

Abstract
Shigella flexneri uses its type III secretion system (T3SS) to promote invasion of human intestinal epithelial cells as the first step in causing shigellosis, a life threatening form of dysentery. The Shigella type III secretion apparatus (T3SA) consists of a basal body that spans the bacterial envelope and an exposed needle that injects effector proteins into target cells. The nascent Shigella T3SA needle is topped with a pentamer of the needle tip protein invasion plasmid antigen D (IpaD). Bile salts trigger recruitment of the first hydrophobic translocator protein, IpaB, to the tip complex where it senses contact with a host membrane. In the bacterial cytoplasm, IpaB exists in a complex with its chaperone IpgC. Several structures of IpgC have been solved and we recently reported the 2.1-Å crystal structure of the N-terminal domain (IpaB74.224) of IpaB. Like IpgC, the IpaB N-terminal domain exists as a homodimer in solution. We now report that when the two are mixed, these homodimers dissociate and form heterodimers having a nanomolar dissociation constant. This is consistent with the equivalent complexes co-purified after being co-expressed in E. coli. Fluorescence data presented here also indicate that the N-terminal domain of IpaB possesses two regions that appear to contribute additively to chaperone binding. It is also likely that the IpaB N terminus adopts an alternative conformation as a result of chaperone binding. The importance of these findings within the functional context of these proteins is discussed.
Type III secretion systems (T3SS) are conserved virulence factors with diverse pathogen-specific roles used by a wide range of Gram-negative bacterial pathogens (1, 2). T3SSs are used to translocate effector proteins directly into a target cell’s cytoplasm where they modulate a variety of host cell functions for the benefit of the pathogen (3). Shigella flexneri, the causative agent of shigellosis, uses its T3SS to invade human colonic epithelial cells as an initial step in establishing infection (3). Invasion is directed by the injection of effector proteins into the host cell cytoplasm that culminate in cytoskeletal rearrangements that ultimately result in pathogen uptake. Once inside the host cell, Shigella quickly lyses the vacuole to enter the cytoplasm where it replicates and spreads to adjacent cells by actin-based motility (1–3).
The type III secretion apparatus (T3SA) is comprised of a basal body that spans the inner and outer bacterial membranes, a surface-exposed needle consisting of a polymer of the protein MxiH, and a secretion-controlling tip complex (4–6). The nascent tip complex is composed of a pentameric ring of invasion plasmid antigen D (IpaD) at the exposed end of the T3SA needle (4, 7, 8). Upon exposure to bile salts, IpaD undergoes a conformational change that leads to recruitment of the first hydrophobic translocator protein, IpaB, to the T3SA needle tip where it localizes distal to IpaD (9). Induction of the T3SS occurs upon subsequent incubation with cholesterol-rich liposomes, which leads to recruitment of the second hydrophobic translocator protein, IpaC, to the needle tip where it associates with IpaB to ultimately form a translocon pore within the host cell membrane (10). The newly formed translocon completes formation of a unidirectional conduit and promotes translocation of effectors from the bacterium directly into the host cell cytoplasm by the T3SS (10).
Prior to their secretion, the translocator proteins IpaB and IpaC independently associate with their cognate class II chaperone IpgC in the Shigella cytoplasm (11). Like all T3SS class II chaperones, IpgC is a small (17.8 kDa), highly α-helical protein with an acidic pI that possesses a tetratricopeptide repeat (TPR) fold (11–15). Cytoplasmic association of the translocators with IpgC presumably occurs to prevent premature association of IpaB with IpaC and/or deleterious interaction of the translocators with the bacterial membrane (11). Expression of the translocators in either Shigella or E. coli in the absence of IpgC results in their rapid degradation (11, 16). Following delivery of the translocators to the ATPase of the T3SA secretion platform in the Shigella cytoplasm, the released IpgC has been reported to form a dimer that associates with MxiE (11). This MxiE-IpgC complex then acts as a transcription factor by binding DNA sequences at MxiE-boxes to promote expression of late T3SS effector protein genes (11).
