

ABSTRACT
Remodeling of the host cytoskeleton is a common strategy employed by bacterial pathogens. Although there is vigorous investigation of the cell biology underlying these bacterially mediated cytoskeleton modifications, knowledge of the plasticity and dynamics of the bacterial signaling networks that regulate the expression of genes necessary for these phenotypes is lacking. Enterohemorrhagic Escherichia coli attaches to enterocytes, forming pedestal-like structures. Pedestal formation requires the expression of the locus-of-enterocyte-effacement (LEE) and espFu genes. The LEE encodes a molecular syringe, a type III secretion system (T3SS) used by pathogens to translocate effectors such as EspFu into the host cell. By using a combination of genetic, biochemical, and cell biology approaches, we show that pedestal formation relies on posttranscriptional regulation by two small RNAs (sRNAs), GlmY and GlmZ. The GlmY and GlmZ sRNAs are unique; they have extensive secondary structures and work in concert. Although these sRNAs may offer unique insights into RNA and posttranscriptional biology, thus far, only one target and one mechanism of action (exposure of the ribosome binding site from the glmS gene to promote its translation) has been described. Here we uncovered new targets and two different molecular mechanisms of action of these sRNAs. In the case of EspFu expression, they promote translation by cleavage of the transcript, while in regard to the LEE, they promote destabilization of the mRNA. Our findings reveal that two unique sRNAs act in concert through different molecular mechanisms to coordinate bacterial attachment to mammalian cells.
IMPORTANCE
Pathogens evolve by horizontal acquisition of pathogenicity islands. We describe here how two sRNAs, GlmY and GlmZ, involved in cellular metabolism and cellular architecture, through the posttranscriptional control of GlmS (the previously only known target of GlmY and GlmZ), which controls amino sugar synthesis, have been coopted to modulate the expression of virulence. These sRNAs quickly allow for plasticity in gene expression in order for enterohemorrhagic Escherichia coli to fine-tune the expression of its complex type III secretion machinery and its effectors to promote bacterial attachment and subsequent actin rearrangement on host cells. Pedestal formation is a very dynamic process. Many of the genes necessary for pedestal formation are located within the same operon to evolutionarily guarantee that they are inherited together. However, it is worth noting that within these operons, several genes need to yield more proteins than others and that these differences cannot be efficiently regulated at the transcriptional level.
INTRODUCTION
Exploitation of the host cytoskeleton by bacterial pathogens is an essential feature of bacterium-host associations. Actin remodeling promotes bacterial invasion of nonphagocytic cells, survival within cells, cell-to-cell spread and locomotion, and colonization at the interface of the host epithelium (1). Enterohemorrhagic Escherichia coli (EHEC) O157:H7 is a deadly pathogen that attaches to enterocytes, forming attaching and effacing (AE) lesions characterized by the formation of a pedestal-like structure beneath the bacterium (2). To induce pedestal formation on epithelial cells, EHEC employs a type III secretion system (T3SS), a needle-like structure that translocates bacterial effectors directly into host cells. The genes for this T3SS and several other genes necessary for AE lesion formation are located within a chromosomal pathogenicity island named the locus of enterocyte effacement (LEE) (3, 4). The LEE region contains five major operons, LEE1 to LEE5 (5–7), which encode the components of the T3SS (4), an adhesin (intimin) (8) and its receptor Tir, which is itself translocated through the T3SS to the host cell, where, upon its insertion into the cell membrane, it serves as a receptor for the bacterial adhesin intimin (9) and other effector proteins (10–14). The LEE-encoded T3SS also translocates effector proteins encoded outside the LEE region, including EspFu/TccP, which is important for efficient AE lesion formation (15–18).
Expression of the LEE and espFu genes is governed through complex multilayered signaling cascades in response to many environmental cues, including human hormones (epinephrine [Epi] and norepinephrine [NE]), bacterial small signaling molecules (autoinducer-3 [AI-3], indole, acyl homoserine lactones), carbon and nitrogen sources, and stress responses, among others. This regulation occurs at both the transcriptional and posttranscriptional levels (19–29). In bacteria, there are many different mechanisms of posttranscriptional regulation of genes (30). One of the more abundant classes is that of trans-acting small RNAs (sRNAs). The majority of these sRNAs require the RNA chaperone Hfq and act by directly binding to mRNAs at the ribosome binding site (RBS) to repress translation, cause direct degradation of the mRNA by recruitment of nucleases, or activate translation by relieving a hairpin that blocks the RBS. While genes of the LEE are known to be posttranscriptionally regulated, no sRNAs responsible for this have been identified to date.
The AI-3/Epi/NE interkingdom signaling cascade activates the expression of virulence genes in EHEC (21, 31–33). The host hormones Epi and NE are specifically sensed by two membrane-bound histidine sensor kinases, QseC and QseE, which are the first bacterial adrenergic receptors identified (34, 35). QseE is downstream of QseC in this signaling cascade, given that the transcription of qseE is activated through QseC (36). In addition to sensing these host hormones, QseC also senses the bacterial signal AI-3 (32). QseE, however, does not sense AI-3, thereby discriminating between host- and bacterium-derived signals (35). Upon sensing their respective signals, QseC and QseE autophosphorylate to activate virulence gene expression and pathogenesis in vitro and in vivo in EHEC (32, 35, 37). QseC transfers its phosphate to three response regulators (RRs), QseB, QseF, and KdpE, which, upon phosphorylation, are activated and function as transcription factors (38). QseE transfers its phosphate only to QseF (39). The concerted action of these RRs activates the EHEC virulence repertoire, including the LEE and espFu genes (Fig. 1A). The QseF RR is necessary for the expression of EspFu (36), and it is known to regulate the sRNA GlmY, located immediately upstream from the qseEGFglnB operon (Fig. 1B) (40). This sRNA is known to act as a molecular mimic (41), stabilizing another sRNA (GlmZ), which directly binds to the mRNA of the gene encoding glucosamine synthetase (glmS) and activates its translation by breaking a hairpin loop and revealing the RBS (42).
FIG 1 .

QseF and QseB regulation of glmY. (A) Schematic representation of the QseC and QseE transduction signaling systems. QseC responds to AI-3, epinephrine, and norepinephrine (Nor), and QseE responds to epinephrine (Epi), sulfate (SO4), and phosphate (PO4). Upon sensing their signals, these histidine sensor kinases autophosphorylate and then transfer their phosphate to their response regulators. QseC phosphotransfers to KdpE (activates the LEE genes), QseB (regulates the flagellum regulon), and QseF (regulates espFu). QseE phosphotransfers only to QseF. (B) Schematic representation of glmY and the qseEGFglnB operon depicting the locations of the σ54 and σ70 promoters. (C) Diagram showing the layout of the glmY regulatory region. QseF binding sites are red, QseB binding sites are blue, and the σ54 and σ70 promoters are magenta and yellow, respectively. (D) β-Galactosidase assay of the glmY::lacZ transcriptional fusion in the WT strain and the ΔqseB and ΔqseF mutant strains. The plasmid contains bp −250 to +20 from the transcription start site. (E) Northern blot assay with a glmY probe of RNAs from the WT strain and the ΔqseB and ΔqseF mutant strains (left) and Northern blot assay of a 5S rRNA probe of the same RNAs as a loading control (right). (F) β-Galactosidase assay of the glmZ::lacZ transcriptional fusion in the WT strain and the ΔqseB and ΔqseF mutant strains. (G) EMSAs of the glmY promoter with increasing amounts of QseF or QseB protein in the presence of acetyl phosphate (left) and EMSAs of the kan promoter with QseF and QseB as a negative control (right). The plasmid contains bp −246 to −20 from the transcription start site. The unpaired Student t test was used to determine statistical significance. A P value of ≤0.05 was considered significant.
Here we show that both the QseB and QseF RRs directly activate the expression of glmY. GlmY and GlmZ coordinate LEE and EspFu expression posttranscriptionally through two different mechanisms. GlmY and GlmZ posttranscriptional regulation of the LEE and EspFu ensures the correct timing and dynamics of AE lesion formation by EHEC on epithelial cells. We propose that sRNA-mediated posttranscriptional regulation is responsible for the dynamic rewiring of the expression of different components of bacterial complex machineries that allow successful interactions with mammalian cells.
RESULTS
Transcriptional regulation of glmY and glmZ.
The QseC/QseE signaling system controls a plethora of virulence genes in EHEC that have to be coordinately expressed to ensure optimal AE lesion formation on epithelial cells, leading to host infection (37, 38, 43, 44). AE lesion formation is a dynamic process that requires plasticity and rapid adaptation of bacterial gene expression. Coupling of transcriptional and posttranscriptional regulation within a signaling transduction cascade in the bacterial cell is key to ensuring fine-tuning and rapid adaptation of gene expression toward the regulation of complex processes such as AE lesion formation. Upstream of the qseEGFglnB operon is glmY (Fig. 1B). The glmY gene is known to have two overlapping promoters, one that is driven by a σ70 RNA polymerase (the homeostatic form of this enzyme) and another that is driven by a σ54 RNA polymerase (Fig. 1B and C). Transcription of glmY is known to be regulated by the σ54-dependent transcriptional activator QseF (40). Additionally, a sequence matching the known consensus sequence of QseB (45), another RR involved in interkingdom signaling, was identified in silico in this promoter region (Fig. 1C).
