

ABSTRACT
Sulfur is an important element in sustaining microbial communities present in hydrothermal vents. Sulfur oxidation has been extensively studied due to its importance in chemosynthetic pathways in hydrothermal fields; however, less is known about sulfate reduction. Here, the metagenomes of hydrothermal chimneys located on the ultraslow-spreading Southwest Indian Ridge (SWIR) were pyrosequenced to elucidate the associated microbial sulfur cycle. A taxonomic summary of known genes revealed a few dominant bacteria that participated in the microbial sulfur cycle, particularly sulfate-reducing Deltaproteobacteria. The metagenomes studied contained highly abundant genes related to sulfur oxidation and reduction. Several carbon metabolic pathways, in particular the Calvin-Benson-Bassham pathway and the reductive tricarboxylic acid cycles for CO2 fixation, were identified in sulfur-oxidizing autotrophic bacteria. In contrast, highly abundant genes related to the oxidation of short-chain alkanes were grouped with sulfate-reducing bacteria, suggesting an important role for short-chain alkanes in the sulfur cycle. Furthermore, sulfur-oxidizing bacteria were associated with enrichment for genes involved in the denitrification pathway, while sulfate-reducing bacteria displayed enrichment for genes responsible for hydrogen utilization. In conclusion, this study provides insights regarding major microbial metabolic activities that are driven by the sulfur cycle in low-temperature hydrothermal chimneys present on an ultraslow midocean ridge.
IMPORTANCE
There have been limited studies on chimney sulfides located at ultraslow-spreading ridges. The analysis of metagenomes of hydrothermal chimneys on the ultraslow-spreading Southwest Indian Ridge suggests the presence of a microbial sulfur cycle. The sulfur cycle should be centralized within a microbial community that displays enrichment for sulfur metabolism-related genes. The present study elucidated a significant role of the microbial sulfur cycle in sustaining an entire microbial community in low-temperature hydrothermal chimneys on an ultraslow spreading midocean ridge, which has characteristics distinct from those of other types of hydrothermal fields.
INTRODUCTION
Deep-sea hydrothermal vents are complex dynamic habitats that feature steep thermal and chemical gradients (1). Previous studies of microbial communities have indicated that the microbial ecosystems in hydrothermal fields are mostly sustained by energy derived from inorganic redox reactions between electron donors (e.g., H2, H2S, Fe2+, and CH4) and acceptors (e.g., O2, NO3−, Fe3+, SO42−, and CO2) (2). Furthermore, diverse microbial populations participate in many important biogeochemical processes simultaneously, such as the nitrogen, sulfur, and carbon cycles.
Within the microbial sulfur cycle, sulfur oxidation has been studied extensively as one of the most important microbial chemosynthetic pathways in hydrothermal vent systems, while information about sulfate reduction remains limited. The available information regarding sulfate reduction includes the isolation of sulfate-reducing bacteria and archaea from deep-sea hydrothermal habitats (3, 4) and the study of heterotrophic sulfate reduction rates in Middle Valley hydrothermal vents (5). The ecological role of sulfate-reducing processes coupled with sulfur oxidation in hydrothermal fields remains unclear. Microbial sulfur cycling has been proposed as a driving force for bacterial survival in surface sediments of the ultramafic-hosted Logatchev hydrothermal field that are heated via conduction, although high levels of dissolved methane and hydrogen have been observed in this region (6). It is anticipated that evidence obtained from functional genes will support the potential coupling of sulfur oxidation and sulfate reduction processes in hydrothermal chimneys.
Using a metagenomic approach, the dominant Thiomicrospira-like group, consisting of sulfur-oxidizing chemolithoautotrophs, was observed in the carbonate chimney at the serpentinite-hosted Lost City Hydrothermal Field on the slow-spreading Mid-Atlantic Ridge (7). This finding indicated that sulfur oxidation plays an important role in energy-generating processes, although information regarding sulfate-reducing processes remains scarce (7, 8). Similarly, despite a metagenomics study examining the black smoker chimney of the Mothra Hydrothermal Vent Field on the intermediate-spreading Juan de Fuca Ridge, information about microbial sulfur cycling is limited to sulfur oxidation (9). To date, the mechanism by which a microbial community can be fueled by sulfate reduction in some hydrothermal fields is unclear. In particular, metagenomic approaches have not been widely applied in studies of energy generation by the microbial sulfur cycle in hydrothermal systems.
In hydrothermal vent systems, short-chain alkanes are formed over relatively short geologic time scales via thermogenic processes, and they are often present at high concentrations. For example, elevated concentrations of methane, short-chain alkanes, and formate have been observed as products of the reaction between ultramafic rocks and water during serpentinization in the Lost City Hydrothermal Field (10). Sulfate reduction could be coupled with short-chain alkane oxidation over time and across temperatures (11). The pyrosequencing results for 16S ribosomal RNA suggest that deltaproteobacterial phylotypes were the phylotypes responsible for mediating oxidation of C2-to-C4 alkanes, particularly a novel sulfate-reducing lineage (11). However, there is no in situ evidence from hydrothermal fields examining the association between sulfate reduction and short-chain alkane oxidation. Further efforts are necessary to identify the functional genes responsible for carbon utilization by sulfate-reducing bacteria.
Potential hydrothermal activity on an ultraslow midocean ridge was first reported in the deep sea on the Southwest Indian Ridge (SWIR) between 58°25′E and 60°15′E (Fig. 1) (12). The first active hydrothermal vent (37°47′S, 49°39′E) was discovered on the SWIR, which features low temperatures and enrichment for chimney sulfides, such as pyrite, marcasite, sphalerite, and chalcopyrite (13–16). The presence of a hydrothermal field in such an ultraslow-spreading ridge provides an unprecedented opportunity to better understand the sulfur cycle mediated by microbes in low-temperature hydrothermal systems. Segments 27 to 29 between the Indomed and Gallieni transform faults have been affected by magma increase since 8 to 10 Ma (million years ago), as described previously (14). The detection of weak redox potential (Eh) and H2S anomalies in segment 27 indicates the presence of low-temperature hydrothermal fields (14). In the present study, the metagenomes of chimneys made of sulfides on the SWIR were pyrosequenced to clarify sulfur cycle-related microbial community structures, as well as microbial linkages with the cycles of other elements, such as carbon and nitrogen.
FIG 1 .

Map of the sampling sites on the Southwest Indian Ridge (SWIR) (modified from Zhu et al. [15] with permission). (a) Location of the SWIR, close to the Central Indian Ridge (CIR) and the Southeast Indian Ridge (SEIR). (b) Magnified view of the bathymetric map of the location indicated by the red box in panel a.
RESULTS
Geochemical features.
X-ray diffraction (XRD) analysis revealed that chalcopyrite (CuFeS2), kusachiite (CuBi2O4), and pyrite (FeS2) were abundant in sample S32, whereas sample S35 contained pyrite and manganese phosphate hydrate Fig. S1, consistent with the results of a previous study conducted in the same region (16). The concentrations of inorganic nitrogen compounds and dissolved organic carbon in the pore water of S32 were higher than those in S35 (Table 1). A remarkably high concentration of sulfate was also recorded in S32 (Table 1).
