

ABSTRACT
Exploiting mechanisms of utilizing the sugar d-galactose in Escherichia coli as a model system, we explored the consequences of accumulation of critical intermediates of the d-galactose metabolic pathways by monitoring cell growth, metabolites, and transcript profiles. These studies revealed both metabolic network changes far from the d-galactose pathway and changes in the global gene regulatory network. The concentration change of a critical intermediate disturbs the equilibrium state, generating a ripple effect through several metabolic pathways that ends up signaling up- or downregulation of specific sets of genes in a programmed manner to cope with the imbalance. Such long-range effects on metabolites and genetic regulatory mechanisms not only may be a common feature in bacteria but very likely operate during cellular development and differentiation in higher organisms as well as in disease cells, like cancer cells.
IMPORTANCE
Metabolite accumulation can create adverse intracellular conditions that are relieved by compensatory immediate changes of metabolite pools and later changes of transcript levels. It has been known that gene expression is normally regulated by added catabolic substrates (induction) or anabolic end products (repression). It is becoming apparent now that change in the concentration of metabolic intermediates also plays a critical role in genetic regulatory networks for metabolic homeostasis. Our study provides new insight into how metabolite pool changes transduce signals to global gene regulatory networks.
INTRODUCTION
Intracellular accumulation of sugar phosphate intermediates causes “cytotoxicity” (1–5). It has been shown that such intracellular conditions created by accumulation or depletion of certain metabolites change the cellular transcript profile in a specific manner (5, 6). Recently it was proposed that Escherichia coli can measure its glycolytic flux with the Cra transcription factor and uses this signal for metabolic regulation (7). These programmed changes occur to fix or minimize the adverse conditions and restore homeostasis.
The Leloir pathway of d-galactose metabolism (8), conserved from E. coli to humans, is shown in Fig. 1. d-Galactose sensitivity of specific gal mutant cells has been known for more than five decades both in bacteria and in yeast (Saccharomyces cerevisiae) (1, 9–11). A galT defect causes the disease galactosemia in humans (12–15). When given d-galactose, E. coli galE mutants accumulate the intermediates of d-galactose catabolism, including UDP-galactose, that bring about changes in cellular transcript profile, some of which help maintain homeostasis (5). In yeast, the effects of d-galactose on a gal7 (galT homolog) mutant were studied by transcript analysis (6). It was observed that inositol metabolic genes were highly induced in d-galactose-intoxicated gal7-deficient cells, although any relationship between d-galactose metabolism and inositol gene expression was not explored.
FIG 1 .
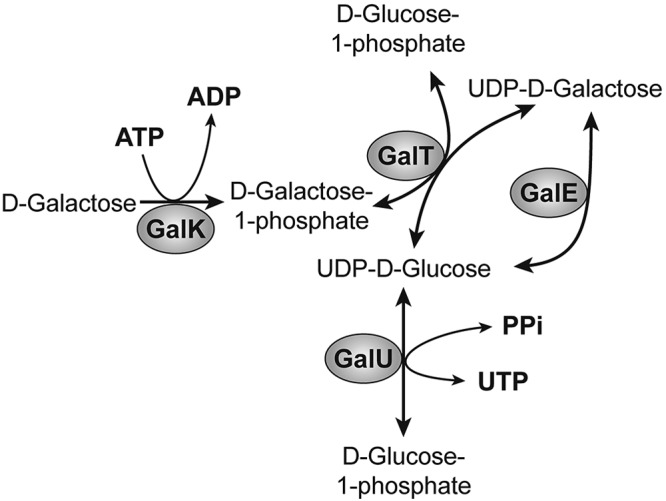
Leloir pathway of d-galactose metabolism in E. coli.
In this article, we report investigation of the cause and effect of cytotoxicity induced by the addition of d-galactose to galT mutant E. coli cells growing on another carbon source and monitoring of their responses in terms of cell growth, metabolite changes, and global gene expression patterns. We discuss the reasons for growth inhibition of galT mutant cells by d-galactose and show how signals are sent from metabolite levels to genetic regulatory systems.
RESULTS
d-Galactose-induced bacteriostasis in galT mutant cells.
When galT mutant cells take in external d-galactose, growth of E. coli cells in any of several carbon sources is retarded, presumably because of accumulation of galactose-1-phosphate (5). Inhibitions of cell growth by accumulation of sugar phosphates are not uncommon (16, 17). We routinely used glycerol or fructose minimal medium to study the effect of d-galactose on growth in a galT mutant. The growth inhibition is severe when d-galactose is added to cells growing in minimal medium containing glycerol. The results describe the effects of d-galactose on galT mutant cells growing in glycerol or fructose minimal media after d-galactose addition.
NTP pool analysis of galT mutants.