Transcriptional β-galactosidase reporters of the promoters of both glmY and glmZ were constructed. As previously reported, in the qseF mutant, glmY expression is starkly decreased and almost ablated. Meanwhile, the qseB mutant, while still expressing glmY, expressed significantly less than the wild type (WT) (Fig. 1D). This result was confirmed by Northern blot analysis for the GlmY RNA (Fig. 1E). Neither RR had any effect on glmZ expression (Fig. 1F). The almost complete ablation of glmY expression in the ΔqseF mutant is due to the σ54 RNA polymerase acting as a repressor in the absence of QseF (40). The σ54 RNA polymerase cannot promote the formation of the DNA open complex to initiate transcription by itself; it requires a σ54 RR, such as QseF, for this process (46). Because the σ54 promoter overlaps the σ70 promoter and in the absence of QseF, the σ54 RNA polymerase occupying the σ54 promoter prevents access to the σ70 promoter by the σ70 RNA polymerase (40).
Electrophoretic mobility shift assays (EMSAs) demonstrated that both QseB and QseF directly bind to the glmY regulatory region (Fig. 1G). Interestingly, while the QseB consensus sequence within the glmY regulatory region differs slightly among EHEC strain 86-24, E. coli K-12 strain MG1655, and enteropathogenic E. coli (EPEC) strain E2348/69, all are capable of binding to QseB (see Fig. S1 in the supplemental material).
Insights into GlmY and GlmZ regulation of EspFu.
Because the QseF RR controls AE lesion formation by indirectly promoting the expression of the EspFu T3SS effector (36), next we investigated whether GlmY and/or GlmZ also play a role in EspFu expression. EspFu interacts with another effector, Tir, through IRTKS and acts as an Nck mimic to recruit N-WASP and Arp2/3 to the site of bacterial attachment, causing the formation of the characteristic actin-rich pedestal (Fig. 2A) (15, 16, 47). The expression of this effector is dependent on QseF, with the qseF mutant having the same phenotype as the ΔespFu mutant, which is the almost complete lack of AE lesion formation on HeLa cells (Fig. 2B and 3A) (15, 16, 36).
FIG 2 .

Posttranscriptional regulation of EspFu. (A) Schematic representation of AE lesion formation. (B) FAS of HeLa cells infected with the WT strain, the ΔqseF mutant, or the ΔqseF mutant complemented with pGlmY or pGlmZ. (C) Northern blot assay with an espFu probe of RNA from the WT and ΔqseF and ΔespFu mutant strains (top) and Northern blot assay with a 5S rRNA probe with the same RNAs as a loading control (bottom). (D) RT-PCR of cDNA from the WT and the ΔqseF mutant with primer sets to the entire espJ-espFu region or just espFu. (E) Schematic representation of the espJ-espFu region and the 2,100-bp espJ-espFu and 1,100-bp espFu transcripts. (F) β-Galactosidase assay of the espFu::lacZ transcriptional fusion in the WT and ΔqseF mutant strains with the plasmid containing bp −461 to +48 from the translation start site. ns, no statistically significant difference. (G) β-Galactosidase assay of an EspFu::LacZ translation reporter plasmid in WT, the ΔqseF mutant, and the complemented ΔqseF mutant. The translational fusion contains the region from −170 bp downstream of the translation start site to +48 bp upstream of it. (H) Diagram of the espJ-espFu WT and deletion constructs. To create p1, the region stretching from 20 bp downstream of the espJ stop codon to 20 bp upstream of the espFu start codon was deleted. To create p2, the region stretching from 20 bp downstream of the espJ stop codon to 170 bp upstream of the espFu start codon was deleted. To create p3, the region stretching from 170 bp upstream of the espFu start codon to 20 bp upstream of the espFu start codon was deleted. p4 contains the region stretching from 262 bp within the espJ gene to the end of espFu. (I) Western blot assays of WT EHEC expressing the espJ-espFu WT and FLAG-tagged deletion constructs probed with an anti-FLAG antibody. Western blot assays with anti-RpoA antibody were used as loading controls. The unpaired Student t test was used to determine statistical significance. A P value of ≤0.05 was considered significant.
FIG 3 .

GlmY and GlmZ regulation of AE lesion formation and the LEE genes. (A) FAS of HeLa cells infected with the WT strain, the ΔespFu mutant, the ΔglmY mutant, the ΔglmY mutant complemented with pGlmY or pGlmZ, the ΔglmZ mutant, or the ΔglmZ mutant complemented with pGlmY or pGlmZ. (B) Quantification of the FAS experiment measuring the average number of bacteria attached per cell. Knockouts were compared to WT EHEC, while the complemented strains were compared to the respective mutants. (C) qPCR of ler, espA, eae, and stx2a with RNA extracted from the WT, the WT overexpressing pGlmY, and the WT overexpressing pGlmZ. (D) Schematic representation of the LEE region depicting the LEE1 to LEE5 and grlRA operons. Ler is encoded by the first gene in the LEE1 operon and is the transcriptional activator of all of the LEE genes. The unpaired Student t test was used to determine statistical significance. A P value of ≤0.05 was considered significant.
Since the regulation of espFu by QseF is known to be indirect (36), we tested the ability of its known target, GlmY, as well as its downstream target, GlmZ, to complement a qseF mutant. The glmY and glmZ genes were cloned under the control of an inducible promoter and transformed into the ΔqseF mutant strain. These strains were then used to infect HeLa cells to perform the fluorescein actin staining (FAS) test to visualize AE lesions. In the FAS assay, the HeLa cytoskeleton was stained green with fluorescein isothiocyanate (FITC)-labeled phalloidin, the bacteria and nuclei were stained red with propidium iodide (PI), and pedestals were visualized as brilliant patches of green underneath a red bacterium. Both sRNAs were able to rescue AE lesion formation in the qseF mutant, indicating that these sRNAs are the intermediaries between QseF and espFu (Fig. 2B).
Since sRNAs act posttranscriptionally, we investigated espFu mRNA levels by using Northern blot assays. EspFu is located outside the LEE within a prophage. Upstream of the espFu gene is the espJ gene, which encodes another T3SS effector (Fig. 2E) (48). In the WT, there is a major band the size of the predicted espFu transcript (1,100 bp), as well as a much fainter upper band 2,100 nucleotides in length. In the qseF mutant, the lower band is still present; however, the upper band is much more pronounced. This upper band corresponds to the expected size of an espJ-espFu transcript (Fig. 2C and E). There are 320 bp between these two genes, and previous work suggested that they are not cotranscribed. However, since only RNA from WT bacteria was used in those experiments, it is possible that the less abundant larger transcript was not detected (36). To confirm that this larger transcript is espJ-espFu, reverse transcription (RT)-PCR was performed with primers spanning this region. While a very faint band corresponding to the espJ-espFu (2,100-bp) transcript was observed in WT EHEC, it was much more pronounced in the qseF mutant (Fig. 2D). Hence, the lack of EspFu expression in a qseF mutant is due not to the absence of its transcript but to the lack of a processing event of the espJ-espFu transcript necessary for EspFu expression.
Transcriptional and translational reporters of espFu were constructed. As expected, the qseF mutant had no defect in espFu transcription (Fig. 2F); however, it is required for the translation of EspFu (Fig. 2G). To determine the regions of espJ-espFu required for this regulation, various deletions in the intergenic region between these genes were constructed. These deletions were cloned into a vector with a FLAG tag at the C terminus of espFu. The following four deletion mutants were constructed by using the previously identified 5′ untranslated region (UTR) of the espFu transcript as a reference point (36): p1, which lacks the entire intergenic region; p2, which lacks the espJ 3′ UTR; p3, which lacks the espFu 5′ UTR; and p4, which does not have the espJ gene but still has the intergenic region (Fig. 2H). Western blot assays with anti-FLAG antiserum were performed with whole-cell lysates of EHEC expressing the WT espJ-espFu-FLAG plasmid and each of these four deletion constructs. The p2 and p4 constructs expressed levels of EspFu::FLAG similar to those of the WT plasmid, while p1 expressed more protein and p3 did not express EspFu (Fig. 2I). These data indicate that the 3′ UTR of espJ acts negatively on the translation of espFu and that this QseF/GlmY/GlmZ-mediated processing event is required for the translation of espFu since the resulting transcript lacks the 3′ UTR of espJ. Additionally, the presence of EspFu::FLAG from the p4 plasmid indicates that in addition to being cotranscribed with espJ, espFu also has its own promoter (Fig. 2I).
The roles of GlmY and GlmZ in pedestal formation.