Full-length IpaB can be produced in large amounts as a recombinant protein in E. coli, but only when co-expressed with IpgC (16). Although the resulting IpaB-IpgC complex can be disassociated by incubation with certain mild detergents, this dissociation is not reversible. This could be a result of IpaB adopting a new conformation upon release from the chaperone since it forms discrete multimeric complexes in the absence of IpgC (data not shown). Some groups have proposed that a short peptide-length region of IpaB accounts for much of its interaction with IpgC and gives rise to a complex containing one IpaB and two IpgC molecules (12, 17). In this study, we examined the interaction of the N-terminal domain of IpaB with IpgC and used information on the structure of this IpaB domain to more precisely define the molecular basis for its chaperone binding. The hydrophobic nature of full-length IpaB has made a high-resolution structure elusive, however, the stabilizing effect of IpgC on IpaB has allowed us to determine the structure of its N-terminal domain (18). Using this structure, we have found a useful starting point for understanding the intricacies of IpaB structure and the effect that IpgC binding has on it. In return, we have also been able to identify how IpaB binding influences the physical state of IpgC in solution. The implications of these findings in light of the known roles of IpaB and IpgC following induction of type III secretion are discussed.
Experimental Procedures
Cloning, expression and purification of recombinant proteins
The full-length ipaB gene was cloned into pT7HMT (19). This vector was co-transformed with ipgC in pACYC-DUET (16) into E. coli BL21(DE3) and grown in Terrific Broth containing kanamycin (50 μg/ml) and chloramphenicol (30 μg/ml) at 37°C to an absorbance at 600 nm (A600) of about 0.8. Protein co-expression and purification was as previously described (18).
Individual DNA fragments encoding specific N-terminal fragments of IpaB (residues 1–94, 1–226 and 28–226, respectively) were designed, amplified by PCR, and subcloned into the pT7HMT plasmid. Each fragment of IpaB was also overexpressed from a modified version of the pT7HMT vector, described previously (20), which encodes a Cys residue as the first (N-terminal) amino acid of the polypeptide and a second version that encodes Cys as the last (C-terminal) residue. Over-expression and purification of the translocator fragments was carried out as previously described for IpaB/IpgC (18), with the exception that recombinant TEV protease was used to digest the fusion affinity tag from each target protein (21). Proteins were dialyzed against 10 mM phosphate (pH 7.4) with 150 mM NaCl (PBS) and used immediately or stored at −80°C.
Fluorescence labeling of proteins
IpaBN28.226, IpaBC28.226, and IpaBN1.226 contained a single Cys residue at either the N or C terminus. These IpaB fragments were dialyzed against labeling buffer (50 mM Hepes (pH 7.0), 150 mM NaCl, and 5 mM tris (2-carboxyethyl)phosphine (TCEP)). The TCEP concentration was then reduced to 1 mM by dialysis prior to labeling with a ten-fold molar excess of AlexaFluor-350 maleimide (Alexa350) or fluorescein maleimide (FM) dissolved in dimethylsulfoxide (DMSO) or N′,N′-dimethylformamide (DMF), respectively. Reactions were stirred under nitrogen for 2 h at 25°C. Unreacted dye was removed by gel filtration using a Superdex-75 gel filtration column (GE Lifesciences) equilibrated with PBS. Alexa350-IpaBC1.226 was unstable and could not be used in this study
Far-UV circular dichroism (CD) spectroscopy
Far-UV CD spectra were collected on a Jasco Model J-815 spectropolarimeter equipped with a Peltier temperature controller (Jasco Inc., Easton, MD) (22). Spectra were acquired using a 0.1 cm path length quartz cuvette at 10°C with a spectral resolution of 1.0 nm, a scanning rate of 50 nm/min, and a 2-sec data integration time. All spectra are an average of three measurements. Secondary structure thermal stability was monitored at 222 nm over a temperature range from 10 to 85°C. Data were acquired every 2.5°C and the temperature ramp rate was 15°C/h. The protein concentration was 0.3 mg/ml for IpaB and IpaB/IpgC in 20 mM citrate-phosphate buffer (pH 7.0) with 150mM NaCl. CD signals were converted to mean residue molar ellipticities [θ]R and thermal transitions were analyzed using the Jasco Spectral Manager™.