It is expected from the metabolic pathway of d-galactose utilization that addition of d-galactose to a galT mutant would cause accumulation of galactose-1-phosphate and likely depletion of ATP (Fig. 1). We analyzed the ribonucleotide triphosphate pools in cells grown in glycerol after d-galactose addition. The ATP pool immediately decreased but gradually recovered (Fig. 2A). Even a 20% reduction in the level of ATP, the most critical nucleoside triphosphate (NTP) in the cell, is expected to have a strong effect on cellular metabolism (18). The ATP recovery is expected because of a variety of ATP-regenerating systems in the cell, including the presence of a robust ATP synthase in E. coli (19). The GTP level did not change significantly; if anything, like ATP it decreased slightly but increased later. The guanosine tetraphosphate (ppGpp) level gradually decreased after d-galactose addition, the cause of which is not clear (18). In contrast, UTP and CTP levels increased in galT mutant cells 5 min after d-galactose addition. As expected, in the absence of the GalT enzyme, UDP-glucose and galactose-1-phosphate accumulate in the cell. UDP-glucose would be converted back to UTP by reverse action of the GalU enzyme leading to accumulation of the latter (Fig. 1), and the fate of accumulated galactose-1-phosphate is mentioned below. The gradual increase in the CTP level is justified because CTP is synthesized from UTP by CTP synthetase (20). Overall, purine nucleoside triphosphate pools transiently decreased and pyrimidine nucleoside triphosphate pools permanently increased after d-galactose addition to galT mutant cells (Fig. 2B).
FIG 2 .
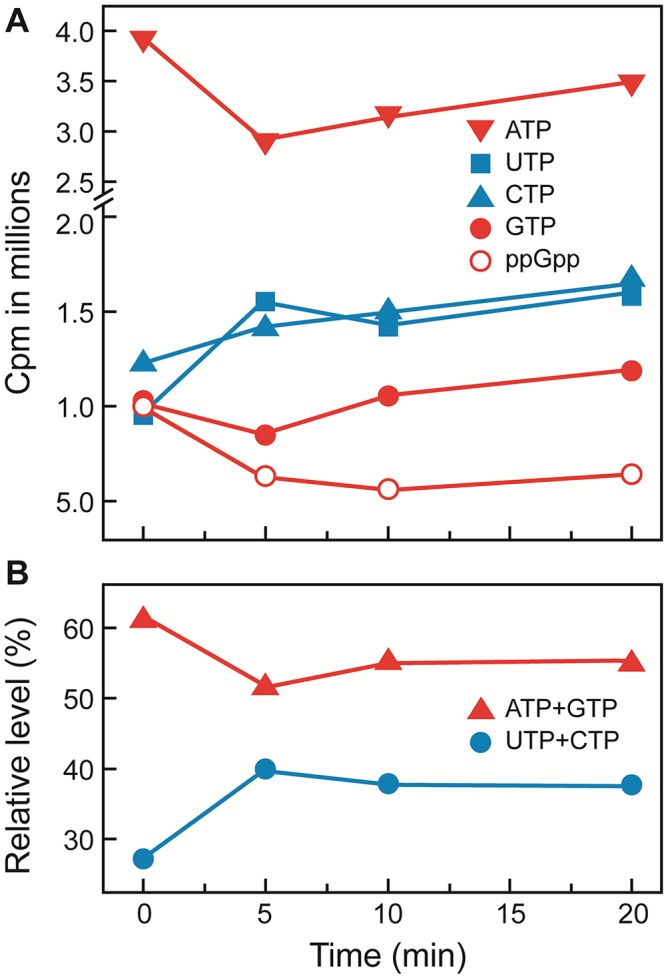
Quantification of thin-layer chromatography of 32P-labeled nucleotides of galT mutants in the presence of d-galactose by ImageQuant. Numbers indicate time (min) after d-galactose addition in the cells.
The metabolite profile of galT mutants.
To confirm the expected accumulation of galactose-1-phosphate, to monitor any ripple effect on equilibrium state of the metabolites, and to fully understand the changes in the NTP pools mentioned above, we analyzed the metabolite profiles in all gal mutants, including the galT mutant, grown in d-fructose with and without d-galactose. The effects of d-galactose on galT mutant cells are apparent on analysis of galactose-1-phosphate-related metabolites. Figure 3 shows the metabolite levels in the E. coli galT mutant strain by plotting the natural log of mean values of ion counts in ionized samples made in the presence versus absence of d-galactose during mass spectrometry analysis (Fig. 3; see Table S2 in the supplemental material). Only P values of <0.1 are shown. (i) We confirmed the expected accumulation of d-galactose and galactose-1-phosphate in the presence of d-galactose in the galT mutant. The level of d-galactose appears to be higher than the level of galactose-1-phosphate; this could be due to many things. First, the induced level of GalK enzyme, which phosphorylates d-galactose, is overwhelmed by excess d-galactose. Second, the product galactose-1-phosphate is subject to dephosphorylation by phosphatases (21). Third, GalK could be product inhibited or even catalyze the reverse reaction when Gal-1-P levels increase. (ii) Glucose, mannose, maltose, maltotriose, and several other hexoses, as well as fructose-6-phosphate and fructose-1,6-bisphosphate, were significantly increased, presumably by anabolic gluconeogenesis. (iii) We also observed a dramatic decrease in the level of precursors of de novo pyrimidine biosynthesis: e.g., N-carbamoylaspartate, orotate, and dihydroorotate in galT mutant cells grown in the presence of d-galactose. Additionally, pyrimidine nucleoside triphosphate precursors or related metabolites (5,6-dihydrouracil, uridine, and cytidine 5′-monophosphate) decreased. The decreases in pyrimidine metabolites are discussed below.
