI] [PubMed] [Google Scholar]
- 5. Garneau JE, Dupuis MÈ, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, Moineau S. 2010. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468:67–71. 10.1038/nature09523 [DOI] [PubMed] [Google Scholar]
- 6. Sinkunas T, Gasiunas G, Waghmare SP, Dickman MJ, Barrangou R, Horvath P, Siksnys V. 2013. In vitro reconstitution of Cascade-mediated CRISPR immunity in Streptococcus thermophilus. EMBO J. 32:385–394. 10.1038/emboj.2012.352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gasiunas G, Barrangou R, Horvath P, Siksnys V. 2012. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U. S. A. 109:E2579–E2586. 10.1073/pnas.1208507109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. 2011. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9:467–477. 10.1038/nrmicro2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, van der Oost J, Brouns SJ, Severinov K. 2011. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U. S. A. 108:10098–10103. 10.1073/pnas.1104144108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mojica FJ, Díez-Villaseñor C, García-Martínez J, Almendros C. 2009. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155:733–740. 10.1099/mic.0.023960-0 [DOI] [PubMed] [Google Scholar]
- 11. Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31:233–239. 10.1038/nbt.2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wiedenheft B, van Duijn E, Bultema JB, Bultema J, Waghmare SP, Waghmare S, Zhou K, Barendregt A, Westphal W, Heck AJ, Heck A, Boekema EJ, Boekema E, Dickman MJ, Dickman M, Doudna JA. 2011. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc. Natl. Acad. Sci. U. S. A. 108:10092–10097. 10.1073/pnas.1102716108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marraffini LA, Sontheimer EJ. 2010. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463:568–571. 10.1038/nature08703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stern A, Keren L, Wurtzel O, Amitai G, Sorek R. 2010. Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet. 26:335–340. 10.1016/j.tig.2010.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aklujkar M, Lovley DR. 2010. Interference with histidyl-tRNA synthetase by a CRISPR spacer sequence as a factor in the evolution of Pelobacter carbinolicus. BMC Evol. Biol. 10:230. 10.1186/1471-2148-10-230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paez-Espino D, Morovic W, Sun CL, Thomas BC, Ueda K, Stahl B, Barrangou R, Banfield JF. 2013. Strong bias in the bacterial CRISPR elements that confer immunity to phage. Nat. Commun 4:1430. 10.1038/ncomms2440 [DOI] [PubMed] [Google Scholar]
- 19. Manica A, Zebec Z, Teichmann D, Schleper C. 2011. In vivo activity of CRISPR-mediated virus defence in a hyperthermophilic archaeon. Mol. Microbiol. 80:481–491. 10.1111/j.1365-2958.2011.07586.x [DOI] [PubMed] [Google Scholar]
- 20. Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183. 10.1016/j.cell.2013.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Edgar R, Qimron U. 2010. The Escherichia coli CRISPR system protects from λ lysogenization, lysogens, and prophage induction. J. Bacteriol. 192:6291–6294. 10.1128/JB.00644-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yosef I, Goren MG, Kiro R, Edgar R, Qimron U. 2011. High-temperature protein G is essential for activity of the Escherichia coli clustered regularly interspaced short palindromic repeats (CRISPR)/Cas system. Proc. Natl. Acad. Sci. U. S. A. 108:20136–20141. 10.1073/pnas.1113519108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA. 2013. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 41:7429–7437. 10.1093/nar/gkt520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bikard D, Hatoum-Aslan A, Mucida D, Marraffini LA. 2012. CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe 12:177–186. 10.1016/j.chom.2012.06.003 [DOI] [PubMed] [Google Scholar]