Fluorescence polarization (FP)
FM-IpaBN28.226, FM-IpaBC28.226, FM-IpaBN1.226, and FM-IpaBC1.226 were maintained at concentration of 80 nM in PBS while IpgC was titrated in at concentrations ranging from 0 to 1 μM IpgC. After a 1 hr incubation at room temperature, FP of fluorescein was measured using an excitation wavelength of 485 nm and an emission of 520 nm in a Molecular Devices SpectraMax M5 plate reader. FP of W105 was measured using an excitation at 295 nm and an emission of 340 nm.
Forster resonance energy transfer and the calculation of intramolecular distances
Fluorescence spectra were collected using a Jobin-Yvon FluoroMax-4 spectrofluorometer based on nonradiative transfer of donor excitation energy from the native W105 to the Alexa350 acceptor of Alexa350-IpaBN28.226, Alexa350-IpaBC28.226, and Alexa350-IpaBN1.226 fragments. Identical concentrations of donor only (D) samples (without Alexa350 probe) or donor plus acceptor (DA) samples (with Alexa350 probe) were excited at 295 nm and emission spectra were collected from 300–400 nm. Excitation and emission slits were set to 2.5 nm, integration time was 1 sec. Emission spectra of samples containing 1 μM Alexa350-IpaBN28.226, Alexa350-IpaBC28.226, and Alexa350-IpaBN1.226 and either 0 or 1 μM IpgC and were then collected. For the reported experiments, acceptor labeling efficiency approached 100%.
The distance between the Trp donors and the Alexa350 acceptors was calculated based on determined FRET efficiency values, which were measured spectrophotometrically and calculated by the quenching of donor fluorescence emission. The donor only spectra in each case were obtained using the IpaB fragment lacking an Alexa350 acceptor. The FRET efficiencies were calculated by:
| (23) | Eq.1 |
where E is the energy transfer efficiency, FDA is the peak fluorescence intensity of the donor emission in the presence of the acceptor and FD is the peak fluorescence intensity of the donor emission in the absence of the acceptor fluorophore. From the energy transfer efficiencies, the distances single donor-acceptor pairs can be calculated by:
| (23) | Eq.2 |
where r is the distance between the FRET pair fluorophores, E is the measured energy transfer efficiency calculated from Eq.1, and R0 is the Förster distance specific for the FRET pair. The Ro value for Trp as the donor and Alexa350 as the acceptor is reported to be 21 Å (24). Though possible donor-acceptor distances for the IpaB N-terminal domain are discussed here, reliable determinations were only possible when the fluorescent IpaB species were not dimerized since the presence of two D and two A probes within the same protein complex compromises calculating a distance with a high level of confidence.
Use of isothermal titration calorimetry to monitor the association of the IpaB fragments with IpgC
ITC experiments were carried out using a VP-ITC instrument (MicroCal) at 25°C. To study the interaction between IpgC and IpaB1.226, 30 injections of 6 μl of IpaB1.226 (600 μM) were titrated into 1.456 ml of IpgC (30 μM). Experiments studying the interaction between IpgC and IpaB28.226 were performed in a similar manner, except that 17 injections of 6 μl of IpaB28.226 (1.569 mM) were titrated into 1.456 ml of IpgC (48 μM). Each set of experiments were performed in triplicate and the final values are derived from the average of all three runs per experiment. A single-site binding model was used to fit the corrected binding isotherm and derive the thermodynamic binding parameters using the ORIGIN ITC software (OriginLab).