Given that EspFu is involved in pedestal formation, we further investigated the roles of GlmY and GlmZ in AE lesion formation by constructing ΔglmY and ΔglmZ mutant strains and performing FAS assays with them. Since they were both capable of rescuing the qseF phenotype, we expected that both mutants would have a decreased ability to form pedestals, similarly to the ΔqseF and ΔespFu mutant strains. Surprisingly, both sRNA mutants attached to and formed pedestals on HeLa cells at levels far higher than that of the WT strain. Both the glmY and glmZ plasmids were capable of complementing the glmY mutant, which is an expected result (Fig. 3A and B). The GlmY sRNA is known to stabilize the GlmZ sRNA (42), and GlmZ, in an Hfq-dependent manner, exposes the RBS of the glmS mRNA to promote its translation (42). Thus far, the only known target of GlmY and GlmZ regulation in E. coli was glmS. The “effector” sRNA that base pairs with the glmS mRNA is GlmZ, and thus, the effect of GlmY on glmS is indirect and attributed solely to its stabilization of GlmZ (42). However, the glmY plasmid was also capable of complementing the ΔglmZ mutant, which suggests that GlmY may have additional functions besides preventing the degradation of GlmZ (Fig. 3A and B). Because, in addition to espFu, AE lesion formation also requires the expression of the LEE genes, we assessed LEE regulation by these sRNAs. The expression of the stx2a gene, which encodes Shiga toxin, and of ler (LEE1 operon), the master regulator of the LEE, was unchanged, but the expression of the LEE4 (espA) and LEE5 (eae) operons was decreased in the WT strain expressing GlmY or GlmZ on a plasmid (Fig. 3C and D), suggesting that these sRNAs decrease LEE4 and LEE5 expression posttranscriptionally. These data offer an explanation for why the deletion of the genes encoding these sRNAs increases AE lesion formation. In the absence of these sRNAs, the transcripts of LEE4 and LEE5 operons (containing many genes essential for pedestal formation) would be stabilized, increasing pedestal formation.
The roles of GlmY and GlmZ in the regulation of AE lesion formation by promoting EspFu translation and destabilizing LEE transcripts (Fig. 2 and 3) seem to be initially confounding. However, AE lesion formation is a dynamic process where pedestals are constantly being formed and unformed during infection (see Movie S1 in the supplemental material), and the precise modulation of the levels of LEE and EspFu expression is important for the efficiency of this phenotype. To better understand the dynamics of pedestal formation responsible for the phenotype of the glmY and glmZ EHEC knockouts, we visualized the cells in real time. The F-actin binding peptide Lifeact (49) proved to be the most effective at visualizing the pedestals formed by EHEC. For ease of experimentation, an HeLa cell line stably expressing Lifeact::green fluorescent protein (GFP) was created. Bacteria were visualized by the expression of mCherry (Fig. 4A to C; see Movies S1 to S3 in the supplemental material). The ΔglmY and ΔglmZ mutant strains attached to and formed pedestals much more efficiently and faster than the WT strain, suggesting that these sRNAs regulate the proper timing and amount of AE lesion formation on epithelial cells.
FIG 4 .

GlmY and GlmZ regulation of AE lesion timing and dynamics. (A) Time-lapse microscopy of Lifeact::GFP-expressing HeLa cells being infected with mCherry-expressing WT EHEC. White arrows show clusters of EHEC AE lesions. (B) Time-lapse microscopy of Lifeact::GFP-expressing HeLa cells being infected with mCherry-expressing ΔglmY EHEC. (C) Time-lapse microscopy of Lifeact::GFP-expressing HeLa cells being infected with mCherry-expressing ΔglmZ EHEC.
GlmY and GlmZ are known to promote the translation of glmS, which encodes the glucosamine synthase enzyme in E. coli K-12 strain MC4100. GlmS is necessary for the synthesis of N-acetylglucosamine-6-P, which is used for cell wall biosynthesis (see Fig. S2A in the supplemental material) (42). A glmS mutant is lethal because it is defective in cell wall biosynthesis. However, the addition of 1% N-acetylglucosamine (GlcNAC) to the medium allows the survival of a glmS mutant because the GlcNAC sugar needed for cell wall synthesis is being provided exogenously (50). To rule out the possibility that the AE lesion phenotype governed by GlmY and GlmZ is due to issues with cell wall synthesis in these sRNA mutants because of the decreased expression of GlmS, FAS assays were repeated in medium containing 1% GlcNAC. The AE lesion phenotypes of the WT and the glmY and glmZ mutants, higher AE lesion formation by both mutants than by the WT, was the same both in the absence and in the presence of GlcNAC (see Fig. S2B and C). Additionally, point mutations in glmZ that abolish the regulation of glmS by GlmZ were created as previously reported, and the mutant GlmZ sRNA was named GlmZ* (see Fig. S2D) (42). GlmZ* was still capable of complementing pedestal formation in the glmZ mutant, indicating that the pedestal formation phenotype is not mediated through downstream effects of diminished GlmS expression or issues with cell wall biosynthesis (see Fig. S2E and F). To further assess the levels of GlmS expression promoted by GlmY and GlmZ in EHEC, Northern blot assays of glmS were performed with RNA from WT EHEC and from WT EHEC expressing GlmY, GlmZ, and GlmZ* on a plasmid. It has been previously reported that in E. coli K-12 strain MC4100, expression of the glmZ mRNA is increased by the expression of these sRNAs on plasmids because of the more efficient translation of glmS (42). In EHEC, however, the expression of both of these sRNAs on plasmids did not affect the levels of the glmS transcript under the conditions we assayed (see Fig. S3A). We also constructed a translational reporter of GlmS. Previous studies indicate that overexpression of either glmY or glmZ should lead to an increase in β-galactosidase activity with this reporter construct. However, in EHEC, overexpression of glmY or glmZ did not change GlmS::LacZ expression (see Fig. S3B). This same reporter plasmid was then assayed in MC4100, the E. coli K-12 strain used in previous studies of glmS (40, 42), and it behaved as previously reported (see Fig. S3C). Since the sequences of both glmS and glmZ that interact are invariant between these two strains, it is likely that there is another level of regulation that is masking the regulation of glmS by GlmZ in EHEC 86-24 that is not present in strain MC4100.
Posttranscriptional regulation of LEE5 and LEE4 by GlmY and GlmZ.
The LEE5 operon in EHEC consists of three genes that encode the translocated intimin receptor (Tir), its chaperone (cesT), and the bacterial adhesin intimin (eae) with which Tir interacts (Fig. 2A and 5A) (5, 7). While this operon is transcribed by a single promoter upstream of tir (5, 51, 52), there is a processing event that results in the separation of cesT-eae from tir (Fig. 5A to D). Inasmuch as GlmY and GlmZ overexpression decreased eae transcript levels (Fig. 3A), we investigated the mRNA levels of each gene in this operon by Northern blot assay (Fig. 5B to D). Overexpression of both sRNAs decreased the levels of the cesT-eae transcript (3,300 bp) (Fig. 5C and D), while the tir transcript was largely unaffected (1,600 bp) (Fig. 5B). The levels of transcription of the entire LEE5 operon (4,900 bp) were also decreased by these sRNAs (Fig. 5B to D). Since the first gene of this operon is unaffected, GlmY and GlmZ must be acting posttranscriptionally. One of the primary ways in which sRNAs affect gene stability is blocking of translation by binding to the RBS (53). An mRNA being translated is largely protected from nucleases by the ribosomes, so blocking of translation can lead to degradation of the transcript. To test this possibility, translational LacZ reporters of all three genes of LEE5 were constructed and β-galactosidase assays were performed (Fig. 5E to G). Neither the knockout of glmY and glmZ nor their overexpression had any effect on the translation of any of the three reporter proteins, suggesting that they are not acting through this mechanism.
FIG 5 .

GlmY and GlmZ regulation of the LEE5 operon. (A) Schematic depiction of the LEE5 operon and its transcripts. (B) Northern blot assays with a probe for tir with RNA from WT bacteria, bacteria overexpressing glmY, and bacteria overexpressing glmZ with the 5S rRNA serving as a loading control. (C) Northern blot assays with a probe for cesT with RNA from WT bacteria, bacteria overexpressing glmY, and bacteria overexpressing glmZ with the 5S rRNA serving as a loading control. (D) Northern blot assays with a probe for eae with RNA from WT bacteria, bacteria overexpressing glmY, and bacteria overexpressing glmZ with the 5S rRNA serving as a loading control. (E) β-Galactosidase assays of the Tir::LacZ translational fusion in WT, WT/pGlmY, WT/pGlmZ, ΔglmY mutant, and ΔglmZ mutant cells. The plasmid contained the region stretching from bp −137 to +90 from the translation start site. (F) β-Galactosidase assays of the Eae::LacZ translational fusion in WT, WT/pGlmY, WT/pGlmZ, ΔglmY mutant, and ΔglmZ mutant cells. The plasmid contained the region stretching from bp −62 to +90 from the translation start site. (G) β-Galactosidase assays of the CesT::LacZ translational fusion in WT, WT/pGlmY, WT/pGlmZ, ΔglmY mutant, and ΔglmZ mutant cells. The plasmid contained the region stretching from bp −71 to +90 from the translation start site. The unpaired Student t test was used to determine statistical significance. A P value of ≤0.05 was considered significant. ns, not statistically significant.