TABLE 1 .
Geophysical features of sampling sites, physicochemical characteristics of samples, and general information about the metagenomic data
| Characteristic | S32 | S35 |
|---|---|---|
| Location | 37°66′S, 50°46′E | 37°78′S, 50°65′E |
| Sampling date | 14 December 2008 | 15 December 2008 |
| Depth (m) | 1,744 | 2,783 |
| Description | Brown polymetallic ooze | Black sulfide |
| NH4+ (µM) | 166.71 | 91.74 |
| NO2− (µM) | 8.79 | 1.73 |
| NO2− plus NO3− (µM) | 9.2 | 2.56 |
| DOC (mg/liter) | 106.3 | 60.6 |
| TN (mg/liter) | 10.32 | 1.97 |
| SO42− (mM) | 27.91 | 14.69 |
| Amt of cells/g (wet wt) | (1.93 ± 0.40) × 105 | (8.35 ± 0.26) × 104 |
| Metal concn (mg/kg) | ||
| Al | 130 | 7,581.2 |
| Ca | 337.3 | 6,697.1 |
| Co | 647.7 | 199.1 |
| Cr | 446.7 | 377.5 |
| Cu | 11,145.1 | 38,944.1 |
| Fe | 274,811.5 | 336,762.8 |
| Mg | 356.8 | 5184.8 |
| Mn | ND. | 340.3 |
| Ni | 51.3 | 69.4 |
| Pb | 80.1 | 1006.1 |
| V | 69.8 | 558 |
| Zn | 1,721.8 | 6,018.8 |
| Metagenome | ||
| Size (Mb) | 450.9 | 447.2 |
| No. of reads | 938,875 | 879,022 |
| Avg length (bp) | 480 | 508 |
| GC content (%) | 43 | 43 |
| No. of contigs | 15,082 | 13,257 |
| Avg contig size (bp) | 1,522 | 1,326 |
| Largest contig (kb) | 73.5 | 69.4 |
| N50 | 1,480 | 1,365 |
Taxonomic compositions by metagenomes.
General information about the two pyrosequenced metagenomes is presented in Table 1. In total, 984 and 1,143 reads >100 bp long were identified as 16S rRNA genes in S32 and S35, respectively. Although a potential effect of multiple copies of the 16S rRNA gene from the same organism on the evaluation of abundance has been raised, this definitely needs much more in-depth sequencing efforts or single-cell genomics methods to resolve. The RDP classifier assigned most of the 16S rRNA genes to the Bacteria, whereas only 18 reads in each metagenome were assigned to the Archaea. Proteobacteria and Bacteroidetes were identified as dominant groups with respect to both sulfides, although the Firmicutes were only dominant in S35 (Fig. 2A). Delta- and Gammaproteobacteria represented the most abundant classes for both sulfides, followed by Epsilon-, Alpha- and Zetaproteobacteria. At the genus level, most of the gammaproteobacterial reads were unclassified, while the Thiomicrospira was the most abundant genus among the classified Gammaproteobacteria (Fig. 2B). Similarly, the deltaproteobacterial communities were dominant in both metagenomes, based on unclassified reads belonging to the family Desulfobulbaceae. In contrast, classified reads were well represented by the genus Desulfobulbus (Fig. 2C).
FIG 2 .
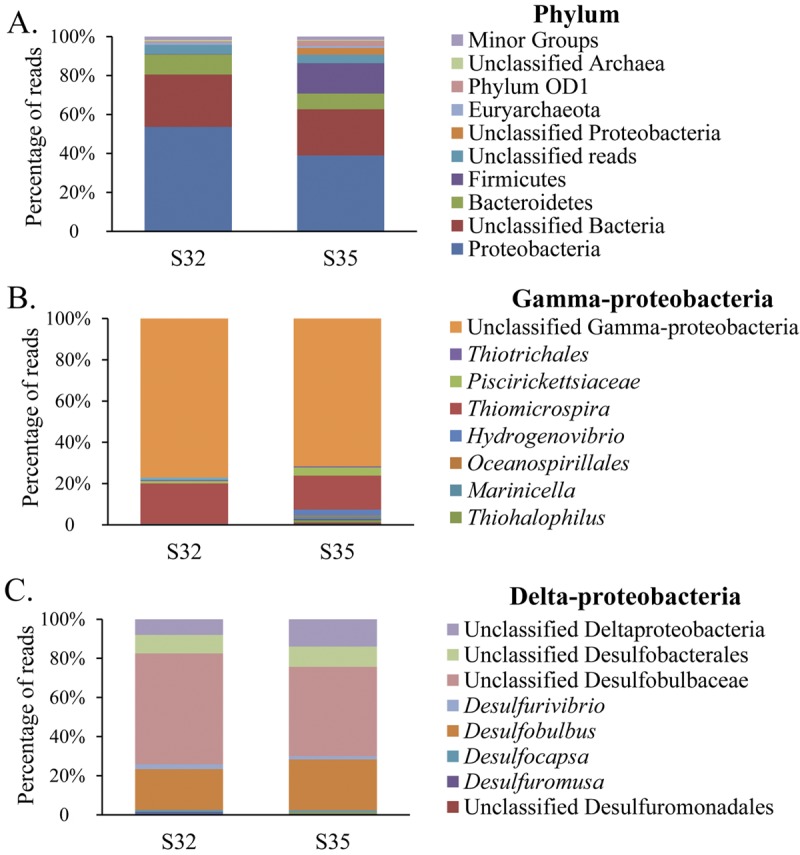
Taxonomic compositions of microbial communities in hydrothermal sulfide chimneys based on 16S rRNA gene sequences retrieved from metagenomes using Meta-RNA software. (A) Phylum-level taxonomic compositions of samples S32 and S35. (B and C) Genus-level taxonomic compositions of the two dominant gamma- and deltaproteobacterial communities, respectively.
Based on the protein-coding sequences, most of the reads were assigned to the Bacteria (96.12% for S32 and 95.61% for S35), consistent with the results obtained for the 16S rRNA genes. Archaea accounted for 1.36% and 1.48% of the reads in S32 and S35, respectively. Less than 1% (0.47% for S32 and 0.78% for S35) of the reads for both sulfides were assigned to the Eukaryota. BLAST searches against the NCBI-nr database provided no results for more than one-third of the reads (37.98% for S32 and 36.52% for S35), and therefore, these reads could not be assigned to a taxonomic designation.
In the bacterial group, Alpha-, Gamma-, and Deltaproteobacteria and Bacteroidetes were highly abundant, consistent with the 16S rRNA gene analysis. In contrast, the Epsilonproteobacteria represented approximately 5% of the annotated reads in both metagenomes. At the genus level, the most abundant reads were closely related to the Desulfobulbus (Fig. 3). There was an abundance of sulfate-reducing bacteria (e.g., Desulfobacterium and Desulfurivibrio), sulfur-oxidizing bacteria (e.g., Thiomicrospira, Sulfurimonas, and Thioalkalivibrio), and iron-oxidizing or -reducing bacteria (e.g., Mariprofundus and Geobacter), which indicated that sulfur and iron play an important role in fueling the microbes present in these habitats (Fig. 3). Additionally, there was a relatively high abundance of bacteria associated with nitrogen metabolism, such as Nitrosococcus (Fig. 3). In contrast to their levels of abundance in S32 and S35, the genera Thiomicrospira and Nitrosococcus were the most abundant groups in the metagenomes from the Lost City and the Juan de Fuca Ridge, respectively (Fig. 3).