FIG 3 .

Metabolomic profile of galT mutant cells grown in M63 minimal medium containing d-fructose with (w/) and without (w/o) d-galactose (Gal). Abbreviations: FBP, fructose-1,6-bisphosphate; F6P, fructose-6-phosphate; G1P, galactose-1-phosphate. Because of very slow cell growth in glycerol when d-galactose is present in the galT mutant, d-fructose was used as a carbon source in these experiments. Identification of metabolites and determination of their relative amounts were performed by ultrahigh-performance liquid chromatography-mass spectrometry. The values in the graph represent ion counts from a metabolite after ionization of the compound. The analysis was done in three replicates, and any changes reported are statistically significant with a P value of <0.05.
Transcript profile of galT mutant cells.
In order to understand the metabolite changes created by d-galactose in galT mutant cells and how the metabolite changes correlate to the gene expression, we analyzed the transcripts of galT mutant cells grown in glycerol minimal medium with and without d-galactose. The results with or without d-galactose were compared graphically (Fig. 4). The figure shows the RNA levels of all of the genes in E. coli by plotting the model-based analysis of tiling array (MAT) score in the presence versus absence of d-galactose. We observed that 42 genes are upregulated and 187 genes downregulated in the presence of d-galactose compared to those in the absence of d-galactose in galT mutant cells. Incidentally, the levels of expression of these genes are not affected by the presence of d-galactose in wild-type cells, except for the upregulation of the gal regulon members. A list of the 18 most upregulated genes and 72 most downregulated genes outside ±2 standard deviations (SD) is shown in Table S1 in the supplemental material. Although the cause of the up- or downregulation of many of these genes remains to be understood, several features, including the severe effect of d-galactose on glycerol metabolism in galT mutant cells, are discussed below.
FIG 4 .
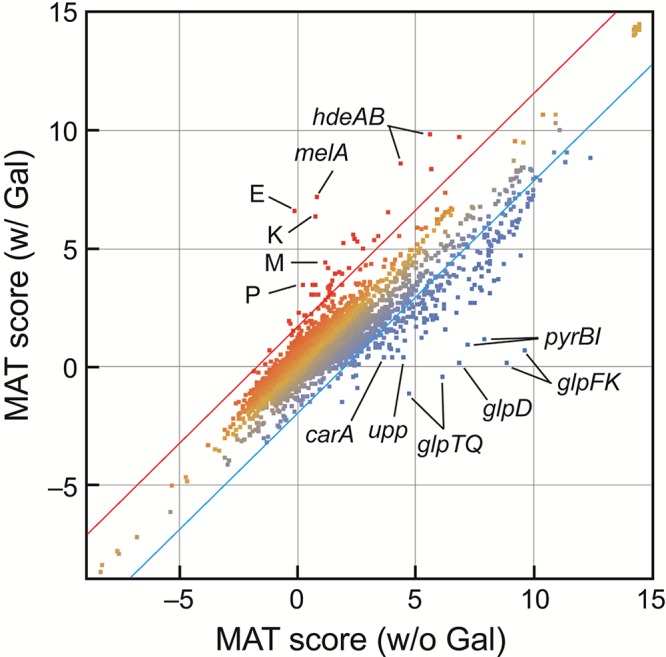
MAT analysis of the transcriptome of galT mutant cells grown in M63 minimal medium containing glycerol in the presence and absence of d-galactose. gal regulon members were clearly induced by d-galactose. E, K, M, and P represent the galE, galK, galM, and galP genes, respectively. Pyrimidine biosynthetic genes (pyrBI, carA, upp, and codBA) as well as glp regulon members are strikingly downregulated by d-galactose. Red and blue lines represent the means ± 2SD, which were used for the selection of the outliers listed in Table S1 in the supplemental material.
The gal regulon.
As mentioned, gal regulon members (galE, galK, galM, and galP) were clearly induced by d-galactose. We note that galT mutant transcripts were not observed because the galT mutation is an in-frame deletion of the entire gene.
Stress and stringent response.
It was previously known and confirmed above that creation of imbalances of critical metabolites brings about what is called cellular stress (1, 5, 22, 23). A cellular stress is defined as a condition that induces synthesis of typical stress-responsive genes, including the hdeA, hdeB, and hdeD genes (24, 25) and the gadA, gadB, and gadC genes (26, 27). We found that galactose-1-phosphate accumulation in the galT mutant upregulates the hde genes as well as the gad genes, although we do not know whether the upregulation is direct or indirect (see Table S1 in the supplemental material). We suggest that galactose-1-phosphate accumulation causes stress intracellularly and thus the hyperinduction of the stress-responsive genes. A striking feature of this stress was substantial downregulation of almost all ribosomal protein genes when the mutant cells are grown in d-galactose and glycerol minimal medium, suggesting onset of a stringent response as well as some stress response perhaps as a result of cessation of growth. In this context, we note that the ppGpp level always decreases, as we observed (Fig. 2A) when pyrimidine or purine metabolites are low (18). Note that under normal stringent conditions, the ppGpp level increases (18, 28).