The LEE4 operon encodes the SepL regulator of effector translocation into host cells; the EspA protein, which forms a filament that creates a sheath around the T3SS needle; EspBD, which create a pore through the eukaryotic cell membrane; a chaperone, CesD2; the EscF structural protein of the needle; an uncharacterized protein, Orf29; and the effector EspF (6, 7, 18, 54–56). The LEE4 operon has only one promoter upstream of the sepL gene and no internal promoters (6, 51, 57–59). The first gene of this operon (sepL) is processed from this operon in an RNase E-dependent manner (24), and there is a terminator in the cesD2 gene that leads to lower expression of the last three genes (6) (Fig. 6A). Similarly to our studies concerning the posttranscriptional regulation of LEE5 (Fig. 5), Northern blot assays were performed for the LEE4 genes in the WT, the WT overexpressing glmY or glmZ, and the glmY and glmZ mutants (Fig. 6B). Similarly to LEE5, the overexpression of both sRNAs led to lower levels of the espA-cesD2 transcript, while the sepL transcript was mostly unaffected. The level of the espA-cesD2 transcript was noticeably higher in the sRNA mutants (Fig. 6B). The transcript of the last three genes of the LEE4 operon could not be detected by Northern blot assay because of their much lower expression due to the transcription terminator in cesD2, so a quantitative PCR (qPCR) was performed, and the results demonstrated that they were also downregulated by the overexpression of glmY and glmZ (Fig. 6C). Translational fusions of SepL, EspA, EspD, and EspB were constructed and assayed, and again the expression of these reporters was unaffected in the ΔglmY and ΔglmZ mutant strains and under the overexpression of the sRNAs (Fig. 6D to G). In a scenario similar to the posttranscriptional regulation of LEE5 (Fig. 5), GlmY and GlmZ are not destabilizing the LEE4 transcripts by blocking translation (Fig. 6).
FIG 6 .

GlmY and GlmZ regulation of the LEE4 operon. (A) Schematic depiction of the LEE4 operon and its transcripts. (B) Northern blot assays with probes for sepL and espA with RNA from WT, WT/pGlmY, WT/pGlmZ, ΔglmY mutant, and ΔglmZ mutant cells, with the 5S rRNA serving as a loading control. (C) qPCR of escF and espF with cDNA from the WT or the WT overexpressing glmY or glmZ. (D) β-Galactosidase assays of the SepL::LacZ translational fusion in WT, WT/pGlmY, WT/pGlmZ, ΔglmY mutant, and ΔglmZ mutant cells. The plasmid contained the region stretching from bp −82 to +90 from the translation start site. (E) β-Galactosidase assays of the EspA::LacZ translational fusion in WT, WT/pGlmY, WT/pGlmZ, ΔglmY mutant, and ΔglmZ mutant cells. The plasmid contained the region stretching from bp −107 to +90 from the translation start site. (F) β-Galactosidase assays of the EspD::LacZ translational fusion in WT, WT/pGlmY, WT/pGlmZ, ΔglmY mutant, and ΔglmZ mutant cells. The plasmid contained the region stretching from bp −61 to +90 from the translation start site. (G) β-Galactosidase assays of the EspB::LacZ translational fusion in WT, WT/pGlmY, WT/pGlmZ, ΔglmY mutant, and ΔglmZ mutant cells. The plasmid contained the region stretching from bp −68 to +90 from the translation start site. (H) Sequence and secondary structure of the GlmZ sRNA depicting the regions that interact with the glmS and LEE4 mRNAs. (I) Schematics showing the predicted interaction between GlmZ and LEE4, the point mutations made in GlmZ to abolish binding, and the compensatory mutations made in LEE4. (J) qRT-PCR of espA with cDNA from WT/pLEE4, ΔglmZ/pLEE4, ΔglmZ/pGlmZ/pLEE4, ΔglmZ/pGlmZ→CG/pLEE4, and ΔglmZ/pGlmZ→CG/pLEE4→GC cells. (K) FAS of HeLa cells infected with WT, ΔglmZ, ΔglmZ/pGlmZ, and ΔglmZ/pGlmZ→CG cells. The unpaired Student t test was used to determine statistical significance. A P value of ≤0.05 was considered significant. ns, not statistically significant.
Some sRNAs bind directly to the coding region of their mRNA target transcript and cause its degradation through the recruitment of an RNase (53). The IntaRNA software (60) was used to predict potential sites of GlmY and GlmZ binding to the coding regions of LEE5 and LEE4, and then a point mutation that would affect binding was created for each prediction. The resulting mutants were then assayed for the ability to complement the glmY and glmZ mutants in FAS tests and quantitative RT-PCR (qRT-PCR) assays of target genes. While we have been unable to find any site (either in silico or empirically) of direct GlmZ or GlmY binding to the LEE5 transcript, one prediction of GlmZ binding to the orf29 region of the LEE4 operon was promising (Fig. 6H and I). We generated a GlmZ mutant by changing two C residues to G to prevent base pairing with the LEE4 operon (Fig. 6I). This GlmZ mutant, named GlmZ-CG, was unable to complement the pedestal formation of the glmZ knockout (Fig. 6K) or to complement the expression of the LEE4 gene espA (Fig. 6J). To confirm this result, we made the corresponding compensatory mutations in a plasmid containing the entire LEE4 operon, so that in the presence of both GlmZ-CG and LEE4-GC, the interaction would be restored (Fig. 6J). This was tested by qPCR for espA. The glmZ-GC plasmid was unable to complement the increased levels of espA in the ΔglmZ mutant; however, LEE4-GC restored this regulation (Fig. 6J). This indicates that GlmZ is interacting with LEE4 at this predicted site and that this interaction is responsible for the regulation of LEE4 by GlmZ.
Since GlmY- and GlmZ-mediated regulation of LEE4 and LEE5 does not act through repression of translation, it is likely that they recruit a nuclease that then degrades these transcripts. The major E. coli enzyme RNase E is known to be recruited to many sRNA-mRNA complexes, and it has been previously shown to mediate the processing of the LEE4 transcript (24). While rne is an essential gene, there is a well-characterized temperature-sensitive E. coli K-12 mutant that we used to test whether RNase E is involved in the GlmZ-dependent posttranscriptional regulation of LEE4 and LEE5. Since this is a K-12 strain, plasmids containing the entire LEE4 or LEE5 operon along with the glmZ overexpression plasmid were transformed into the rneWT (WT) and rnets (temperature-sensitive mutant) strains. The bacteria were heat shocked for 15 min before RNA was extracted. This was sufficient to stop the RNase E-mediated processing of sepL from espA as previously reported (24); however, it had no effect on the GlmZ downregulation of the espA-cesD2 transcript (see Fig. S4 in the supplemental material). The processing of tir from cesT-eae is also RNase E dependent, but GlmZ again is able to function in the absence of RNase E (see Fig. S4). These data show that, in both cases, LEE4 and LEE5 are processed by RNase E; however, GlmZ does not act by recruiting this nuclease. Another potential nuclease known to be recruited by sRNAs is RNase III. While the rnc gene is not essential, its knockout produces severe growth defects in EHEC, preventing its characterization.
DISCUSSION
By asking the question of how an extracellular bacterial pathogen rapidly and precisely coordinates the expression of its molecular circuitry to engage the expression of an array of genes necessary to encode the molecular structures and effectors that rearrange host actin dynamics, we uncovered three new targets and two different molecular mechanisms of action of the GlmY and GlmZ sRNAs. Our data establish that both the LEE and espFu genes that are necessary for pedestal formation on epithelial cells are posttranscriptionally regulated by the GlmY/GlmZ sRNAs through two different mechanisms. Several previous reports recognized that the LEE region is highly posttranscriptionally regulated (19, 23, 24, 29, 61, 62). However, no sRNA has yet been shown to be responsible for this regulation. One system known to be involved is the RNA binding protein and global regulator CsrA, which directly binds to the LEE4 operon and regulates a wide array of virulence factors through indirect means (19). Additionally, the RNA chaperone Hfq is required for the proper expression of many virulence genes (22, 23, 63), which suggested that trans-acting sRNAs are involved at some level of regulation. Here we describe the first sRNAs known to regulate the LEE, GlmY and GlmZ. Previous to this work, GlmY and GlmZ had only one target, the glmS mRNA, and one known molecular mechanism of action. GlmY was described as a molecular mimic of GlmZ, protecting GlmZ from degradation and allowing GlmZ to base pair with the glmS mRNA to expose the RBS and promote the translation of this gene (41, 42). Our data unraveled two new mechanisms of action for these sRNAs. GlmY and GlmZ promote cleavage of the intergenic region between espJ and espFu to allow the translation of EspFu. We do not know whether this is a direct effect of these sRNAs in the espJ-espFu transcript or an indirect effect through QseF regulation of other regulatory elements controlling EspFu expression (Fig. 2). Moreover, through destabilization of the LEE4 and LEE5 transcripts, these sRNAs fine-tune LEE expression (Fig. 7). One of the key advantages of posttranscriptional regulation of the LEE by GlmY and GlmZ is that it also allows for the differential regulation of gene expression within the LEE4 and LEE5 operons. This decoupling of the regulation of the genes of a polycistronic mRNA from each other enables a much more varied pattern of gene expression. GlmZ specifically downregulates the downstream portion of the LEE4 operon, including the filament EspA, the pore EspDB, and the needle EscF, while leaving SepL unaffected (Fig. 6). SepL is an important regulator of the translocation of effectors (64) and is likely to be required for effector delivery to host cells even when many of the structural proteins of the T3SS translocon are not. The posttranscriptional regulation of LEE4 mediated by GlmZ enables EHEC to tightly regulate the process of AE lesion formation (Fig. 2 to 7). The role of GlmY and GlmZ in the regulation of AE lesion formation by promoting EspFu translation and destabilizing LEE transcripts, seems to be initially confounding. However, AE lesion formation is a dynamic process, and the precise modulation of the levels of LEE and EspFu expression are important for the efficiency of this phenotype. Coupling of transcriptional and posttranscriptional regulation within a signaling transduction cascade in the bacterial cell is key to ensuring fine-tuning and rapid adaptation of gene expression toward the regulation of complex processes such as AE lesion formation.