- 25. Vercoe RB, Chang JT, Dy RL, Taylor C, Gristwood T, Clulow JS, Richter C, Przybilski R, Pitman AR, Fineran PC. 2013. Cytotoxic chromosomal targeting by CRISPR/Cas systems can reshape bacterial genomes and expel or remodel pathogenicity islands. PLOS Genet. 9 e1003454. 10.1371/journal.pgen.1003454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. 2008. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964. 10.1126/science.1159689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pul U, Wurm R, Arslan Z, Geissen R, Hofmann N, Wagner R. 2010. Identification and characterization of E. coli CRISPR-cas promoters and their silencing by H-NS. Mol. Microbiol. 75:1495–1512. 10.1111/j.1365-2958.2010.07073.x [DOI] [PubMed] [Google Scholar]
- 28. Jiang W, Maniv I, Arain F, Wang Y, Levin BR, Marraffini LA. 2013. Dealing with the evolutionary downside of CRISPR Immunity: bacteria and beneficial plasmids. PLoS Genet. 9:e1003844. 10.1371/journal.pgen.1003844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karvelis T, Gasiunas G, Miksys A, Barrangou R, Horvath P, Siksnys V. 2013. crRNA and tracrRNA guide Cas9-mediated DNA interference in Streptococcus thermophilus. RNA Biol. 10:841–851. 10.4161/rna.24203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Studier FW, Daegelen P, Lenski RE, Maslov S, Kim JF. 2009. Understanding the differences between genome sequences of Escherichia coli B strains REL606 and BL21(DE3) and comparison of the E. coli B and K-12 genomes. J. Mol. Biol. 394:653–680. 10.1016/j.jmb.2009.09.021 [DOI] [PubMed] [Google Scholar]
- 31. McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, Hou S, Layman D, Leonard S, Nguyen C, Scott K, Holmes A, Grewal N, Mulvaney E, Ryan E, Sun H, Florea L, Miller W, Stoneking T, Nhan M, Waterston R, Wilson RK. 2001. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413:852–856. 10.1038/35101614 [DOI] [PubMed] [Google Scholar]
- 32. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sinkunas T, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V. 2011. Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system. EMBO J. 30:1335–1342. 10.1038/emboj.2011.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. 2013. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 31:839–843. 10.1038/nbt.2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. 2013. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31:827–832. 10.1038/nbt.2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cradick TJ, Fine EJ, Antico CJ, Bao G. 2013. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 41:9584–9592. 10.1093/nar/gkt714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. 2013. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31:822–826. 10.1038/nbt.2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fraley RT, Fornari CS, Kaplan S. 1979. Entrapment of a bacterial plasmid in phospholipid vesicles: potential for gene transfer. Proc. Natl. Acad. Sci. U. S. A. 76:3348–3352. 10.1073/pnas.76.7.3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lu TK, Collins JJ. 2007. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl. Acad. Sci. U. S. A. 104:11197–11202. 10.1073/pnas.0704624104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Westwater C, Kasman LM, Schofield DA, Werner PA, Dolan JW, Schmidt MG, Norris JS. 2003. Use of genetically engineered phage to deliver antimicrobial agents to bacteria: an alternative therapy for treatment of bacterial infections. Antimicrob. Agents Chemother. 47:1301–1307. 10.1128/AAC.47.4.1301-1307.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kaiser D, Dworkin M. 1975. Gene transfer to myxobacterium by Escherichia coli phage P1. Science 187:653–654. 10.1126/science.803710 [DOI] [PubMed] [Google Scholar]
- 42. Hagens S, Habel A, von Ahsen U, von Gabain A, Bläsi U. 2004. Therapy of experimental Pseudomonas infections with a nonreplicating genetically modified phage. Antimicrob. Agents Chemother. 48:3817–3822. 10.1128/AAC.48.10.3817-3822.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Westwater C, Schofield DA, Schmidt MG, Norris JS, Dolan JW. 2002. Development of a P1 phagemid system for the delivery of DNA into gram-negative bacteria. Microbiology (Reading, Engl.) 148:943–950 [DOI] [PubMed] [Google Scholar]
- 44. Rai M, Yadav A, Gade A. 2009. Silver nanoparticles as a new generation of antimicrobials. Biotechnol. Adv. 27:76–83. 10.1016/j.biotechadv.2008.09.002 [DOI] [PubMed] [Google Scholar]