Glycerol metabolism.
The effects of d-galactose on growth of E. coli galT mutant cells on glycerol as a carbon source in both liquid and solid agar minimal media are shown in Fig. 5. As shown, when d-galactose is added to the liquid culture exponentially growing in glycerol, the cell growth rate was immediately retarded compared to that of the culture with no d-galactose addition (Fig. 5A). No visible cell growth was observed after 60 h of incubation of cells on agar plates under similar conditions (Fig. 5B). We observed, as listed in Table S1 in the supplemental material, a severe downregulation of glp regulon members (glpFK, glpTQ, glpD, glpA, glpB, glpC, and gapA) involved in glycerol metabolism. This explains why d-galactose-mediated growth inhibition is stronger in glycerol minimal medium than in fructose minimal medium. A metabolite of glycerol, sn-glycerol-3-phosphate, induces the glp regulon by altering the conformation of the cognate GlpR regulatory protein (29). Our results show that the d-galactose or galactose-1-phosphate that accumulates in the galT mutant (see Table S2 in the supplemental material) is an inhibitor of growth. The addition of d-galactose to a galK mutant causes accumulation of only d-galactose and not galactose-1-phosphate (30) and does not inhibit cell growth in glycerol medium (5). We propose two models to explain the growth inhibition in glycerol minimal medium by the presence of d-galactose in a galT mutant. (i) Galactose-1-phosphate is the anti-inducer of the GlpR-mediated repression of the glp regulon, thus preventing the synthesis of glycerol enzymes. (ii) Galactose-1-phosphate inhibits the activity of glycerokinase and prevents the formation of the inducer sn-glycerophosphate, thus keeping the synthesis of the glycerol enzymes repressed. We tested an anti-inducer role (model i) of galactose-1-phosphate by introducing a defective (deletion) glpR gene into the galT mutant strain. We monitored the growth of the galT glpR double mutant cells in both liquid and solid glycerol minimal media with and without d-galactose. As expected, both galT single and galT glpR double mutant cells cannot grow on minimal liquid and agar plates containing only d-galactose as the sole carbon source because of impaired metabolic pathway for d-galactose. galT mutant cells cannot grow on both minimal liquid and agar media due to their inability to express glp genes in the presence of d-galactose, as observed in transcript analysis. However, the presence of the glpR mutation in galT mutant cells allowed cell growth under similar conditions in both liquid and solid media. The slight growth advantage of the glpR mutant in glycerol medium in the presence over in the absence of d-galactose is because both sugars are being used as a carbon source in the former case. These results clearly showed that in the absence of the glpR gene, d-galactose failed to inhibit the expression of glp genes in the galT mutant. Thus, galactose-1-phosphate-mediated repression of the glp expression involves the glpR gene. These results support the anti-inducer role (model i) of galactose-1-phosphate. The glpR mutant results, however, do not completely eliminate the glycerokinase inhibitor role of galactose-1-phosphate (model ii). It is possible that glpR mutation causes synthesis of a very high level of glycerokinase and that the small level of galactose-1-phosphate that accumulated in galT mutant (see Table S2 in the supplemental material) is not sufficient to totally inhibit the synthesis of sn-glycerophosphate by glycerokinase. We are currently confirming the anti-inducer role of galactose-1-phosphate in an in vitro transcription system.
FIG 5 .

Growth of galT, galT glpR, and glpR mutant cells in M63 broth (A) and on M63 minimal agar (B) containing glycerol (0.3%) as the sole carbon source in the presence of d-galactose (0.3%). The agar plates were incubated for 60 h at 37°C.
Pyrimidine metabolism.
A noteworthy finding in the transcript analysis of galT mutant cells was the dramatic change of de novo nucleotide biosynthetic mRNAs (Fig. 4). Pyrimidine biosynthetic regulon members (yjgF, carA, upp, pyrB, and pyrI) were very much downregulated (see Table S1 in the supplemental material). This is because of the observed accumulation of UTP and CTP levels in cells. UTP and CTP accumulation represses the expression of genes involved in the biosynthesis of pyrimidine nucleoside triphosphates (31). Such conditions would be expected to lower the levels of biosynthetic precursors of pyrimidines, precisely what was observed in metabolite analysis mentioned above.
Nucleoside supplementation.