Cross-linking of different IpaB fragments and IpgC
Co-purified IpaB1.94/IpgC and IpaB1.226/IpgC complexes (10 μM in PBS pH 7.4) were subjected to cross-linking using a final concentration of 168 μM dithiobis[succinimidyl propionate] (DSP), which is a thiol-containing homobifunctional cross-linker (16). After 30 min at room temperature, the reaction was quenched with SDS-PAGE sample buffer without reducing agents. The reactions were split into two aliquots with one aliquot receiving DTT to a final concentration of 1 mM to promote cleavage of the cross-linker. The samples were boiled, the proteins separated on a 17% SDS-PAGE gel and then stained with Oriole UV-fluorescent gel stain (BioRad). The interaction between individually purified IpaB28.226 or IpaB1.226 and IpgC at a molar ratio of 1:2 was examined in a similar manner except the proteins were allowed to interact for 1 hr prior to cross-linking for 30 min. These reconstituted complexes were analyzed on 15% SDS-PAGE gels.
Results
Identification, structure and analysis of stable IpaB fragments
We previously identified stable fragments of IpaB based on limited proteolysis of the IpaB/IpgC complex (Supplemental Figure S1) (18). One IpaB fragment containing residues 28 to 226 (IpaB28.226) was found to be highly soluble and stable, even in the absence of chaperone. This fragment was used to provide the first high-resolution structure for a significant portion of IpaB (residues 74–224 were resolved in this crystal structure, see Figure 1) (18). IpaB74.226 formed a highly stable, extended (>100 Å) coiled-coil that shares exceptional structural homology with the coiled-coil region of colicins E3 and Ia (18, 25, 26). Within the context of full-length IpaB, the coiled-coil is immediately followed by a hydrophobic region (a functional schematic for IpaB is shown in Supplemental Figure S2). This presumably allows the IpaB N-terminal domain to present the region needed for membrane penetration outward from the T3SA needle tip (27–29).
Figure 1. The crystal structure of the N-terminal domain of IpaB shows 2.1 Å resolution for residues 74–224.

The native Trp residue within this domain is indicated and Cys residues placed at the N or C terminus of IpaB1.226 and IpaB28.226 would not be visible in this structure, indicating that they are in unstructured regions within the otherwise stable N-terminal domain. Panel A shows a surface rendition of the structure (PDB 3U0C) and panel B shows a ribbon depiction for this region of IpaB (18). The position of Trp residue 105 is indicated with an arrow (W105) and the length of the entire structure is indicated.
Because IpaB N-terminal fragments co-purified with IpgC, we elected to examine IpaB1.226, IpaB28.226 and IpaB1.94 for their solution properties and their ability to interact with IpgC in vitro and in vivo. IpaB1.226 was stably expressed and purified both independently and in complex with IpgC with little to no degradation occurring during the timeline of this study. It is noteworthy that IpaB28.226 contains the previously reported IpgC binding site (12, 30), but the co-purified complex does not remain stably associated during further chromatographic purification steps (data not shown). Thus, no data pertaining to co-purified IpaB28.226 and IpgC could be obtained for this study. In contrast, IpaB1.94 could be purified stably as a complex with IpgC, but was quickly degraded when expressed in the absence of IpgC. Because of this, only data pertaining to the IpaB1.94/IpgC complex is presented here. Despite these limitations, all three IpaB N-terminal fragments, alone or in complex with IpgC, provided a platform for examining the influence of IpgC binding on the structural features of the IpaB N-terminal domain and, conversely, the influence of this region on the oligomeric state of IpgC.
Secondary structure analysis reveals stable, highly 3-helical polypeptides
Far-UV circular dichroism (CD) spectroscopy was first used to determine the secondary structure content and stability of the IpaB N-terminal fragments examined in this study. Previous crystallographic analysis of IpgC and IpaB74.224 has demonstrated that both proteins are rich in α-helical structure (12, 13, 18). Likewise, the CD spectra for IpaB1.94, IpaB1.226 and IpaB28.226 alone or in complex with IpgC indicate that these proteins contain a significant proportion of α-helical structure (interference below 200 nm prevented determining secondary structure ratios) (Supplemental Figure S3A).