FIG 7 .
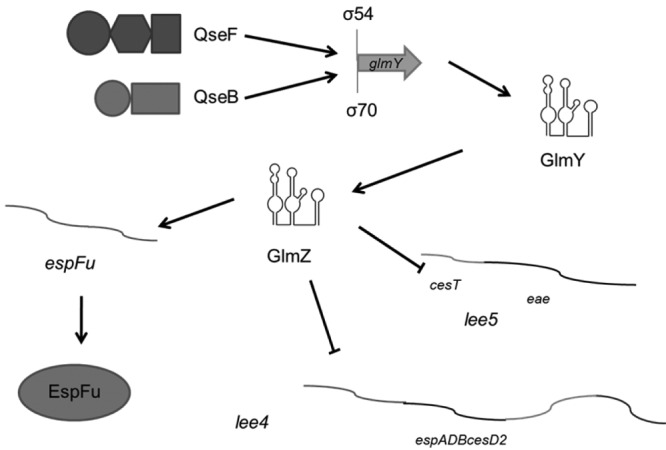
Proposed model of GlmY and GlmZ regulation of LEE4, LEE5, and espFu. Transcription of glmY is activated by both QseF and QseB. GlmY then stabilizes GlmZ, which controls espFu expression at the translational level and destabilizes the LEE4 and LEE5 mRNAs.
Core chromosome-encoded sRNAs that regulate metabolic functions in bacteria have been shown to be coopted to regulate virulence genes that are horizontally acquired by bacterial pathogens (65, 66). Pathogens evolve through the integration of horizontally acquired genetic material that is known to be integrated within existing transcriptional regulatory networks in the recipient cell (67). EHEC integrates the transcription of horizontally acquired virulence genes through the core QseC/QseE signaling system, which controls a plethora of virulence genes in EHEC that have to be coordinately expressed to ensure optimal AE lesion formation on epithelial cells, leading to host infection (37, 38, 43, 44). GlmZ is well characterized as the activator of glmS translation. This core metabolism-regulating sRNA was coopted to regulate the LEE and espFu, both horizontally acquired islands, at some point in evolutionary history. Horizontal acquisition of pathogenicity islands contributes to the virulence of an organism, allowing exploitation of other niches and hosts for colonization (68). Our results suggest that the interplay between ancient and recent evolutionary acquisitions has shaped EHEC pathogenicity. An inverse example of this phenomenon comes from the InvR sRNA from Salmonella enterica (69), where a coopted sRNA that is adjacent to the SPI-1 pathogenicity island regulates many core chromosomal genes.
While we did not directly observe the regulation of glmS by GlmZ under the conditions we assayed in EHEC, we have evidence from the ΔqseF mutant transcriptome that suggests that amino sugar metabolism and cell wall synthesis are still affected at some level in this EHEC mutant (70). The tying of glucosamine synthesis to interkingdom chemical signaling and pathogenesis may reflect the need to integrate both host and bacterial physiological cues to ensure a successful association between these organisms. It is also noteworthy that the LEE-encoded T3SS must span the periplasm and pass through the peptidoglycan layer. Since EHEC creates dozens of these injectisomes, there is likely significant remodeling of the cell wall during host infection.
MATERIALS AND METHODS
Strains and growth medium.
For the bacterial strains and plasmids used in this study, see Tables S1 and S2 in the supplemental material. Strains were grown in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) with low glucose or in Luria-Bertani medium (LB; Invitrogen) at 37°C and 250 rpm unless otherwise stated. Where necessary, the medium was supplemented with antibiotics at the following concentrations: ampicillin, 100 µg ml−1; chloramphenicol, 50 µg ml−1; kanamycin, 50 µg ml−1; streptomycin, 50 µg ml−1. When needed, the medium was also supplemented with 0.2% arabinose or 0.5 mM isopropyl-β-d-thiogalactopyranoside.
Plasmid construction.
The glmY and glmZ genes were cloned into plasmid pBAD33, and the LEE4 and LEE5 operons were cloned into pACYC177, disrupting the ampicillin gene. Site-directed mutagenesis was performed with the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). For transcriptional reporters, the promoter region was cloned into plasmid pRS551. For pCG48 (glmY::lacZ), bp −250 to +20 from the transcription start site were used. For pCG49 (glmZ::lacZ), bp −246 to −20 were used. For pCG50 (espFu::lacZ), bp −461 to +48 from the translation start site were used. The translation reporter system was constructed by cloning lacZ from MG1655 into the vector pBAD24 and then cloning the gene of interest from the transcription start site or processing site to a spot within the coding region, creating an in-frame reporter protein under the control of the arabinose promoter. pCG57 (EspFu::LacZ) contains bp −170 from the translation start site to bp +48. pCG59 (GlmS::LacZ) contains bp −160 to +117. pCG60 (Tir::LacZ) contains bp −137 to +90. pCG61 (CesT::LacZ) contains bp −71 to +90. pCG62 (Eae::LacZ) contains bp −62 to +90. pCG63 (SepL::LacZ) contains bp −82 to +90. pCG64 (EspA::LacZ) contains bp −107 to +90. pCG65 (EspD::LacZ) contains bp −61 to +90. pCG66 (EspB::LacZ) contains bp −68 to +90. The espJ-espFu constructs were created by first cloning the region from the espJ promoter to espFu with a FLAG tag added via a primer and inserting it into the Zero Blunt TOPO vector (Invitrogen). The deletion mutants were then created through sewing PCR. In p1, the region from 20 bp downstream of the espJ stop codon to 20 bp upstream of the espFu start codon was deleted. In p2, the region from 20 bp downstream of the espJ stop codon to 170 bp upstream of the espFu start codon was deleted. In p3, the region from 170 bp upstream of the espFu start codon to 20 bp upstream of the espFu start codon was deleted. In p4, the region starting 262 bp within the espJ gene to the end of espFu was cloned. All plasmids were confirmed through sequencing. For the primers used, see Table S3 in the supplemental material.
Isogenic mutant construction.
Nonpolar glmY and glmZ mutants were constructed through the lambda Red system (71). Briefly, PCR products (obtained with the primers listed in Table S3) were amplified from plasmid pKD3 with flanking regions matching glmY or glmZ and were transformed into EHEC expressing the Red genes from plasmid pKD46. After selection and confirmation, the resistance cassette was resolved with flippase from temperature-sensitive plasmid pCP20, which was then cured through growth at 37°C. This generated nonpolar mutants, which were confirmed by sequencing.
Reporter assays.
Transcriptional and translational β-galactosidase assays were performed by using the same protocol. Bacteria containing the reporter plasmids were grown overnight in LB with the appropriate antibiotic and then diluted 1:100 in clear DMEM supplemented with 0.2% arabinose and grown to an optical density at 600 nm (OD600) of 0.8. These were then assayed for β-galactosidase activity with o-nitrophenyl-β-d-galactopyranoside as previously described (72). For the experiments with the GlmS::LacZ reporter plasmid, bacteria were grown overnight in LB with 0.2% arabinose and specific activity (nM/min/mg protein) was measured by determining protein concentrations by the assay of Lowry et al. (73). The unpaired Student t test was used to determine statistical significance. A P value of ≤0.05 was considered significant.
EMSA.
QseB and QseF proteins were His tagged and purified from BL-21(DE3) and TOP10 cells, respectively, as previously described (36, 45). The glmY promoter and the kanamycin resistance-encoding gene were amplified by PCR (for the primers used, see Table S3 in the supplemental material) and radiolabeled with [γ-32]ATP and T4 polynucleotide kinase (NEB). The labeled DNA was then incubated with increasing amounts of protein and run on a 6% polyacrylamide Tris-Bis gel, dried, and exposed to film overnight.
Operon analysis by RT-PCR.
WT and ΔqseF mutant bacteria were grown to an OD600 of 1.0 in low-glucose DMEM, and RNA was extracted with the RiboPure kit (Ambion). RNA was reverse transcribed and amplified by PCR with different sets of primers (see Table S3 in the supplemental material).
Western blot assays.
Bacteria were grown overnight in LB and then diluted 1:100 in low-glucose DMEM and grown to an OD600 of 1.0. The pellets were then lysed under denaturing conditions, subjected to 12% SDS-PAGE, and transferred to a polyvinylidene difluoride membrane. The membrane was probed with an ant-FLAG (Sigma) or anti-RpoA (Santa Cruz Biotechnology) antibody as the endogenous control with a horseradish peroxidase-conjugated secondary antibody that was visualized by ECL (GE Bioscience) and exposed to film.
Cell culture and FAS.
HeLa cells were maintained in high-glucose DMEM supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin-glutamine and grown at 37°C and 5% CO2. Cells were split into a 12-well plate, grown to confluence, washed, given low-glucose DMEM supplemented with 10% FBS, and then infected with bacteria grown overnight in LB statically at 37°C at a 1:100 dilution. FAS assays were performed as described by Knutton et al. (36, 74) to examine pedestal formation. After 6 h of infection at 37°C in 5% CO2, the coverslips were washed, fixed, and permeabilized. The samples were then treated with FITC-labeled phalloidin and PI to visualize actin accumulation and bacteria, respectively. PI also stained HeLa nuclei red. The coverslips were then mounted on slides and visualized with a Zeiss Axiovert microscope. Pedestal formation was quantified by randomly imaging different fields of view and counting the first 100 cells while recording the number of bacteria attached to each one. Replicate coverslips from multiple experiments were quantified, and statistical analysis was performed with the Student t test. Serially diluted samples of the original bacterial cultures were also plated to confirm that similar CFU ratios were used for infection.