FIG 3 .

The most abundant genera (>1%) retrieved from the two sulfide metagenomes and the abundance of comparable genera from another two metagenomes from hydrothermal fields (LC, lost city; JDF, Juan de Fuca).
Sulfur metabolism-related functional genes.
It is hypothesized that functional genes for sulfur oxidation (Sox) participate in a thiosulfate-oxidizing multienzyme system. These genes include SoxXYZABDEFGH, which is capable of oxidizing various reduced sulfur compounds to sulfate (17). Such a system was potentially identified in S32 and S35 (Fig. 4 and 5A). In the present study, genes encoding SoxB were retrieved (234 reads in S32 and 201 reads in S35) and classified at the genus level (Fig. 5A). Ten assembled soxB gene sequences were used for the phylogenetic analysis, based on the deduced amino acid sequences. Several unique types of soxB with large genetic distances were identified (see Fig. S2 in the supplemental material). A large majority of soxB genes (63 from S32 and 36 from S35) were considered to be similar to one fosmid (Fosmid_sox1, ACZ28657.1) from the Juan de Fuca Ridge, which has been proposed to be close to those harbored in Halothiobacillus hydrothermalis and Halothiobacillus neapolitanus (9). In the present study, this fosmid sequence was grouped with the soxB genes from the Gammaproteobacteria strain HIMB30, Thiomicrospira crunogena, and Thioalkalimicrobium-like bacteria, supported by solid statistical analyses. The large genetic distances suggested by the long branches between the soxB sequences from the present study and other soxB sequences indicated different phylogenetic positions for the sulfur-oxidizing bacteria analyzed herein, designated the SWIR1 group (Fig. 5; Fig. S2). Another fosmid-derived soxB sequence (Fosmid_sox2, ACZ28668.1) was similar to 1 contig retrieved in the present study and to H. neapolitanus (9) (Fig. S2). In addition, several sulfur-oxidizing lineages were identified in the SWIR that exhibited various sulfur oxidation abilities. For example, contig0004 was similar to the soxB genes from Sulfurimonas. Contig0003 exhibited a close relationship with genes from Neptuniibacter, and contig00015 was similar to the soxB genes from Beggiatoa. However, contig0001, contig0005, and contig0008 displayed large genetic differences compared with the sequences collected to date, indicating that they are completely novel lineages (Fig. S2). Within these two metagenomes, several genera with different abundances of soxB were observed, such as Thiomicrospira, Thiorhodococcus, and Halothiobacillus, with a higher abundance in S35 than in S32 (Fig. 5B). Other than Sox, a paucity of sulfite oxidase (EC 1.8.3.1, K00387) and sulfite dehydrogenase (EC 1.8.2.1, K05301) was observed in both metagenomes; however, these 2 enzymes were absent in the metagenome from the Juan de Fuca Ridge (9).
FIG 4 .

Functional genes of sulfur-oxidizing and sulfate-reducing bacteria and their abundance in both metagenomes. (A) Enzymes associated with intracellular sulfur metabolism in sulfur-oxidizing and sulfate-reducing bacteria. A, B, C, J, K, O, and P indicate the subunit for the according enzyme. OM, outer membrane; HYD, hydrogenase; ACL, ATP citrate lyase; sat, sulfate adenylyltransferase; APS, adenosine 5'-phosphosulfate; APR, adenosine 5'-phosphosulfate reductase; DSR, dissimilatory sulfite reductase. (B) Abundance of the genes corresponding to the enzymes shown in panel A. The functions of the genes shown in panel B are based on proposed models from Sheik et al. (18) and Pereira et al. (19).
FIG 5 .

Taxonomic assignments of soxB, cbbL/M, aprA, and acetyl-CoA synthetase genes in chimney sulfides, showing relative abundances as percentages. The genes were extracted from both sulfide metagenomes. The genera of these genes were predicted based on the best hits retrieved via BLAST analysis against the NCBI-nr database (E value, <10−4; Bit score, >60).
An enrichment for genes involved in sulfate reduction was also observed, including the aprA and aprB genes encoding adenylyl-sulfate reductase subunits A and B (EC 1.8.99.2, K00394 and K00395), the sulfate reductase subunits A and B (EC 1.8.99.3, dsrA and dsrB, K11180 and K11181), ATP sulfurylase (EC 2.7.7.4, K00955 to K00958), and adenylyl-sulfate kinase (EC 2.7.1.25, K00860, K00955, and K13811) (Fig. 4 and 5). The most abundant gene retrieved herein, aprA, was classified to the genus level (Fig. 5C). Almost 30% of all reads in both metagenomes were assigned to the genus Desulfobulbus (Fig. 5C), consistent with the phylogenetic analysis (see Fig. S8 in the supplemental material). Based on the phylogenetic tree, most of the assembled dsrB genes clustered with the Desulfobulbus genus (Fig. S3). One lineage was close to the sulfur-oxidizing bacteria (the Thiocapsa genus and some symbionts) (Fig. S3). In addition, several contigs (contigs 2, 5, and 10) displayed a larger genetic distance than the sequences available in GenBank. Moreover, the dsr gene cluster was identified in 1 longer contig in S32 and showed a high identity with genes of Desulfobulbus propionicus (Fig. S4).
Carbon metabolism genes.
In the dark world of deep-sea hydrothermal fields, CO2 fixation pathways, such as the Calvin-Benson-Bassham (CBB) cycle and the reductive tricarboxylic acid (rTCA) cycle (20–22), are critical. A total of 206 and 219 reads for cbbL/M (two forms, cbbL and cbbM), which encodes the key CBB cycle enzyme ribulose-1,5-bisphosphate carboxylase/oxygenase, were identified in S32 and S35, respectively. In S32 and S35, 100 and 109 reads, respectively, were recovered for cbbP, which encodes phosphoribulokinase (EC 2.7.1.19) (Fig. 4 and 5). The genus names, as assigned by the cbbL/M reads, were summarized in both sulfide metagenomes (Fig. 6B). It was not surprising that most of these reads were similar to those retrieved from the hydrothermal fields. The cbbL/M sequences shared a high similarity with those retrieved from purple sulfur bacteria, sulfur-oxidizing bacteria (e.g., Thiothrix, Thimicrospira, Thiomonas, and Thiocapsa) and iron-oxidizing bacteria (e.g., Mariprofundus) (Fig. 5B). This finding was supported by a phylogenetic analysis based on the deduced CbbL/M amino acid sequences (see Fig. S5 in the supplemental material). Two additional lineages of CbbL/M separated by long branches, were identified (Fig. S5). The largest difference between these two metagenomes was the higher abundance of several genera in S35 than in S32, including Hydrogenovibrio, Magnetovibrio, Methanocaldococcus, Methylophaga, Methanothermococcus, Thioalkalicoccus, and Halorhodospira (Fig. 5B).