Since the levels of UTP and CTP were high and those of ATP and GTP were low (with a later recovery) in the presence of d-galactose in galT mutant cells (Fig. 2), we surmised that the growth reduction of galT mutant cells by d-galactose might be alleviated by the addition of purine nucleotide precursors. We added different nucleosides to the culture medium of galT mutant cells in the presence and absence of d-galactose. The growth arrest of galT mutant cells by d-galactose was partially corrected by addition of purine nucleosides (adenosine or guanosine) but not by pyrimidine nucleosides (uridine or cytidine), suggesting that a reduction in purine nucleosides is partly responsible for their reduced growth (Fig. 6). The major reason for the reduced cell growth might be the repression of genes encoding enzymes of glycerol metabolism by galactose-1-phosphate, as discussed above. In galT glpR double mutant cells, we added the different nucleosides to test the recovery of cellular growth. The growth of d-galactose-stressed galT glpR double mutant cells was improved by addition of purine nucleosides (adenosine and guanosine) but not by that of pyrimidine nucleosides (uridine and cytidine) (Fig. 7).
FIG 6 .

Nucleoside effect on growth of galT mutant cells in M63 minimal medium containing glycerol (0.3%) and/or d-galactose (0.3%). Nucleosides (0.1 mM) and d-galactose were added at the same time at an OD of 0.3, as indicated by arrows. Error bars represent standard deviations. Abbreviations: A, adenosine; G, guanosine; U, uridine; C, cytidine; and Gal, d-galactose.
FIG 7 .

Nucleoside effect on growth of galT glpR double mutant cells in M63 minimal medium containing glycerol (0.3%) and/or d-galactose (0.3%). Nucleosides (0.1 mM) and d-galactose were added at the same time at an OD of 0.3, as indicated by arrows. Error bars represent standard deviations. Abbreviations are as described in the legend to Fig. 6.
DISCUSSION
Exploration of the cellular stress created by intermediary metabolites is not easy, because metabolic reactions are very fast and difficult to stop. As an alternative, we can investigate the equilibrium state of cellular factors under a given condition. It is also hard to know which of two correlated effects is the cause and which is the effect. In this study, we investigated the state of the metabolome and transcriptome under stress created by accumulation of a specific d-galactose intermediates, galactose-1-phosphate, in a d-galactose-nonutilizing mutant (galT defective). Although we have not been able to explain numerous changes that took place both at the metabolic as well as the gene transcriptional levels, several interesting features about intracellular response under the stress have been emerged. These features are summarized below as follows.
(i) We explored a biochemical relationship between d-galactose metabolism and nucleotide levels. Since the former process requires a purine nucleoside triphosphate (ATP in the phosphorylation of d-galactose) and a pyrimidine nucleoside triphosphate (UTP in making UDP-glucose), accumulation of galactose-1-phosphate would lower ATP and nonutilization of galactose-1-phosphate would accumulate UTP in a galT mutant cell. However, any depletion of ATP would be ultimately overcome by ATP produced by various ATP-regenerating systems, as mentioned. We also showed that the UTP accumulation signals UTP biosynthetic genes to be turned off and not to make the pyrimidine nucleoside triphosphates any more (31).
(ii) It has been demonstrated that there are several indicators, called stress responses, that show up when cells are subjected to adverse conditions (heat or cold shock, change in osmolality or pH, etc.). The indicators are upregulation of several genes (e.g., the set of hde genes and gad genes) and extensive downregulation of the ribosomal protein genes and not the ribosomal RNA genes. Our results showed that when there is a sudden change in intracellular metabolite concentrations, the stress response genes are expressed at high levels, suggesting that intracellular metabolite changes are recognized by cells as stress. The cells are programmed at the genetic level to adjust to stress and restore a new balance.
(iii) Our study appears to have resolved a long-standing puzzle in glycerol metabolism in bacteria: the failure to metabolize glycerol when utilization of d-galactose present simultaneously is blocked after initial phosphorylation. We showed that the accumulation of the resulting galactose-1-phosphate intermediate may act as an anti-inducer of genes that encode glycerol utilization enzymes. The anti-inducer is postulated to act by antagonizing the inducing action of sn-glycerol-3-phosphate on the GlpR repressor. Consistently, a glpR deletion obviates the d-galactose sensitivity of galT mutant cells growing on glycerol. A single mutation in a metabolic pathway that leads to accumulation or depletion of an apparently innocuous metabolite intermediate can have ripple effect on the metabolic network (32). The resulting concentration of metabolites at the end of a ripple ends up signaling genes for the purpose of restoring the imbalance.
(iv) Finally, our genetic approach to understand metabolic imbalances exposed a plethora of important surprises about metabolic network connections: how ripple effects of a single biochemical can change a large number of metabolites, how the cell is programmed to deal with the change(s), and how metabolites are intricately connected with each other and to the gene regulatory signaling system. It would not be surprising if such connections guide cellular development and differentiations, as well as operate in disease cells. Our results also made us recall the importance of observations made decades ago and reviewed again that the gross alteration in cellular energy metabolism is one of the defining features of cancer (33–35). Cancer cells shut down the tricarboxylic acid (TCA) cycle (aerobic ATP synthesis) and strongly upregulate the glycolytic pathway (substrate-level ATP synthesis), thereby supporting biosynthetic pathways. These changes in cancer may be due to changes in genetic regulatory mechanisms by metabolite changes, as reported here.