- 45. Lu TK, Koeris MS. 2011. The next generation of bacteriophage therapy. Curr. Opin. Microbiol. 14:524–531. 10.1016/j.mib.2011.07.028 [DOI] [PubMed] [Google Scholar]
- 46. Sapranauskas R, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V. 2011. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 39:9275–9282. 10.1093/nar/gkr606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Qi L, Haurwitz RE, Shao W, Doudna JA, Arkin AP. 2012. RNA processing enables predictable programming of gene expression. Nat. Biotechnol. 30:1002–1006. 10.1038/nbt.2355 [DOI] [PubMed] [Google Scholar]
- 48. Ebina H, Misawa N, Kanemura Y, Koyanagi Y. 2013. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 3:2510. 10.1038/srep02510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Swingle B, Markel E, Costantino N, Bubunenko MG, Cartinhour S, Court DL. 2010. Oligonucleotide recombination in gram-negative bacteria. Mol. Microbiol. 75:138–148. 10.1111/j.1365-2958.2009.06976.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang HH, Xu G, Vonner AJ, Church G. 2011. Modified bases enable high-efficiency oligonucleotide-mediated allelic replacement via mismatch repair evasion. Nucleic Acids Res. 39:7336–7347. 10.1093/nar/gkr183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Olsen PA, Randøl M, Luna L, Brown T, Krauss S. 2005. Genomic sequence correction by single-stranded DNA oligonucleotides: role of DNA synthesis and chemical modifications of the oligonucleotide ends. J. Gene Med. 7:1534–1544. 10.1002/jgm.804 [DOI] [PubMed] [Google Scholar]
- 53. Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gibson DG. 2011. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol. 498:349–361. 10.1016/B978-0-12-385120-8.00015-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Russell WM, Klaenhammer TR. 2001. Efficient system for directed integration into the Lactobacillus acidophilus and Lactobacillus gasseri chromosomes via homologous recombination. Appl. Environ. Microbiol. 67:4361–4364. 10.1128/AEM.67.9.4361-4364.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Makarova K, Slesarev A, Wolf Y, Sorokin A, Mirkin B, Koonin E, Pavlov A, Pavlova N, Karamychev V, Polouchine N, Shakhova V, Grigoriev I, Lou Y, Rohksar D, Lucas S, Huang K, Goodstein DM, Hawkins T, Plengvidhya V, Welker D, Hughes J, Goh Y, Benson A, Baldwin K, Lee JH, Díaz-Muñiz I, Dosti B, Smeianov V, Wechter W, Barabote R, Lorca G, Altermann E, Barrangou R, Ganesan B, Xie Y, Rawsthorne H, Tamir D, Parker C, Breidt F, Broadbent J, Hutkins R, O’Sullivan D, Steele J, Unlu G, Saier M, Klaenhammer T, Richardson P, Kozyavkin S, Weimer B, Mills D. 2006. Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. U. S. A. 103:15611–15616. 10.1073/pnas.0607117103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Walker DC, Aoyama K, Klaenhammer TR. 1996. Electrotransformation of lactobacillus acidophilus group A1. FEMS Microbiol. Lett. 138:233–237. 10.1111/j.1574-6968.1996.tb08163.x [DOI] [PubMed] [Google Scholar]
- 58. Lambrecht JA, Schmitz GE, Downs DM. 2013. RidA proteins prevent metabolic damage inflicted by PLP-dependent dehydratases in all domains of life. mBio 4(1):e00033–13. 10.1128/mBio.00033-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Quantitating the removal capacity of genome-targeting CRISPR spacers. A genomic sequence is inserted within the native CRISPR array of E. coli K-12 in the pCRISPR plasmid (see Fig. S2) and transformed into BW25113-T7 cells harboring the two cas-expressing plasmids, pCasA-E and pCas3. The transformation efficiency is then assessed on plates containing l-arabinose, IPTG, and antibiotics that select for all three plasmids. Although this scheme uses three plasmids, more compact schemes are possible, such as using type II systems with a single-guide RNA. Download