FIG 6 .

Two-way clustering analysis of KEGG gene frequencies in metagenomes from the Southwest Indian Ridge Hydrothermal Field and two other metagenomes from hydrothermal fields (LC, Lost City; JDF, Juan de Fuca). Genes were positioned in 4 regions of the heat map (S32, S35, LC, and JDF) and were associated with different KEGG pathways. Numbers in parentheses represent the numbers of genes identified and of the corresponding enzymes in the indicated complete pathway.
We identified ATP-citrate lyase (acl, EC 2.3.3.8, K01648, 52 and 42 reads in S32 and S35, respectively), isocitrate dehydrogenase (NADP+) (EC 1.1.1.42, K00031, 363 and 357 reads in S32 and S35, respectively), 2-oxoglutarate:ferredoxin oxidoreductase (EC 1.2.7.3, K00174-K00177) and pyruvate:ferredoxin oxidoreductase (EC 1.2.7.1, K00169 to K00172), which are involved in the complete rTCA cycle (Fig. 4). Most of the ATP-citrate lyase genes identified herein shared a high identity with those from Sulfurimonas in hydrothermal fields, such as the Mid-Okinawa Trough Hydrothermal Field (23) and the black smoker chimneys on the Mid-Atlantic Ridge (21 of 51 reads in S32; 17 of 41 reads in S35) (24). The majority of these species, classified according to the genes mentioned above, were found to be sulfur oxidizers. This result suggested that autotrophic carbon fixation plays an important role in these species.
Methyl coenzyme M reductase (EC 2.8.4.1, mcrA, K00399, K00401, and K00402) catalyzes the final step in methanogenesis to produce methane. In addition, it can function in reverse in the anaerobic oxidation of methane. However, this enzyme was absent in both metagenomes. In contrast, a higher abundance of reads assigned to acetyl-coenzyme A (CoA) synthetase (EC 6.2.1.1, K01895) was identified in both sulfide metagenomes from the present study (Fig. 5D). The majority of reads could be assigned to sulfate-reducing bacteria at the genus level (e.g., Desulfobulbus and Desulfovibrio) (Fig. 5D). The high abundance of sulfate-reducing bacteria indicated that various short-chain organic compounds could be an important carbon source for these species in this hydrothermal field. A phylogenetic tree based on the deduced acetyl-CoA synthetase amino acid sequence also revealed that sulfate-reducing bacteria represented an abundant group carrying this enzyme (see Fig. S6 in the supplemental material). Additionally, phylogenetic and genus assignment analyses revealed that sulfur-oxidizing bacteria carried this enzyme (Fig. 5D; Fig. S6). Interestingly, 2 contigs grouped with the methylotrophic and ammonia-oxidizing bacteria, as supported by the genus assignment and phylogenetic analyses (Fig. 5D; Fig. S6). Moreover, based on a U test, 2 metabolic pathways, 1,4-dichlorobenzene degradation and tetrachloroethene degradation, were identified in both metagenomes. These 2 pathways had essentially complete genes, as deduced from KEGG gene cluster analysis, in contrast to those from the Lost City and the Juan de Fuca Ridge (Fig. 6).
Denitrification-related genes and hydrogenases.
Denitrification-related genes were identified in both metagenomes in the present study, most of which were similar to those from the sulfur-oxidizing bacteria. For example, nar/nap (ferredoxin-type nitrate reductase, EC 1.7.99.4) is responsible for nitrate reduction. The nar genes are similar to those from Sulfurimonas autotrophica and Thiobacillus denitrificans, whereas the nap genes are closely related to those from Thiomicrospira crunogena, Ralstonia eutropha, and Cupriavidus metallidurans. In the present study, some of the identified NarG proteins displayed an identity of 91.36% with the deduced protein (NarG, ACZ28645) found in the Juan de Fuca Ridge. These NarG sequences were closely related to the one from Thiobacillus denitrificans, which is an obligate chemolithoautotrophic bacterium. Thiobacillus denitrificans is capable of generating energy by coupling the oxidation of reduced sulfur compounds to denitrification (9). The nir products (nitrite reductase) include several types of enzymes that were overrepresented in the present study, in particular EC 1.7.2.1 (NO forming) (K00368; 157 and 130 reads in S32 and S35, respectively), which is similar to the enzymes in Sulfurimonas denitrificans and Sulfurimonas autotrophica. Other abundant types of nir include (i) NAD(P)H reductase (EC 1.7.1.4, K00362 and K00363), similar to the sulfur-oxidizing symbionts “Candidatus Endoriftia persephone” from the East Pacific Rise hydrothermal vent system (25), and (ii) the ferredoxin-nitrite reductase genes (EC 1.7.7.1), closely related to those of Sulfurimonas autotrophica. Nitric oxide reductase genes were also identified, including nor genes (EC 1.7.2.5), which were likely from the magnetotactic Magnetospirillum magneticum, as well as nos (EC 1.7.2.4) genes from the ferrous-oxidizing Sideroxydans lithotrophicus.
In the metagenomes from the SWIR, exceptionally rich reads that aligned hydrogenases were observed. In general, 4 monophyletic groups of [NiFe]-hydrogenases were proposed. The uptake of [NiFe]-hydrogenases in group 1 included membrane-bound respiratory uptake hydrogenases that couple H2 oxidation to a cytochrome, resulting in the pumping of protons across a membrane (26). The enrichment for genes encoding the large subunit of group 1 hydrogenases was consistent with the results from some sulfate-reducing bacteria, such as Desulfotalea psychrophila and Thermodesulfatator indicus, which were isolated from hydrothermal vents on the Central Indian Ridge (27) (see Fig. S7 and Table S1 in the supplemental material). The small subunit-encoding hydrogenase genes were also closely related to those assigned to sulfate-reducing bacteria (Fig. S7 and Table S1). These findings provide direct evidence for the potential hydrogenase activity of sulfate-reducing bacteria and their association with sulfate-reducing processes in chimney sulfides on the SWIR. In addition, some functional genes associated with [NiFe]-hydrogenase group 1 were found to utilize cytochromes, including the cytochrome b and c subunits. Potential genes encoding hydrogenases in both metagenomes and that were associated with the cofactor cytochrome b shared high levels of similarity with those of some sulfur- or sulfite-reducing bacteria, such as Beggiatoa species and Desulfurobacterium thermolithotrophum isolated from hydrothermal fields (Table S1). Group 2 hydrogenases are subdivided into two subgroups: group 2a, which includes cyanobacterial uptake hydrogenases, was absent in the present study; however, group 2b members that were similar to those of some sulfate-reducing bacteria, such as the most abundant genus Desulfobulbus, were detected herein (Table S1). Group 3, the dimeric cytoplasmic hydrogenase module associated with other subunits, can bind soluble cofactors, such as cofactor 420 (F420, 8-hydroxy-5-deazaflavin), NAD, or NADP. In this study, most of the group 3-expressing relatives included sulfur- and metal-related microorganisms (Table S1). However, group 4, which was typically associated with archaea, was not detected in the present study, consistent with the lower abundance of archaea detected in the taxonomic analysis.