MATERIALS AND METHODS
Bacterial strains.
Bacteriophage P1 lysates of the Keio Collection (36) were used to transduce galT and glpR mutations carrying kanamycin markers into the BW25113 wild-type strain to make isogenic mutant strains. P1 transduction was carried out to introduce a glpR mutation into galT mutant cells to make galT glpR double mutant cells. Subsequently, pCP20 (37) was transformed into galT mutants to delete kanamycin markers, to avoid an unwanted transcriptional polarity in the gal operon. After removal of the kanamycin marker, the temperature-sensitive pCP20 plasmid was cured at 42°C.
DNA tiling arrays.
Tiling array DNA chips (E. coli_Tab520346F) were purchased from Affymetrix, Inc. (Santa Clara, CA). The chips had 1,159,908 probes and a 25-mer probe every 8 bp in both strands of the whole E. coli genome. Each DNA probe overlapping 4 bp with the opposite-strand probe was designed to cover the entire E. coli genome. E. coli galT mutant cells were cultivated in 125-ml Corning flasks containing 30 ml of M63 minimal medium plus glycerol (final concentration, 0.3%) at 37°C. At an optical density at 600 nm (OD600) of 0.5, cell cultures were separated into two flasks. d-Galactose (final concentration, 0.3%) was added to one flask, and the cells were grown for 1.5 h further. Cells were then placed on ice, and RNAprotect bacterial reagent (Qiagen) was added to stabilize the RNA. Total RNAs of cells were purified by RNeasy minikit (Qiagen). Subsequent cDNA synthesis, labeling, hybridization, and staining were performed as previously described (5). Stained tiling arrays were scanned using a GeneChip scanner 3000 (Affymetrix). Standardized signals for each probe in the arrays were generated using the model-based analysis of tiling array (MAT) software (38), which provides a model-based, sequence-specific background correction for each sample. A gene-specific score was then calculated for each gene by averaging all MAT scores (natural log) for all probes under the annotated gene coordinates. Gene annotation was from the ASAP database (39), University of Wisconsin—Madison, for E. coli K-12 MG1655 version m56. Differences of 2 log-scale values in the absence or presence of d-galactose were calculated in galT mutants. Then the genes with significant signal difference in tiling arrays were selected and listed in Table S1 in the supplemental material. ArrayStar software (DNASTAR, Inc., Madison, WI) was employed for diagonal visualization of the data in Fig. 4.
TLC assay.
Analysis of nucleoside triphosphate pools was carried out by thin-layer chromatography (TLC), as previously described (40). Cells were grown in MOPS (morpholinepropanesulfonic acid) medium with glycerol (final concentration, 0.3%) and high phosphate (final concentration, 1.3 mM) for 12 h and then transferred to MOPS medium containing low phosphate (final concentration, 0.3 mM). Inorganic phosphate (32P; final concentration, 150 µCi/ml) was added for labeling of nucleotide phosphates at an OD600 of 0.2. d-Galactose (final concentration, 0.3%) was added at OD600 0.4. Ten microliters of samples was taken and mixed with 10 µl of 2 N formic acid and immediately chilled in dry ice prior to at least three freeze-thaw cycles. Four microliters of each lysate was equilibrated to pH 7.0 with Tris-base solutions and was loaded on polyethyleneimine cellulose TLC plates and chromatographed in phosphate buffer. Amounts of nucleotides were quantified using the ImageQuant PhosphorImager (Molecular Dynamics, CA). Relative levels of nucleotides were calculated by amounts of purine and pyrimidine nucleoside triphosphate divided by total nucleotides, including ppGpp.
Nucleoside supplementation test.
All nucleosides were purchased from Sigma-Aldrich and dissolved in dimethyl sulfoxide (DMSO) to make stock solutions (final concentration, 250 mM). Nucleosides (final concentration, 0.1 mM) and d-galactose (final concentration, 0.3%) were added at an OD600 of 0.3 during cell growth in M63 minimal medium (20 ml in 125-ml flask) containing 0.3% glycerol. Cell cultures were diluted 1:3 or 1:5 in the same medium to accurately measure the optical density during subsequent growth. The optical density of cell cultures at 600 nm was monitored for cell growth using an Ultrospec 3100 Pro spectrophotometer (Amersham Biosciences).
Microarray data accession number.
Tiling array data have been deposited in NCBI’s Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) and are accessible through GEO series accession no. GSE42821.
SUPPLEMENTAL MATERIAL
Significantly affected genes in the presence of d-galactose in galT cells grown in glycerol M63 minimal medium.
Metabolomic profile of galactose-stressed galT strain.
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, and was also supported by the Basic Science Research Program through the National Research Foundation of Korea, funded by the Ministry of Education, Science and Technology (NRF-2012R1A1A2042114), and the KRIBB Research Innovative Program, Korea.