Live-cell imaging.
The Lifeact::GFP-expressing cell line was created with the Flip-In System (Invitrogen). HeLa cells were transfected with pLacZ::zeocin by using Fugene 6 (Promega) to create flippase recognition target sites in the genome. Cells were then selected with 100 µg/ml zeocin. Resistant foci were grown and assayed by Southern blot assay for lacZ for single insertions, and then β-galactosidase assays were performed to measure the expression of the inserted locus. High-expression single insertions were then transfected with the flippase helper plasmid pOD44 and Lifeact::GFP cloned into plasmid pFRT. These transfected cells were then selected in 100 µg/ml hygromycin, and resistant foci were visualized by fluorescence microscopy to measure levels of Lifeact::GFP expression. These cells were then maintained in 50 µg/ml hygromycin. When they are split before infection, hygromycin was not added. EHEC cells were transformed with mCherry-expressing plasmid pDP151 and grown overnight statically in LB. The Lifeact::GFP-expressing cell medium was replaced with low-glucose DMEM supplemented with 10% FBS, and the cells were infected with a 1:100 dilution of the overnight culture. Infection was allowed to continue for 2 h at 37°C and 5% CO2, and then the cells are washed three times with DMEM and then visualized by live-cell imaging with a Zeiss microscope. Images were taken every 2 min for 2 h.
Northern blot assays.
Bacteria were grown aerobically in low-glucose DMEM supplemented with 0.2% arabinose at 37°C to an OD600 of 1.0 from a 1:100 dilution of an overnight culture grown in LB. RNA was extracted with the RiboPure kit (Ambion), run on a 1% formaldehyde agarose gel, and transferred overnight to a Zeta-Probe membrane (Bio-Rad). RNA probes were created by PCR amplification of a segment of the gene of interest with the T7 promoter and in vitro transcription with the MAXIscript T7 kit (Ambion) with [α-32P]UTP. The oligoprobe for the 5S endogenous control was labeled with [γ-32P]ATP by using T4 polynucleotide kinase (NEB). The membranes were then hybridized overnight with Ultrahyb (Ambion) at 68ºC for the RNA probes and 37°C for the oligoprobes. The membranes were washed, exposed to a phosphorimager screen overnight, and then visualized with a STORM scanner (GE Healthcare).
RNA extraction and qRT-PCR.
Cultures were grown overnight in LB, diluted 1:100 in low-glucose DMEM with 0.2% arabinose on a 12-well plate, and then grown for 6 h at 37°C under 5% CO2. RNA was extracted from three biological replicates with the RiboPure Bacteria isolation kit according to the manufacturer’s protocols (Ambion). qRT-PCR was performed as described previously (38). Briefly, diluted extracted RNA was mixed with, validated primers (see Table S3 in the supplemental material), RNase inhibitor, and reverse transcriptase (AB). The mixture was used in a one-step reaction utilizing an ABI 7500 sequence detection system. Data were collected with ABI Sequence detection 1.2 software, normalized to endogenous rpoA levels, and analyzed by the comparative critical threshold method. Analyzed data were presented as fold changes over the WT levels. The unpaired Student t test was used to determine statistical significance. A P value of ≤0.05 was considered significant.
Temperature-sensitive RNase E strain.
rneWT strain NL3433 and rnets strain NL3431 were transformed with the pLEE4 or pLEE5 plasmid and pGlmZ. Both were grown overnight in LB at 30°C in the appropriate antibiotics and then grown to an OD600 of 1.0 from a 1:100 dilution in LB at 35°C. Once they reached this OD600, they were shifted to the nonpermissive temperature of 43°C for 15 min and then the samples were treated as for the other Northern blot assays.
SUPPLEMENTAL MATERIAL
Strains used in this study.
Plasmids used in this study.
Primers used in this study.
QseB binding sites in EHEC, EPEC, and K-12 strains. (A) Alignment of the glmY regulatory region depicting the QseB binding site of EHEC strain 86-24 and K-12 strain MG1655. (B) EMSAs of the glmY regulatory region from EHEC strain 86-24, K-12 strain MG155, and EPEC strain E2348/69 with the QseB protein. Download
GlcNAc and GlmS are not involved in the AE lesion GlmY/GlmZ-dependent phenotype. (A) Scheme depicting the mechanism of GlmZ regulation of glmS translation. (B) FAS of HeLa cells infected with WT or ΔglmY or ΔglmZ mutant bacteria in the absence or presence of 1% GlcNAC. (C) Quantification of the FAS of HeLa cells infected with WT or ΔglmY or ΔglmZ mutant bacteria in the absence or presence of 1% GlcNAC depicting the average number of bacteria attached per cell. (D) Sequence and structure of the GlmZ sRNA. Arrows depict the site-directed mutations created to disrupt GlmZ/glmS pairing. This mutant sRNA is named GlmZ*. (E) FAS of HeLa cells infected with the WT, the ΔglmZ mutant, the ΔglmZ mutant complemented with pGlmZ, or the ΔglmZ/pGlmZ* mutant, which produces a GlmZ protein that is incapable of binding to glmS. (F) Quantification of the FAS of HeLa cells infected with the WT, the ΔglmZ mutant, the ΔglmZ mutant complemented with pGlmZ, or the ΔglmZ/pGlmZ* mutant, which produces a GlmZ protein that is incapable of binding to glmS, depicting the average number of bacteria attached per cell. Download
GlmY and GlmZ regulation of glmS in EHEC and K-12 strain MC4100. (A) Northern blot assay for glmS with RNA from WT, ΔglmZ, ΔglmZ/pGlmZ, and ΔglmZ/pGlmZ* cells. (B) β-Galactosidase assay of the pGlmS::LacZ translation reporter in WT EHEC, WT EHEC overexpressing glmY, and WT EHEC overexpressing glmZ. GlmS::LacZ contains the region stretching from bp −160 to +117 from the translation start site (C) β-Galactosidase assay of the pGlmS::Lac translation reporter in strain MC4100, strain MC4100 overexpressing glmY, and strain MC4100 overexpressing glmZ. Download
LEE4 and LEE5 GlmZ-mediated RNA processing does not occur through RNase E. Northern blot assays for sepL, espA, tir, and eae with the 5S rRNA serving as a loading control on RNA from heat-shocked cells of the rneWT strain, the rneWT strain overexpressing glmZ strain, the rnets strain, and the rnets strain overexpressing glmZ. Download
Live-cell microscopy of Lifeact::GFP-expressing HeLa cells being infected with mCherry-expressing WT EHEC. Download
Live-cell microscopy of Lifeact::GFP-expressing HeLa cells being infected with mCherry-expressing ΔglmY mutant EHEC. Download
Live-cell microscopy of Lifeact::GFP-expressing HeLa cells being infected with mCherry-expressing ΔglmZ mutant EHEC. Download
ACKNOWLEDGMENTS
We thank Lora V. Hooper, and Jörg Vogel for reading of the manuscript. We also thank Neal M. Alto and Robert C. Orchard for help with the live-cell imaging, as well as Nicholas K. Conrad and Sara H. Stubbs for help with creation of the Lifeact-expressing cell line. We also thank James B. Kaper for the rnets strain.
This work was supported by NIH grants AI053067 and AI077613 and by the Burroughs Wellcome Fund. C.C.G. was supported by grant T32-AI07520.
Footnotes
Citation Gruber CC, Sperandio V. 2014. Posttranscriptional control of microbe-induced rearrangement of host cell actin. mBio 5(1):e01025-13. doi:10.1128/mBio.01025-13.