DISCUSSION
Although the sulfur cycle could be one of the most important microbial chemosynthetic pathways providing energy for microbial habitats in hydrothermal vents, there is little supporting evidence from functional genes to confirm this hypothesis. Here, a metagenomic study of the chimneys on the Southwest Indian Ridge provided insight about the complete sulfur cycle based on functional gene analysis (Fig. 7 shows details). The accumulation of sulfides at the outer chimney permitted the coupling of sulfide oxidation to electron acceptors present in the marine water, including oxygen and nitrate, supported by retrieval of the functional genes described herein (Fig. 4 and 7). Most of the denitrification-related genes identified in these 2 metagenomes were similar to those identified in the dominant sulfur-oxidizing bacteria (e.g., the nar/nap genes). These findings suggested that the association between sulfur oxidation and denitrification could be an important energy-generating pathway fueling the microbial community at the sulfide-enriched outer chimney. This phenomenon has rarely been reported in previous studies (9)
FIG 7 .

Schematic diagram showing the microbial sulfur cycle in the hydrothermal chimney containing sulfur-oxidizing and sulfate-reducing bacteria, stratified according to the outer and inner regions of the chimney.
Sulfide oxidation has been studied extensively and is considered one of the most important microbial chemosynthetic processes; however, sulfate reduction at hydrothermal vents has been less thoroughly explored. Although sulfate reduction rates and potentially influential environmental factors have been determined in the laboratory, supporting the potential role of sulfate reduction in hydrothermal fields (5), information concerning the sulfate reduction process in situ remains limited. In the present study, microbial classifications based on both 16S rRNA and functional genes suggested a dominant position for sulfate-reducing bacteria in chimney sulfides on the SWIR (Fig. 2 and 3). The presence of Desulfobulbus-like microbes as the most abundant taxa supported their contributions to sulfate reduction in low-temperature hydrothermal chimneys on the SWIR. In contrast, Thermodesulfovibrio-like organisms might play a role in sulfate reduction in warmer environments (5). Sulfate-reducing microorganisms commonly use hydrogen and/or dissolved organic matter as electron donors, both of which have been detected in hydrothermal fluids in similar environments (10). Although hydrogen and dissolved organic matter were not closely monitored herein, a variety of organic compounds, such as long-chain hydrocarbons, fatty acids, and polycyclic aromatic hydrocarbons, have been detected in deep-sea hydrothermal fluids (28). For instance, the geochemical process of serpentinization in the crust of the Lost City could reduce CO2 to a spectrum of organic products, e.g., formate, pentane, and acetate (10). These types of organic compounds could act as a carbon source for the chemosynthetic microbial community, especially the heterotrophic sulfate-reducing bacteria. The key enzyme responsible for the utilization of acetate, acetyl-CoA synthetase, was assigned most commonly to sulfate-reducing bacteria. This finding indicated that low-weight organic compounds, including acetate and other short-chain alkanes, might play an important role in supporting chemosynthetic microorganisms in chimney sulfides on the SWIR (Fig. 7; see also Fig. S6 in the supplemental material). In addition, the identification of components of 2 metabolic pathways, 1,4-dichlorobenzene degradation and tetrachloroethene degradation, indicated that haloalkane could be another carbon source for sulfate-reducing bacteria (Fig. 6).
In addition to reduced, dissolved organic matter, the group 1 and group 2b hydrogenases recovered in this study were similar to those utilized by some sulfate-reducing bacteria, providing direct support for the ability of hydrogen to function as a critical electron donor (Fig. S7; see also Table S1 in the supplemental material). However, the potentially autotrophic Betaproteobacteria belonging to the order Burkholderiales, which are likely to oxidize H2, were observed in the Lost City hydrothermal metagenome from carbonate chimneys in which hydrogen was produced via the serpentinization process (29). Thiomicrospira and anaerobic methane-oxidizing archaea (Lost City Methanosarcinales phylotype) were identified as the most abundant groups in the Lost City carbonate chimney. In contrast, in the SWIR, sulfate-reducing bacteria were the dominant group, with a decreased abundance of methane-oxidizing bacteria and methanogenic and anaerobic methane-oxidizing archaea (Fig. 3) (7). In addition to the concentrations of various metals, the temperature discrepancy between the Lost City carbonate chimney and sulfide chimneys could play an important role in differences in microbial community composition. Temperatures ranging from 40 to 75°C and a pH of 5 were proposed as optimal conditions for sulfate reduction in hydrothermal vents (11), in agreement with the low temperature of the SWIR compared with that of the Lost City.
Metagenomic studies revealed the dominance of remarkably similar microbial communities consisting of sulfate-reducing and sulfur-oxidizing bacteria in both sulfides. This result could be attributed to the low temperature of both sites. However, the sites possess other distinct geophysical conditions, including the depth of the water and the concentrations of metal elements (Fig. 1 and Table 1). Some differences were observed between the metagenomes. For example, Firmicutes was identified as a major group in S35 but a minor group in S32 (Fig. 2). In addition, bacilli and clostridia were proposed in the Firmicutes from S35, although no close relatives were identified. Firmicutes have been documented in hydrothermal fluids collected from a sealed borehole on the flank of the Juan de Fuca Ridge (30) and in chimneys of the Lost City (8). Some members of the bacilli and clostridia were observed in the current study, indicating the potentially high activity of hydrogen in S35. The metal resistance possessed by Firmicutes has been described previously (31), consistent with the higher abundance of Firmicutes in S35 compared with its occurrence in S32 (Table 1). In addition, in the sorting cbbL/M gene analysis, hydrogen-utilizing Hydrogenovibrio and methanogenic or methanotrophic microbes like Methanocaldococcus, Methanothermococcus, and Methylophaga were more abundant in S35 than in S32 (Fig. 5B). This finding suggested the presence of relatively high hydrothermal activity in S35.
The overrepresentation of sulfate-reducing bacteria and sulfur-oxidizing bacteria in the chimney sulfides studied herein led to the proposal of 1 stratified pattern of microbial distribution (Fig. 7). Sulfur-oxidizing bacteria could utilize sulfide as an energy source, leading to oxidization of sulfide to sulfate, followed by the precipitation of sulfate in the outer chimney. Sulfate-reducing bacteria could inhabit the inner chimney and reduce sulfate to sulfide, resulting in the precipitation of metals (Fig. 7). The sulfur cycle could be centralized in an energy-generating manner to support the entire microbial community in these chimneys. As described above, nitrates, metals, and hydrogen could function as electron donors or acceptors for sulfate-reducing and sulfur-oxidizing bacteria. A similar distribution pattern has been proposed for the East Lau Spreading Center (ELSC) and the Valu Fa Ridge (VFR) based solely on 16S rRNA analyses (32). In inactive sulfide structures, sulfur-oxidizing Alphaproteobacteria, Gammaproteobacteria, and Epsilonproteobacteria have been suggested to be dominant in the exterior chimney, while putative sulfur-reducing Deltaproteobacteria are dominant in the interior of the chimney (32). In the present study, functional genes related to the sulfur cycle, especially the overrepresentation of sulfate-reducing bacteria, provided evidence supporting the stratified pattern of microbial distribution and highlighted the important role of sulfate-reducing bacteria in energy-source cycling within less active hydrothermal chimneys. However, further-refined sampling processes are needed to confirm this hypothesis.