We thank Phuoc Le and Hyun Ju Kim for technical help and Edward D. Karoly (Metabolon, Inc.) for assistance with metabolite analysis.
The authors declare they have no conflicts of interest.
Footnotes
Citation Lee SJ, Trostel A, Adhya S. 2014. Metabolite changes signal genetic regulatory mechanisms for robust cell behavior. mBio 5(1):e00972-13. doi:10.1128/mBio.00972-13.
REFERENCES
- 1. Yarmolinsky MB, Wiesmeyer H, Kalckar HM, Jordan E. 1959. Hereditary defects in galactose metabolism in Escherichia coli mutants. II. Galactose-induced sensitivity. Proc. Natl. Acad. Sci. U. S. A. 45:1786–1791. 10.1073/pnas.45.12.1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Irani MH, Maitra PK. 1977. Properties of Escherichia coli mutants deficient in enzymes of glycolysis. J. Bacteriol. 132:398–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Prasad C, Freese E. 1974. Cell lysis of Bacillus subtilis caused by intracellular accumulation of glucose-1-phosphate. J. Bacteriol. 118:1111–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Englesberg E, Anderson RL, Weinberg R, Lee N, Hoffee P, Huttenhauer G, Boyer H. 1962. l-Arabinose-sensitive, l-ribulose 5-phosphate 4-epimerase-deficient mutants of Escherichia coli. J. Bacteriol. 84:137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee SJ, Trostel A, Le P, Harinarayanan R, Fitzgerald PC, Adhya S. 2009. Cellular stress created by intermediary metabolite imbalances. Proc. Natl. Acad. Sci. U. S. A. 106:19515–19520. 10.1073/pnas.0910586106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Slepak T, Tang M, Addo F, Lai K. 2005. Intracellular galactose-1-phosphate accumulation leads to environmental stress response in yeast model. Mol. Genet. Metab. 86:360–371. 10.1016/j.ymgme.2005.08.002 [DOI] [PubMed] [Google Scholar]
- 7. Kochanowski K, Volkmer B, Gerosa L, Haverkorn van Rijsewijk BR, Schmidt A, Heinemann M. 2013. Functioning of a metabolic flux sensor in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 110:1130–1135. 10.1073/pnas.1202582110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Frey PA. 1996. The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J. 10:461–470 [PubMed] [Google Scholar]
- 9. Nikaido H. 1961. Galactose-sensitive mutants of Salmonella. I. Metabolism of galactose. Biochim. Biophys. Acta 48:460–469. 10.1016/0006-3002(61)90044-0 [DOI] [PubMed] [Google Scholar]
- 10. Kalckar HM, Anderson EP, Isselbacher KJ. 1956. Galactosemia, a congenital defect in a nucleotide transferase: a preliminary report. Proc. Natl. Acad. Sci. U. S. A. 42:49–51. 10.1073/pnas.42.2.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mumma JO, Chhay JS, Ross KL, Eaton JS, Newell-Litwa KA, Fridovich-Keil JL. 2008. Distinct roles of galactose-1P in galactose-mediated growth arrest of yeast deficient in galactose-1P uridylyltransferase (GALT) and UDP-galactose 4′-epimerase (GALE). Mol. Genet. Metab. 93:160–171. 10.1016/j.ymgme.2007.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lai K, Elsas LJ, Wierenga KJ. 2009. Galactose toxicity in animals. IUBMB Life 61:1063–1074. 10.1002/iub.262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Riehman K, Crews C, Fridovich-Keil JL. 2001. Relationship between genotype, activity, and galactose sensitivity in yeast expressing patient alleles of human galactose-1-phosphate uridylyltransferase. J. Biol. Chem. 276:10634–10640. 10.1074/jbc.M009583200 [DOI] [PubMed] [Google Scholar]
- 14. Fridovich-Keil JL. 2006. Galactosemia: the good, the bad, and the unknown. J. Cell. Physiol. 209:701–705. 10.1002/jcp.20820 [DOI] [PubMed] [Google Scholar]
- 15. Bosch AM. 2006. Classical galactosaemia revisited. J. Inherit. Metab. Dis. 29:516–525. 10.1007/s10545-006-0382-0 [DOI] [PubMed] [Google Scholar]
- 16. Kadner RJ, Murphy GP, Stephens CM. 1992. Two mechanisms for growth inhibition by elevated transport of sugar phosphates in Escherichia coli. J. Gen. Microbiol. 138:2007–2014. 10.1099/00221287-138-10-2007 [DOI] [PubMed] [Google Scholar]
- 17. Vanderpool CK, Gottesman S. 2007. The novel transcription factor SgrR coordinates the response to glucose-phosphate stress. J. Bacteriol. 189:2238–2248. 10.1128/JB.01689-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Potrykus K, Cashel M. 2008. (p)ppGpp: still magical? Annu. Rev. Microbiol. 62:35–51. 10.1146/annurev.micro.62.081307.162903 [DOI] [PubMed] [Google Scholar]
- 19. Vik SB, Ishmukhametov RR. 2005. Structure and function of subunit a of the ATP synthase of Escherichia coli. J. Bioenerg. Biomembr. 37:445–449. 10.1007/s10863-005-9488-6 [DOI] [PubMed] [Google Scholar]