REFERENCES
- 1. Hicks SW, Galán JE. 2013. Exploitation of eukaryotic subcellular targeting mechanisms by bacterial effectors. Nat. Rev. Microbiol. 11:316–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaper JB, Nataro JP, Mobley HL. 2004. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2:123–140 [DOI] [PubMed] [Google Scholar]
- 3. McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB. 1995. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. U. S. A. 92:1664–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jarvis KG, Girón JA, Jerse AE, McDaniel TK, Donnenberg MS, Kaper JB. 1995. Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc. Natl. Acad. Sci. U. S. A. 92:7996–8000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Elliott SJ, Hutcheson SW, Dubois MS, Mellies JL, Wainwright LA, Batchelor M, Frankel G, Knutton S, Kaper JB. 1999. Identification of CesT, a chaperone for the type III secretion of Tir in enteropathogenic Escherichia coli. Mol. Microbiol. 33:1176–1189 [DOI] [PubMed] [Google Scholar]
- 6. Mellies JL, Elliott SJ, Sperandio V, Donnenberg MS, Kaper JB. 1999. The per regulon of enteropathogenic Escherichia coli: identification of a regulatory cascade and a novel transcriptional activator, the locus of enterocyte effacement (LEE)-encoded regulator (Ler). Mol. Microbiol. 33:296–306 [DOI] [PubMed] [Google Scholar]
- 7. Elliott SJ, Wainwright LA, McDaniel TK, Jarvis KG, Deng YK, Lai LC, McNamara BP, Donnenberg MS, Kaper JB. 1998. The complete sequence of the locus of enterocyte effacement (LEE) from enteropathogenic Escherichia coli E2348/69. Mol. Microbiol. 28:1–4 [DOI] [PubMed] [Google Scholar]
- 8. Jerse AE, Yu J, Tall BD, Kaper JB. 1990. A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc. Natl. Acad. Sci. U. S. A. 87:7839–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kenny B, DeVinney R, Stein M, Reinscheid DJ, Frey EA, Finlay BB. 1997. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91:511–520 [DOI] [PubMed] [Google Scholar]
- 10. McNamara BP, Donnenberg MS. 1998. A novel proline-rich protein, EspF, is secreted from enteropathogenic Escherichia coli via the type III export pathway. FEMS Microbiol. Lett. 166:71–78 [DOI] [PubMed] [Google Scholar]
- 11. Kenny B, Jepson M. 2000. Targeting of an enteropathogenic Escherichia coli (EPEC) effector protein to host mitochondria. Cell. Microbiol. 2:579–590 [DOI] [PubMed] [Google Scholar]
- 12. Elliott SJ, Krejany EO, Mellies JL, Robins-Browne RM, Sasakawa C, Kaper JB. 2001. EspG, a novel type III system-secreted protein from enteropathogenic Escherichia coli with similarities to VirA of Shigella flexneri. Infect. Immun. 69:4027–4033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tu X, Nisan I, Yona C, Hanski E, Rosenshine I. 2003. EspH, a new cytoskeleton-modulating effector of enterohaemorrhagic and enteropathogenic Escherichia coli. Mol. Microbiol. 47:595–606 [DOI] [PubMed] [Google Scholar]
- 14. Kanack KJ, Crawford JA, Tatsuno I, Karmali MA, Kaper JB. 2005. SepZ/EspZ is secreted and translocated into HeLa cells by the enteropathogenic Escherichia coli type III secretion system. Infect. Immun. 73:4327–4337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Campellone KG, Robbins D, Leong JM. 2004. EspFU is a translocated EHEC effector that interacts with Tir and N-WASP and promotes Nck-independent actin assembly. Dev. Cell 7:217–228 [DOI] [PubMed] [Google Scholar]
- 16. Garmendia J, Phillips AD, Carlier MF, Chong Y, Schüller S, Marches O, Dahan S, Oswald E, Shaw RK, Knutton S, Frankel G. 2004. TccP is an enterohaemorrhagic Escherichia coli O157:H7 type III effector protein that couples Tir to the actin-cytoskeleton. Cell. Microbiol. 6:1167–1183 [DOI] [PubMed] [Google Scholar]
- 17. Tobe T, Beatson SA, Taniguchi H, Abe H, Bailey CM, Fivian A, Younis R, Matthews S, Marches O, Frankel G, Hayashi T, Pallen MJ. 2006. An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. Proc. Natl. Acad. Sci. U. S. A. 103:14941–14946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deng W, Puente JL, Gruenheid S, Li Y, Vallance BA, Vázquez A, Barba J, Ibarra JA, O’Donnell P, Metalnikov P, Ashman K, Lee S, Goode D, Pawson T, Finlay BB. 2004. Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc. Natl. Acad. Sci. U. S. A. 101:3597–3602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bhatt S, Edwards AN, Nguyen HT, Merlin D, Romeo T, Kalman D. 2009. The RNA binding protein CsrA is a pleiotropic regulator of the locus of enterocyte effacement pathogenicity island of enteropathogenic Escherichia coli. Infect. Immun. 77:3552–3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Njoroge JW, Nguyen Y, Curtis MM, Moreira CG, Sperandio V. 2012. Virulence meets metabolism: Cra and KdpE gene regulation in enterohemorrhagic Escherichia coli. mBio 3(5):e00280-00212. 10.1128/mBio.00280-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sperandio V, Torres AG, Jarvis B, Nataro JP, Kaper JB. 2003. Bacteria-host communication: the language of hormones. Proc. Natl. Acad. Sci. U. S. A. 100:8951–8956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kendall MM, Gruber CC, Rasko DA, Hughes DT, Sperandio V. 2011. Hfq virulence regulation in enterohemorrhagic Escherichia coli O157:H7 strain 86-24. J. Bacteriol. 193:6843–6851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shakhnovich EA, Davis BM, Waldor MK. 2009. Hfq negatively regulates type III secretion in EHEC and several other pathogens. Mol. Microbiol. 74:347–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lodato PB, Kaper JB. 2009. Post-transcriptional processing of the LEE4 operon in enterohaemorrhagic Escherichia coli. Mol. Microbiol. 71:273–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mellies JL, Barron AM, Carmona AM. 2007. Enteropathogenic and enterohemorrhagic Escherichia coli virulence gene regulation. Infect. Immun. 75:4199–4210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hughes DT, Terekhova DA, Liou L, Hovde CJ, Sahl JW, Patankar AV, Gonzalez JE, Edrington TS, Rasko DA, Sperandio V. 2010. Chemical sensing in mammalian host-bacterial commensal associations. Proc. Natl. Acad. Sci. U. S. A. 107:9831–9836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kendall MM, Gruber CC, Parker CT, Sperandio V. 2012. Ethanolamine controls expression of genes encoding components involved in interkingdom signaling and virulence in enterohemorrhagic Escherichia coli O157:H7. mBio 3(3):e00050-12. 10.1128/mBio.00050-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pacheco AR, Curtis MM, Ritchie JM, Munera D, Waldor MK, Moreira CG, Sperandio V. 2012. Fucose sensing regulates bacterial intestinal colonization. Nature 492:113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bhatt S, Romeo T, Kalman D. 2011. Honing the message: post-transcriptional and post-translational control in attaching and effacing pathogens. Trends Microbiol. 19:217–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Storz G, Vogel J, Wassarman KM. 2011. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell 43:880–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sperandio V, Torres AG, Giron JA, Kaper JB. 2001. Quorum sensing is a global regulatory mechanism in enterohemorrhagic Escherichia coli O157:H7. J. Bacteriol. 183:5187–5197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clarke MB, Hughes DT, Zhu C, Boedeker EC, Sperandio V. 2006. The QseC sensor kinase: a bacterial adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A. 103:10420–10425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Walters M, Sircili MP, Sperandio V. 2006. AI-3 synthesis is not dependent on luxS in Escherichia coli. J. Bacteriol. 188:5668–5681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clarke MB, Hughes DT, Zhu C, Boedeker EC, Sperandio V. 2006. The QseC sensor kinase: a bacterial adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A. 103:10420–10425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reading NC, Rasko DA, Torres AG, Sperandio V. 2009. The two-component system QseEF and the membrane protein QseG link adrenergic and stress sensing to bacterial pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 106:5889–5894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reading NC, Torres AG, Kendall MM, Hughes DT, Yamamoto K, Sperandio V. 2007. A novel two-component signaling system that activates transcription of an enterohemorrhagic Escherichia coli effector involved in remodeling of host actin. J. Bacteriol. 189:2468–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rasko DA, Moreira CG, Li DR, Reading NC, Ritchie JM, Waldor MK, Williams N, Taussig R, Wei S, Roth M, Hughes DT, Huntley JF, Fina MW, Falck JR, Sperandio V. 2008. Targeting QseC signaling and virulence for antibiotic development. Science 321:1078–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hughes DT, Clarke MB, Yamamoto K, Rasko DA, Sperandio V. 2009. The QseC adrenergic signaling cascade in enterohemorrhagic E. coli (EHEC). PLoS Pathog. 5:e1000553. 10.1371/journal.ppat.1000553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yamamoto K, Hirao K, Oshima T, Aiba H, Utsumi R, Ishihama A. 2005. Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli. J. Biol. Chem. 280:1448–1456 [DOI] [PubMed] [Google Scholar]
- 40. Reichenbach B, Göpel Y, Görke B. 2009. Dual control by perfectly overlapping sigma 54- and sigma 70-promoters adjusts small RNA GlmY expression to different environmental signals. Mol. Microbiol. 74:1054–1070 [DOI] [PubMed] [Google Scholar]