MATERIALS AND METHODS
Sampling and physicochemical analyses.
Two chimney sulfides were sampled from segment 27 of the SWIR (14) during cruise DY115-20 of Research Vessel Da-Yang Yi-Hao in 2008. Chimney sample S32 was collected at 37°66′S, 50°46′E from a depth of 1,744 m and characterized as brown polymetallic ooze. Black sulfide chimney sample S35 was sampled at 37°78′S, 50°65′E from a depth of 2,738 m (Fig. 1). Both samples were collected on segment 27, which is magmatically the most robust spreading segment on the SWIR. Sample S32 was located in the shallow segment center, while S35 was in the deeper margin area of the segment close to a deep-sea basin (Fig. 1). The samples were sealed and stored at −80°C. Major and trace elements were analyzed using inductively coupled plasma atomic emission spectroscopy (ICP-AES) following treatment with the strong acid HNO3−HClO4 (pseudo-total digestion method) (33). Simultaneously, in the pore water, ammonia, nitrate, and nitrite were quantified using the spectrophotometric method. The concentrations of total dissolved organic carbon (DOC) and total nitrogen (TN) were determined using the combustion method with the TNM-I analyzer (Shimadzu, Kyoto, Japan). The concentration of sulfate was measured using a QuantiChrom sulfate assay kit (BioAssay Systems, Hayward, CA, USA). X-ray diffraction (XRD) analysis, using PANalytical’s X’Pert PRO X-ray diffractometer machine (PANalytical BV, Almelo, Netherlands), was conducted for both sulfides to identify major and trace elements.
Pyrosequencing of metagenomes and bioinformatics analyses.
For each sample, crude genomic DNA was extracted and purified from 10 g (wet weight) of sulfide chimney sample using the MO BIO PowerMax soil DNA isolation kit (Solana Beach, CA, USA). The quantity and quality of the DNA were checked using a Nanodrop ND2000 device (Thermo Scientific, Wilmington, DE, USA) and gel electrophoresis. For each sample, approximately 500 ng of DNA was subjected to pyrosequencing using a Roche 454 FLX Titanium platform. Reference metagenomes included those from a carbonate chimney sample collected from the Lost City Hydrothermal Vent Field (90°C, pH 9 to 11) (8) and a black smoker chimney sample located in the Mothra Hydrothermal Vent Field at the Juan de Fuca Ridge (>300°C) (9).
The NGS QC toolkit, version 2.3, was used for quality control analyses of the raw data (34). Artificial replicate sequences at a 98% sequence identity threshold were identified by cdhit-454 (35, 36) and then discarded. The 16S rRNA gene fragments were identified using Meta-RNA (37) and classified with the Ribosomal Database Project (RDP) classifier (release 10.30) with an 80% confidence threshold (38).
The reads generated using 454 sequencing were assembled using the de novo Newbler assembler (version 2.6) from Roche, with default parameters. Scaffolds and singletons were processed using FragGeneScan, which combines sequencing error models and codon usage in a Hidden Markov Model (HMM) to improve the prediction of protein-coding regions in short reads (39). All of the predicted open reading frames (ORFs) were annotated by comparison with the revised in-house KEGG database (http://www.genome.jp/kegg) (40) and the Clusters of Orthologous Groups (COG) sequence database (41) within the STRING database (version 9.0) (http://string-db.org), using BLAST searches with an E value cutoff of 10−5 (42). The Enzyme Commission (EC) numbers were retrieved from annotations of the KEGG genes. The KEGG metabolic pathway and module summaries of each sample were performed as previously described (43). Comparisons between 2 samples were conducted using the Mann-Whitney U test (44). Raw counts of COG and KEGG functional annotations for the 4 metagenomes were normalized as previously described (43). To generate heat maps of COG and KEGG genes, the normalized numbers were standardized to Z scores and then analyzed using Cluster 3 (http://rana.lbl.gov/EisenSoftware.htm) (45).
In addition, a BLAST search (42) of all reads against the nonredundant protein database in NCBI (NCBI-nr) (updated in July 2012) was performed. All of the hits obtained from the BLAST searches were retained, and taxonomic affiliations were determined using MEGAN4 (46, 47) with bit-score values of >60. KEGG and SEED annotations in MEGAN4 were referenced. The taxonomic compositions of each metagenome and comparisons between them were determined using MEGAN4.
The soxB dsrA, aprA, cbbL/M, and acetyl-CoA synthetase gene fragments were retrieved from both metagenomes and assembled de novo using the Newbler assembler. For each assembled gene, long contigs were selected for translation into amino acid sequences and aligned with other protein sequences collected from NCBI using MUSCLE3.5 (48). The alignments were checked manually and then used to reconstruct neighbor-joining (NJ) trees with 1,000 bootstrap replicates using MEGA5 (49). A maximum-likelihood (ML) phylogenetic tree was reconstructed based on 500 bootstrap replicates using the consensus NJ tree in MEGA5.
BioProject accession number.
Two metagenomes were deposited in GenBank under the BioProject accession number PRJNA226255.
SUPPLEMENTAL MATERIAL
Main minerals in both chimney sulfides based on X-ray diffraction (XRD) analyses. Download
Phylogenetic tree based on deduced protein sequences of SoxB, including the contigs assembled in the present study (contig length, >361 amino acids). Download
Phylogeny of protein sequences of DsrB deduced from the contigs of >199 amino acids assembled herein. Download
The dsr gene cluster in a contig assembled from the S32 metagenome (this study) and corresponding genes in the closely related Desulfobulbus propionicus DSM 2032 based on the identity of the deduced protein sequences. Download
Phylogenetic tree constructed based on the protein sequences of CbbL/M deduced from the contigs with lengths of >263 amino acids assembled in the present study. Download
Phylogeny of the protein sequences of acetyl-CoA deduced from the contigs with lengths of >271 amino acids assembled herein. Download
Numbers of reads enriched in the sulfide metagenomes that mapped to genes encoding hydrogenases. Download
Phylogenetic tree of the amino acid sequences of AprA deduced from the contigs with lengths of >210 amino acids assembled in the present study, constructed using the maximum-likelihood method. Download
Hydrogenase-encoding genes enriched in both metagenomes and their relatives in different bacterial species based on the identity of deduced protein sequences.
ACKNOWLEDGMENTS
This study was supported by a grant from the National Basic Research Program of China (973 Program, no. 2012CB417304), a grant (DY125-15-R-01) from the China Ocean Mineral Resources Research & Development Association (COMRRDA12SC02) , and awards from the Sanya Institute of Deep-sea Science and Engineering of (SIDSSE-201206), Chinese Academy of Science (CAS), and the King Abdullah University of Science and Technology in Saudi Arabia to P.-Y.Q. (SA-C0040/UK-C0016).
We thank the Research Vessel Da-Yang Yi-Hao crews for assisting with sample collection, Xiang-Dong Li of Hong Kong Polytechnic University for assisting with the analyses of major and trace elements, and the HPC Center of the Kunming Institute of Botany, CAS, China, for computational support.