To further analyze the solution properties of the IpaB N-terminal fragments, the thermal unfolding profiles for these polypeptides alone and in complex with IpgC were determined by monitoring the increase in signal for the dominant α-helix minimum at 222 nm (Table 1 and Supplemental Figure S3B). IpgC displayed a midpoint of thermal unfolding near 45°C while IpaB1.226 and IpaB28.226 unfolded at slightly higher temperatures. The IpaB1.226/IpgC complex displayed two thermal transitions at 45°C and 60°C, perhaps indicating complex separation at a lower temperature followed by individual unfolding of IpgC and IpaB1.226 at respective higher melting temperatures. Interestingly, the IpaB1.94/IpgC complex had a thermal unfolding transition at 62.5°C, suggesting that strong interactions occur between the extreme N-terminus of IpaB and the IpgC chaperone. These data indicate that association of the IpaB N-terminal fragments with the chaperone IpgC influences the solution properties of both proteins.
Table 1.
Thermal unfolding temperatures for different IpaB N-terminal domain proteins with and without IpgC.
| Protein | Tm (°C)a |
|---|---|
| IpgC | 45.0 |
| IpaB28.226 | 57.5 |
| IpaB1.226 | 57.5 |
| IpaB1.226/IpgC | 45.0/60.0b |
| IpaB1.94/IpgC | 62.5 |
The CD signal at 222 nm was monitored for each protein as a function of temperature from 10–85°C. Spectra were collected every 2.5°C with 5 min allowed for temperature equilibration.
The midpoint of thermal unfolding (given as Tm) was determined as an indicator of transition from a folded to unfolded state. The standard error in each case is less than 2°C.
This protein pair appeared to have two unfolding transitions, which in this case could represent a separation of the two proteins followed by observation of their individual unfolding transitions.
IpaB1.226 and IpaB28.226 interact with IpgC in vitro
Following co-expression in E. coli, full-length IpaB (580 residues) co-purifies with IpgC and this is the only reliable method for purifying this hydrophobic translocator protein (16). Once IpaB is separated from IpgC by the addition of detergent, it forms stable multimers, which do not allow the reformation of IpaB-IpgC complex in vitro (data not shown). Therefore, we chose to determine whether the soluble IpaB N-terminal domain could be prepared alone and then used to form a complex with IpgC in vitro. Fluorescence polarization (FP) was used to monitor protein-protein interactions between unlabeled IpgC and IpaB N-terminal fragments labeled with fluorescein maleimide (FM) on a Cys residue placed at the N or C terminus of the IpaB fragment (FM-IpaBN1.226, FM-IpaBC1.226, FM-IpaBN28.226 and FM-IpaBC28.226). Upon adding IpgC to the labeled IpaB N-terminal fragment, a sharp rise in polarization was seen (Figure 2). Based on the observed binding curves, apparent Kd values were estimated to be in the sub-micromolar range (Table 2), consistent with a strong interaction between these binding partners. These Kd values are based on a single-site saturation ligand binding equation, which fits well to the obtained FP data with a correlation coefficient of ≥ 0.94 in all cases. Comparison of the estimated Kd values suggests that the interaction between IpaB1.226 and IpgC is stronger than that observed between IpaB28.226 and IpgC, regardless of whether the FM is located at the N or C terminus. These data indicate that the extreme N-terminus of IpaB has an important role in this translocator’s association with IpgC and that the extreme N-terminus, though not physically present in the published crystal structure of the IpaB N-terminal domain, is required for maximal binding affinity.
Figure 2. Fluorescence polarization (FP) measurements for the binding of IpaB1.226 and IpaB28.226.

Panel A) N-terminus FM-labeled IpaBN28.226 (open circles, R2=0.99) and IpaBN1.226 (closed circles, R2=0.94) or Panel B) C-terminus FM-labeled IpaBC28.226 (open circles, R2= 0.99) and IpaBC1.226 (closed circles, R2=0.99) were held at a constant concentration as increasing concentrations of IpgC were added. A single-site ligand binding saturation equation was fit to these data. Data shown are representative of three independent experiments with at least 3 technical replicates per experiment. Error bars represent standard deviation.