- 41. Göpel Y, Papenfort K, Reichenbach B, Vogel J, Görke B. 2013. Targeted decay of a regulatory small RNA by an adaptor protein for RNase E and counteraction by an anti-adaptor RNA. Genes Dev. 27:552–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Urban JH, Vogel J. 2008. Two seemingly homologous noncoding RNAs act hierarchically to activate glmS mRNA translation. PLoS Biol. 6:e64. 10.1371/journal.pbio.0060064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Clarke MB, Hughes DT, Zhu C, Boedeker EC, Sperandio V. 2006. The QseC sensor kinase: a bacterial adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A. 103:10420–10425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Njoroge J, Sperandio V. 2012. Enterohemorrhagic Escherichia coli virulence regulation by two bacterial adrenergic kinases, QseC and QseE. Infect. Immun. 80:688–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Clarke MB, Sperandio V. 2005. Transcriptional regulation of flhDC by QseBC and sigma (FliA) in enterohaemorrhagic Escherichia coli. Mol. Microbiol. 57:1734–1749 [DOI] [PubMed] [Google Scholar]
- 46. Gyaneshwar P, Paliy O, McAuliffe J, Jones A, Jordan MI, Kustu S. 2005. Lessons from Escherichia coli genes similarly regulated in response to nitrogen and sulfur limitation. Proc. Natl. Acad. Sci. U. S. A. 102:3453–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vingadassalom D, Campellone KG, Brady MJ, Skehan B, Battle SE, Robbins D, Kapoor A, Hecht G, Snapper SB, Leong JM. 2010. Enterohemorrhagic E. coli requires N-WASP for efficient type III translocation but not for EspFU-mediated actin pedestal formation. PLoS Pathog. 6:e1001056. 10.1371/journal.ppat.1001056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marchès O, Covarelli V, Dahan S, Cougoule C, Bhatta P, Frankel G, Caron E. 2008. EspJ of enteropathogenic and enterohaemorrhagic Escherichia coli inhibits opsono-phagocytosis. Cell. Microbiol. 10:1104–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. 2008. Lifeact: a versatile marker to visualize F-actin. Nat. Methods 5:605–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vogler AP, Trentmann S, Lengeler JW. 1989. Alternative route for biosynthesis of amino sugars in Escherichia coli K-12 mutants by means of a catabolic isomerase. J. Bacteriol. 171:6586–6592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sperandio V, Mellies JL, Nguyen W, Shin S, Kaper JB. 1999. Quorum sensing controls expression of the type III secretion gene transcription and protein secretion in enterohemorrhagic and enteropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 96:15196–15201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Abe A, de Grado M, Pfuetzner RA, Sánchez-Sanmartín C, Devinney R, Puente JL, Strynadka NC, Finlay BB. 1999. Enteropathogenic Escherichia coli translocated intimin receptor, Tir, requires a specific chaperone for stable secretion. Mol. Microbiol. 33:1162–1175 [DOI] [PubMed] [Google Scholar]
- 53. Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136:615–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sekiya K, Ohishi M, Ogino T, Tamano K, Sasakawa C, Abe A. 2001. Supermolecular structure of the enteropathogenic Escherichia coli type III secretion system and its direct interaction with the EspA-sheath-like structure. Proc. Natl. Acad. Sci. U. S. A. 98:11638–11643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Knutton S, Rosenshine I, Pallen MJ, Nisan I, Neves BC, Bain C, Wolff C, Dougan G, Frankel G. 1998. A novel EspA-associated surface organelle of enteropathogenic Escherichia coli involved in protein translocation into epithelial cells. EMBO J. 17:2166–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Crane JK, McNamara BP, Donnenberg MS. 2001. Role of EspF in host cell death induced by enteropathogenic Escherichia coli. Cell. Microbiol. 3:197–211 [DOI] [PubMed] [Google Scholar]
- 57. Elliott SJ, Sperandio V, Girón JA, Shin S, Mellies JL, Wainwright L, Hutcheson SW, McDaniel TK, Kaper JB. 2000. The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 68:6115–6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McNally A, Roe AJ, Simpson S, Thomson-Carter FM, Hoey DE, Currie C, Chakraborty T, Smith DG, Gally DL. 2001. Differences in levels of secreted locus of enterocyte effacement proteins between human disease-associated and bovine Escherichia coli O157. Infect. Immun. 69:5107–5114 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59. Roe AJ, Yull H, Naylor SW, Woodward MJ, Smith DG, Gally DL. 2003. Heterogeneous surface expression of EspA translocon filaments by Escherichia coli O157:H7 is controlled at the posttranscriptional level. Infect. Immun. 71:5900–5909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Busch A, Richter AS, Backofen R. 2008. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 24:2849–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lodato PB, Hsieh PK, Belasco JG, Kaper JB. 2012. The ribosome binding site of a mini-ORF protects a T3SS mRNA from degradation by RNase E. Mol. Microbiol. 86:1167–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Campellone KG, Roe AJ, Lobner-Olesen A, Murphy KC, Magoun L, Brady MJ, Donohue-Rolfe A, Tzipori S, Gally DL, Leong JM, Marinus MG. 2007. Increased adherence and actin pedestal formation by dam-deficient enterohaemorrhagic Escherichia coli O157:H7. Mol. Microbiol. 63:1468–1481 [DOI] [PubMed] [Google Scholar]
- 63. Hansen AM, Kaper JB. 2009. Hfq affects the expression of the LEE pathogenicity island in enterohaemorrhagic Escherichia coli. Mol. Microbiol. 73:446–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Deng W, Li Y, Hardwidge PR, Frey EA, Pfuetzner RA, Lee S, Gruenheid S, Strynakda NC, Puente JL, Finlay BB. 2005. Regulation of type III secretion hierarchy of translocators and effectors in attaching and effacing bacterial pathogens. Infect. Immun. 73:2135–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Papenfort K, Podkaminski D, Hinton JC, Vogel J. 2012. The ancestral SgrS RNA discriminates horizontally acquired Salmonella mRNAs through a single G-U wobble pair. Proc. Natl. Acad. Sci. U. S. A. 109:E757–E764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Papenfort K, Sun Y, Miyakoshi M, Vanderpool CK, Vogel J. 2013. Small RNA-mediated activation of sugar phosphatase mRNA regulates glucose homeostasis. Cell 153:426–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Navarre WW, Porwollik S, Wang Y, McClelland M, Rosen H, Libby SJ, Fang FC. 2006. Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science 313:236–238 [DOI] [PubMed] [Google Scholar]
- 68. Ochman H, Lawrence JG, Groisman EA. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304 [DOI] [PubMed] [Google Scholar]
- 69. Vogel J. 2009. A rough guide to the non-coding RNA world of Salmonella. Mol. Microbiol. 71:1–11 [DOI] [PubMed] [Google Scholar]
- 70. Reading NC, Rasko D, Torres AG, Sperandio V. 2010. A transcriptome study of the QseEF two-component system and the QseG membrane protein in enterohaemorrhagic Escherichia coli O15:H7. Microbiology 156:1167–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 73. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275 [PubMed] [Google Scholar]
- 74. Knutton S, Baldwin T, Williams PH, McNeish AS. 1989. Actin accumulation at sites of bacterial adhesion to tissue culture cells: basis of a new diagnostic test for enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 57:1290–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Strains used in this study.
Plasmids used in this study.
Primers used in this study.
QseB binding sites in EHEC, EPEC, and K-12 strains. (A) Alignment of the glmY regulatory region depicting the QseB binding site of EHEC strain 86-24 and K-12 strain MG1655. (B) EMSAs of the glmY regulatory region from EHEC strain 86-24, K-12 strain MG155, and EPEC strain E2348/69 with the QseB protein. Download
GlcNAc and GlmS are not involved in the AE lesion GlmY/GlmZ-dependent phenotype. (A) Scheme depicting the mechanism of GlmZ regulation of glmS translation. (B) FAS of HeLa cells infected with WT or ΔglmY or ΔglmZ mutant bacteria in the absence or presence of 1% GlcNAC. (C) Quantification of the FAS of HeLa cells infected with WT or ΔglmY or ΔglmZ mutant bacteria in the absence or presence of 1% GlcNAC depicting the average number of bacteria attached per cell. (D) Sequence and structure of the GlmZ sRNA. Arrows depict the site-directed mutations created to disrupt GlmZ/glmS pairing. This mutant sRNA is named GlmZ*. (E) FAS of HeLa cells infected with the WT, the ΔglmZ mutant, the ΔglmZ mutant complemented with pGlmZ, or the ΔglmZ/pGlmZ* mutant, which produces a GlmZ protein that is incapable of binding to glmS. (F) Quantification of the FAS of HeLa cells infected with the WT, the ΔglmZ mutant, the ΔglmZ mutant complemented with pGlmZ, or the ΔglmZ/pGlmZ* mutant, which produces a GlmZ protein that is incapable of binding to glmS, depicting the average number of bacteria attached per cell. Download
GlmY and GlmZ regulation of glmS in EHEC and K-12 strain MC4100. (A) Northern blot assay for glmS with RNA from WT, ΔglmZ, ΔglmZ/pGlmZ, and ΔglmZ/pGlmZ* cells. (B) β-Galactosidase assay of the pGlmS::LacZ translation reporter in WT EHEC, WT EHEC overexpressing glmY, and WT EHEC overexpressing glmZ. GlmS::LacZ contains the region stretching from bp −160 to +117 from the translation start site (C) β-Galactosidase assay of the pGlmS::Lac translation reporter in strain MC4100, strain MC4100 overexpressing glmY, and strain MC4100 overexpressing glmZ. Download
LEE4 and LEE5 GlmZ-mediated RNA processing does not occur through RNase E. Northern blot assays for sepL, espA, tir, and eae with the 5S rRNA serving as a loading control on RNA from heat-shocked cells of the rneWT strain, the rneWT strain overexpressing glmZ strain, the rnets strain, and the rnets strain overexpressing glmZ. Download
Live-cell microscopy of Lifeact::GFP-expressing HeLa cells being infected with mCherry-expressing WT EHEC. Download
Live-cell microscopy of Lifeact::GFP-expressing HeLa cells being infected with mCherry-expressing ΔglmY mutant EHEC. Download
Live-cell microscopy of Lifeact::GFP-expressing HeLa cells being infected with mCherry-expressing ΔglmZ mutant EHEC. Download