Footnotes
Citation Cao H, Wang Y, Lee OO, Zeng X, Shao Z, Qian P. 2014. Microbial sulfur cycle in two hydrothermal chimneys on the Southwest Indian Ridge. mBio 5(1):e00980-13. doi:10.1128/mBio.00980-13.
REFERENCES
- 1. Jannasch HW, Mottl MJ. 1985. Geomicrobiology of deep-sea hydrothermal vents. Science 229:717–725. 10.1126/science.229.4715.717 [DOI] [PubMed] [Google Scholar]
- 2. Orcutt BN, Sylvan JB, Knab NJ, Edwards KJ. 2011. Microbial ecology of the dark ocean above, at, and below the Seafloor. Microbiol. Mol. Biol. Rev. 75:361–422. 10.1128/MMBR.00039-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Houghton JL, Seyfried WE, Jr, Banta AB, Reysenbach AL. 2007. Continuous enrichment culturing of thermophiles under sulfate and nitrate-reducing conditions and at deep-sea hydrostatic pressures. Extremophiles 11:371–382. 10.1007/s00792-006-0049-7 [DOI] [PubMed] [Google Scholar]
- 4. Audiffrin C, Cayol JL, Joulian C, Casalot L, Thomas P, Garcia JL, Ollivier B. 2003. Desulfonauticus submarinus gen. nov., sp. nov., a novel sulfate-reducing bacterium isolated from a deep-sea hydrothermal vent. Int. J. Syst. Evol. Microbiol. 53:1585–1590. 10.1099/ijs.0.02551-0 [DOI] [PubMed] [Google Scholar]
- 5. Frank KL, Rogers DR, Olins HC, Vidoudez C, Girguis PR. 2013. Characterizing the distribution and rates of microbial sulfate reduction at Middle Valley hydrothermal vents. ISME J. 7:1391–1401. 10.1038/ismej.2013.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schauer R, Røy H, Augustin N, Gennerich HH, Peters M, Wenzhoefer F, Amann R, Meyerdierks A. 2011. Bacterial sulfur cycling shapes microbial communities in surface sediments of an ultramafic hydrothermal vent field. Environ. Microbiol. 13:2633–2648. 10.1111/j.1462-2920.2011.02530.x [DOI] [PubMed] [Google Scholar]
- 7. Brazelton WJ, Baross JA. 2010. Metagenomic comparison of two Thiomicrospira lineages inhabiting contrasting deep-sea hydrothermal environments. PLoS One 5:e13530. 10.1371/journal.pone.0013530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brazelton WJ, Baross JA. 2009. Abundant transposases encoded by the metagenome of a hydrothermal chimney biofilm. ISME J. 3:1420–1424. 10.1038/ismej.2009.79 [DOI] [PubMed] [Google Scholar]
- 9. Xie W, Wang F, Guo L, Chen Z, Sievert SM, Meng J, Huang G, Li Y, Yan Q, Wu S, Wang X, Chen S, He G, Xiao X, Xu A. 2011. Comparative metagenomics of microbial communities inhabiting deep-sea hydrothermal vent chimneys with contrasting chemistries. ISME J. 5:414–426. 10.1038/ismej.2010.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lang SQ, Butterfield DA, Schulte M, Kelley DS, Lilley MD. 2010. Elevated concentrations of formate, acetate and dissolved organic carbon found at the Lost City hydrothermal field. Geochim. Cosmochim. Acta 74:941–952. 10.1016/j.gca.2009.10.045 [DOI] [Google Scholar]
- 11. Adams MM, Hoarfrost AL, Bose A, Joye SB, Girguis PR. 2013. Anaerobic oxidation of short-chain alkanes in hydrothermal sediments: potential influences on sulfur cycling and microbial diversity. Front. Microbiol. 4:110. 10.3389/fmicb.2013.00110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. German CR, Baker ET, Mevel C, Tamaki K, Fuji Science Team 1998. Hydrothermal activity along the southwest Indian ridge. Nature 395:490–493. 10.1038/26730 [DOI] [Google Scholar]
- 13. Tao C, Li H, Huang W, Han X, Wu G, Su X, Zhou N, Lin J, He Y, Zhou J. 2011. Mineralogical and geochemical features of sulfide chimneys from the 49°39′E hydrothermal field on the Southwest Indian Ridge and their geological inferences. Chin. Sci. Bull. 56:2828–2838. 10.1007/s11434-011-4619-4 [DOI] [Google Scholar]
- 14. Tao CH, Lin J, Guo SQ, Chen YJ, Wu GH, Han XQ, German CR, Yoerger DR, Zhou N, Li HM, Su X, Zhu J. 2012. First active hydrothermal vents on an ultraslow-spreading center: Southwest Indian Ridge. Geology 40:47–50. 10.1130/G32389.1 [DOI] [Google Scholar]
- 15. Zhu JA, Lin JA, Chen YSJ, Tao CH, German CR, Yoerger DR, Tivey MA. September 2010. A reduced crustal magnetization zone near the first observed active hydrothermal vent field on the Southwest Indian Ridge. Geophys. Res. Lett. 37:L18303. 10.1029/2010GL043542 [DOI] [Google Scholar]
- 16. Peng XT, Chen S, Zhou HY, Zhang LX, Wu ZJ, Li JT, Li JW, Xu HC. 2011. Diversity of biogenic minerals in low-temperature Si-rich deposits from a newly discovered hydrothermal field on the ultraslow spreading Southwest Indian Ridge. J. Geophys. Res. Biogeosci. 116:G03030. 10.1029/2011JG001691 [DOI] [Google Scholar]
- 17. Friedrich CG, Bardischewsky F, Rother D, Quentmeier A, Fischer J. 2005. Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 8:253–259. 10.1016/j.mib.2005.04.005 [DOI] [PubMed] [Google Scholar]
- 18. Sheik CS, Jain S, Dick GJ. 2014. Metabolic flexibility of enigmatic SAR324 revealed through metagenomics and metatranscriptomics. Environ. Microbiol. 16:304–317. 10.1111/1462-2920.12165 [DOI] [PubMed] [Google Scholar]
- 19. Pereira IA, Ramos AR, Grein F, Marques MC, da Silva SM, Venceslau SS. 2011. A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front. Microbiol. 2:69. 10.3389/fmicb.2011.00069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Campbell BJ, Cary SC. 2004. Abundance of reverse tricarboxylic acid cycle genes in free-living microorganisms at deep-sea hydrothermal vents. Appl. Environ. Microbiol. 70:6282–6289. 10.1128/AEM.70.10.6282-6289.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakagawa S, Takai K. 2008. Deep-sea vent chemoautotrophs: diversity, biochemistry and ecological significance. FEMS Microbiol. Ecol. 65:1–14. 10.1111/j.1574-6941.2008.00502.x [DOI] [PubMed] [Google Scholar]
- 22. Hügler M, Sievert SM. 2011. Beyond the Calvin cycle: autotrophic carbon fixation in the ocean. Ann. Rev. Mar. Sci. 3:261–289. 10.1146/annurev-marine-120709-142712 [DOI] [PubMed] [Google Scholar]
- 23. Sikorski J, Munk C, Lapidus A, Ngatchou Djao OD, Lucas S, Glavina Del Rio T, Nolan M, Tice H, Han C, Cheng JF, Tapia R, Goodwin L, Pitluck S, Liolios K, Ivanova N, Mavromatis K, Mikhailova N, Pati A, Sims D, Meincke L, Brettin T, Detter JC, Chen A, Palaniappan K, Land M, Hauser L, Chang YJ, Jeffries CD, Rohde M, Lang E, Spring S, Göker M, Woyke T, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Kyrpides NC, Klenk HP. 2010. Complete genome sequence of Sulfurimonas autotrophica type strain (OK10). Stand. Genomic Sci. 3:194–202. 10.4056/sigs.1173118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Voordeckers JW, Do MH, Hügler M, Ko V, Sievert SM, Vetriani C. 2008. Culture dependent and independent analyses of 16S rRNA and ATP citrate lyase genes: a comparison of microbial communities from different black smoker chimneys on the Mid-Atlantic Ridge. Extremophiles 12:627–640. 10.1007/s00792-008-0167-5 [DOI] [PubMed] [Google Scholar]