Design and insertion of spacer sequences into the pCRISPR plasmid. The protospacer is selected as the 32 nt immediately downstream of a protospacer-adjacent motif, or PAM (AAG, AGG, GAG, and ATG for E. coli K-12 MG1655). The sequence and selected spacer within the ftsA gene used in Fig. 2 are shown at the top. The first 30 bp of the protospacer are integrated into two chemically synthesized oligonucleotides. The last two nucleotides of the spacer (outlined in blue) are fixed because they are part of the XhoI overhang. The overhangs of the annealed oligonucleotides allow insertion into the pCRISPR plasmid digested with KpnI and XhoI. Insertion recreates the KpnI and XhoI sites at the 5′ end and destroys the XhoI site (XhoI′) at the 3′ end. Recreating the KpnI and XhoI sites allows the sequential insertion of additional repeat-spacer pairs into the CRISPR array as long as the 30th nt in each spacer is not a C. The base CRISPR array is contained within the gBlock shown in Table S2. Download
Removal of bacteria using native CRISPR-Cas systems. (A) Removal of Escherichia coli through the native type I-E system. Pure cultures of the wild-type strain (WT) (BW25113) or the hns deletion strain (Δhns; JW1225-2) were transformed with the α-ftsA CRISPR plasmid depicted in Fig. 2A. Transformed cells were plated on LB agar supplemented with l-arabinose and ampicillin. See Fig. 2B for an explanation of the transformation efficiency. Values represent the geometric means and SEM of data from three independent experiments. (B) Removal of Streptococcus thermophilus through the native type II systems. The two systems, termed CRISPR1 and CRISPR3, possess distinct repeat sequences, Cas proteins, and PAMs. Strain LMD-9, harboring the pTRK669 plasmid, was transformed with the pORI28 plasmid, encoding a spacer targeting PAM-flanking protospacers within the lacZ gene (STER_1366; protein ID ABJ66539.1). See the legend for Fig. 2B for an explanation of the transformation efficiency; only the original pORI28 plasmid served as the control plasmid. The gray background indicates the limit of detection of the transformation assay. Values represent the arithmetic means and SEM of data from five independent experiments. The arithmetic means and SEM were employed because many of the experiments resulted in zero colonies. Download
Analysis of viable transformants that escaped removal by the α-ftsA CRISPR plasmid. Plasmid DNA was isolated from viable colonies following transformation with the α-ftsA plasmid. The CRISPR locus then was sequenced using upstream and downstream primers, resulting in the depicted truncations. Red lines designate deletions. NR, no read from sequencing starting upstream and downstream of the CRISPR locus, suggestive of the absence of the entire locus. Download
Compensatory mutations within the ftsA coding region. The three mutations (m2, m5, and m7) were selected to minimize changes to the amino acid sequence of FtsA. The first two mutations (m2 and m5) are silent, whereas the third mutation (m7) changes a valine to an alanine. Numbers above each amino acid designate the location within the coding region of ftsA. The short black bar above the wild type (WT) sequence designates the seed region. Download
Transformation efficiencies with an incomplete type I CRISPR-Cas system in E. coli. Transformations paralleled those shown in Fig. 3, except that the cells harbored pCasA-E′ (− casABCDE) and pCas3 (+ cas3). See the legend for Fig. 2B for an explanation of the transformation efficiency. Download
Impact of simultaneously targeting multiple sites on the transformation efficiency in E. coli K-12. A plasmid encoding a synthetic CRISPR array targeting four different endogenous genes (asd, msbA, ftsA, and nusB) was used. See the legend for Fig. 2B for an explanation of the transformation efficiency. Values represent the geometric means and SEM of data from three independent experiments. In comparison to the α-ftsA plasmid (Fig. 2B), targeting the three additional sites did not significantly alter the relative transformation efficiency (P = 0.48; n = 3). Download
E. coli strains BW25113-T7 and BL21(DE3) can be readily distinguished on LB agar with IPTG and X-Gal. The lacZ gene is disrupted in BW25113-T7 and is intact in BL21(DE3). As a result, BW25113-T7 colonies remain white, whereas BL21(DE3) colonies turn blue on LB agar supplemented with IPTG and X-Gal. Download
Protospacer sequences
Strains, plasmids, and oligonucleotides used in this work