- 20. Long CW, Pardee AB. 1967. Cytidine triphosphate synthetase of Escherichia coli B. I. purification and kinetics. J. Biol. Chem. 242:4715–4721 [PubMed] [Google Scholar]
- 21. Diepenbrock F, Heckler R, Schickling H, Engelhard T, Bock D, Sander J. 1992. Colorimetric determination of galactose and galactose-1-phosphate from dried blood. Clin. Biochem. 25:37–39. 10.1016/0009-9120(92)80043-G [DOI] [PubMed] [Google Scholar]
- 22. Klauck E, Hengge R. 2012. Sigmas-controlling networks in Escherichia coli, p 1–26 In Filloux AAM, Bacterial regulatory networks. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 23. Ayala JA, Cava F, de Pedro MA. 2012. Cell wall stress-sensing regulatory systems in gram-negative bacteria, p 1–18 In Requena JM, Stress response in microbiology. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 24. Gajiwala KS, Burley SK. 2000. HDEA, a periplasmic protein that supports acid resistance in pathogenic enteric bacteria. J. Mol. Biol. 295:605–612. 10.1006/jmbi.1999.3347 [DOI] [PubMed] [Google Scholar]
- 25. Tucker DL, Tucker N, Conway T. 2002. Gene expression profiling of the pH response in Escherichia coli. J. Bacteriol. 184:6551–6558. 10.1128/JB.184.23.6551-6558.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Biase D, Tramonti A, Bossa F, Visca P. 1999. The response to stationary-phase stress conditions in Escherichia coli: role and regulation of the glutamic acid decarboxylase system. Mol. Microbiol. 32:1198–1211. 10.1046/j.1365-2958.1999.01430.x [DOI] [PubMed] [Google Scholar]
- 27. Wei Y, Lee JM, Richmond C, Blattner FR, Rafalski JA, LaRossa RA. 2001. High-density microarray-mediated gene expression profiling of Escherichia coli. J. Bacteriol. 183:545–556. 10.1128/JB.183.2.545-556.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schneider DA, Gaal T, Gourse RL. 2002. NTP-sensing by rRNA promoters in Escherichia coli is direct. Proc. Natl. Acad. Sci. U. S. A. 99:8602–8607. 10.1073/pnas.132285199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin EC. 1976. Glycerol dissimilation and its regulation in bacteria. Annu. Rev. Microbiol. 30:535–578. 10.1146/annurev.mi.30.100176.002535 [DOI] [PubMed] [Google Scholar]
- 30. Wu HC, Kalckar HM. 1966. Endogenous induction of the galactose operon in Escherichia coli K12. Proc. Natl. Acad. Sci. U. S. A. 55:622–629. 10.1073/pnas.55.3.622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Turnbough CL, Jr, Switzer RL. 2008. Regulation of pyrimidine biosynthetic gene expression in bacteria: repression without repressors. Microbiol. Mol. Biol. Rev. 72:266–300. 10.1128/MMBR.00001-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Van Dyk TK, LaRossa RA. 1987. Involvement of ack-pta operon products in alpha-ketobutyrate metabolism by Salmonella typhimurium. Mol. Gen. Genet. 207:435–440. 10.1007/BF00331612 [DOI] [PubMed] [Google Scholar]
- 33. Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144:646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 34. Warburg O, Posener K, Negelein E. 1924. Über den Stoffwechsel der Carcinomzelle. Biochem. Zeitschr. 152:309–344 [Google Scholar]
- 35. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. 2008. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7:11–20. 10.1016/j.cmet.2007.10.002 [DOI] [PubMed] [Google Scholar]
- 36. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. 10.1016/0378-1119(95)00193-A [DOI] [PubMed] [Google Scholar]
- 38. Johnson WE, Li W, Meyer CA, Gottardo R, Carroll JS, Brown M, Liu XS. 2006. Model-based analysis of tiling-arrays for ChIP-chip. Proc. Natl. Acad. Sci. U. S. A. 103:12457–12462. 10.1073/pnas.0601180103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Glasner JD, Liss P, Plunkett G, III, Darling A, Prasad T, Rusch M, Byrnes A, Gilson M, Biehl B, Blattner FR, Perna NT. 2003. ASAP, a systematic annotation package for community analysis of genomes. Nucleic Acids Res. 31:147–151. 10.1093/nar/gkg125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cashel M, Lazzarini RA, Kalbacher B. 1969. An improved method for thin-layer chromatography of nucleotide mixtures containing 32 P-labelled orthophosphate. J. Chromatogr. 40:103–109. 10.1016/S0021-9673(01)96624-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Significantly affected genes in the presence of d-galactose in galT cells grown in glycerol M63 minimal medium.
Metabolomic profile of galactose-stressed galT strain.