- 25. Markert S, Arndt C, Felbeck H, Becher D, Sievert SM, Hügler M, Albrecht D, Robidart J, Bench S, Feldman RA, Hecker M, Schweder T. 2007. Physiological proteomics of the uncultured endosymbiont of Riftia Pachyptila. Science 315:247–250. 10.1126/science.1132913 [DOI] [PubMed] [Google Scholar]
- 26. Vignais PM, Billoud B. 2007. Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev. 107:4206–4272. 10.1021/cr050196r [DOI] [PubMed] [Google Scholar]
- 27. Moussard H, L’Haridon S, Tindall BJ, Banta A, Schumann P, Stackebrandt E, Reysenbach AL, Jeanthon C. 2004. Thermodesulfatator indicus gen. nov., sp. nov., a novel thermophilic chemolithoautotrophic sulfate-reducing bacterium isolated from the Central Indian Ridge. Int. J. Syst. Evol. Microbiol. 54:227–233. 10.1099/ijs.0.02669-0 [DOI] [PubMed] [Google Scholar]
- 28. McCollom TM, Seewald JS. 2007. Abiotic synthesis of organic compounds in deep-sea hydrothermal environments. Chem. Rev. 107:382–401. 10.1021/cr0503660 [DOI] [PubMed] [Google Scholar]
- 29. Brazelton WJ, Nelson B, Schrenk MO. 2012. Metagenomic evidence for h(2) oxidation and h(2) production by serpentinite-hosted subsurface microbial communities. Front. Microbiol. 2:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cowen JP, Giovannoni SJ, Kenig F, Johnson HP, Butterfield D, Rappé MS, Hutnak M, Lam P. 2003. Fluids from aging ocean crust that support microbial life. Science 299:120–123. 10.1126/science.1075653 [DOI] [PubMed] [Google Scholar]
- 31. Papp-Wallace KM, Maguire ME. 2006. Manganese transport and the role of manganese in virulence. Annu. Rev. Microbiol. 60:187–209. 10.1146/annurev.micro.60.080805.142149 [DOI] [PubMed] [Google Scholar]
- 32. Sylvan JB, Sia TY, Haddad AG, Briscoe LJ, Toner BM, Girguis PR, Edwards KJ. 2013. Low temperature geomicrobiology follows host rock composition along a geochemical gradient in lau basin. Front. Microbiol. 4:61. 10.3389/fmicb.2013.00061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee CS, Li X, Shi W, Cheung SC, Thornton I. 2006. Metal contamination in urban, suburban, and country park soils of Hong Kong: a study based on GIS and multivariate statistics. Sci. Total Environ. 356:45–61. 10.1016/j.scitotenv.2005.03.024 [DOI] [PubMed] [Google Scholar]
- 34. Patel RK, Jain M. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. 10.1371/journal.pone.0030619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gomez-Alvarez V, Teal TK, Schmidt TM. 2009. Systematic artifacts in metagenomes from complex microbial communities. ISME J. 3:1314–1317. 10.1038/ismej.2009.72 [DOI] [PubMed] [Google Scholar]
- 36. Niu B, Fu L, Sun S, Li W. 2010. Artificial and natural duplicates in pyrosequencing reads of metagenomic data. BMC Bioinformatics 11:187. 10.1186/1471-2105-11-187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang Y, Gilna P, Li W. 2009. Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 25:1338–1340. 10.1093/bioinformatics/btp161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37:D141–D145. 10.1093/nar/gkp353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rho M, Tang H, Ye Y. 2010. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 38:e191. 10.1093/nar/gkq747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kanehisa M, Goto S. 2000. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30. 10.1093/nar/28.7.e27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tatusov RL, Galperin MY, Natale DA, Koonin EV. 2000. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28:33–36. 10.1093/nar/28.1.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 43. Wang Y, Cao H, Zhang G, Bougouffa S, Lee OO, Al-Suwailem A, Qian PY. 2013. Autotrophic microbe metagenomes and metabolic pathways differentiate adjacent Red Sea brine pools. Sci. Rep. 3:1748. 10.1038/srep01748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang Y, Yang J, Lee OO, Dash S, Lau SC, Al-Suwailem A, Wong TY, Danchin A, Qian PY. 2011. Hydrothermally generated aromatic compounds are consumed by bacteria colonizing in Atlantis II Deep of the Red Sea. ISME J. 5:1652–1659. 10.1038/ismej.2011.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Eisen MB, Spellman PT, Brown PO, Botstein D. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U. S. A. 95:14863–14868. 10.1073/pnas.95.25.14863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huson DH, Auch AF, Qi J, Schuster SC. 2007. MEGAN analysis of metagenomic data. Genome Res. 17:377–386. 10.1101/gr.5969107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC. 2011. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21:1552–1560. 10.1101/gr.120618.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Main minerals in both chimney sulfides based on X-ray diffraction (XRD) analyses. Download
Phylogenetic tree based on deduced protein sequences of SoxB, including the contigs assembled in the present study (contig length, >361 amino acids). Download
Phylogeny of protein sequences of DsrB deduced from the contigs of >199 amino acids assembled herein. Download
The dsr gene cluster in a contig assembled from the S32 metagenome (this study) and corresponding genes in the closely related Desulfobulbus propionicus DSM 2032 based on the identity of the deduced protein sequences. Download
Phylogenetic tree constructed based on the protein sequences of CbbL/M deduced from the contigs with lengths of >263 amino acids assembled in the present study. Download
Phylogeny of the protein sequences of acetyl-CoA deduced from the contigs with lengths of >271 amino acids assembled herein. Download
Numbers of reads enriched in the sulfide metagenomes that mapped to genes encoding hydrogenases. Download
Phylogenetic tree of the amino acid sequences of AprA deduced from the contigs with lengths of >210 amino acids assembled in the present study, constructed using the maximum-likelihood method. Download
Hydrogenase-encoding genes enriched in both metagenomes and their relatives in different bacterial species based on the identity of deduced protein sequences.