Table 2.
Calculated dissociation constants for N-terminal IpaB fragments with IpgC based on fluorescence polarization.
| Protein | Kd (nM)a |
|---|---|
| IpaBN28.226 | 410.9 ± 14.2 |
| IpaBC28.226 | 500.7 ± 77.1 |
| IpaBN1.226 | 199.4 ± 62.6 |
| IpaBC1.226 | 59.5 ± 5.6 |
Dissociation constants were determined for the different protein interactions by fitting the data with a single-site saturation ligand binding algorithm using SigmaPlot.
Isothermal titration calorimetry (ITC) demonstrates both endothermic, entropically-driven interaction between translocator and chaperone
While the Kd can be calculated with isothermal titration calorimetry (ITC) or FP data, ITC has the added advantage of being fluorophore-independent as well as deriving several other relevant binding parameters (stoichiometry, ΔH and ΔS). Therefore, we used ITC to examine the interaction between translocator fragments and the cognate chaperone (Table 3). The Kd for the interaction between IpaB1.226 and IpgC as determined by ITC was 208 nM ± 51 nM, which agrees well with the Kd values obtained from the FP data (Tables 2 and 3). Likewise, reduced affinity (nearly 14-fold) was seen based on the Kd value for IpaB28.226 binding to IpgC (Kd = 2.96 ± 0.5 μM) relative to the binding of IpaB1.226. This is largely in agreement with the FP data presented above (Figures 2 and 3, Tables 2 and 3). The positive ΔS and ΔH values associated with both interactions (Table 3) indicates that they are endothermic and entropically-driven in nature. Significantly, both of the IpaB polypeptides associate with IpgC at approximately an equimolar ratio (Table 3). These and the FP data not only indicate that the IpaB N-terminal domain can reversibly associate with IpgC, but that the interaction occurs at an equimolar ratio and is dependent upon the IpaB N-terminus for maximal affinity.
Table 3.
Thermodynamic parameters for the binding of different IpaB N-terminal domain fragments with IpgC.
| Parameter | IpaB1.226 | IpaB28.226 |
|---|---|---|
| Na | 1.03 ± 0.01 | 0.88 ± 0.02 |
| ΔH (kcal/mol) | 8.68 ± 0.14 | 3.55 ± 0.11 |
| ΔS (J/K) | 59.7 | 37.2 |
| Kdb (μM) | 0.208 ± 0.05 | 2.96 ± 0.53 |
Thermodynamic parameters of the indicated protein interactions as measured by isothermal titration calorimetry. Data were fit to a single-site model to determine the binding properties.
N indicates the approximate stoichiometry for IpaB N-terminal domain with IpgC.
Kd is the determined dissociation constant.
Figure 3. Isothermal titration calorimetry (ITC) measurements for IpgC and IpaB fragments.

Titrations of IpgC with IpaB1.226 (left panel) and IpaB28.226 (right panel) as monitored by ITC. The upper panel of each section depicts the thermograms obtained during the course of each injection series. The corresponding integrated enthalpy changes (after background correction) are shown in each lower panel. A solid line is depicted in the lower panel that fits the data according to a single-site binding model. Experimentally derived thermodynamic values are listed in Table 3.
Cross-linking analyses show that the IpaB N-terminal domain associates with IpgC to form a heterodimer
Previously reported data have indicated that IpgC exists as a homodimer in solution (13). There are, however, conflicting data as to whether IpgC is a monomer (16) or dimer (12, 17) upon association with either translocator (IpaB or IpaC). Because the ITC data presented above suggest an equimolar incorporation of IpaB and IpgC into this complex, we chose to determine the final content of the IpaB N-terminal domain-IpgC complex using chemical cross-linking analysis. Similar analyses originally indicated that full-length IpaB co-expressed with IpgC bound as a 1:1 heterodimer (16). The co-purified IpaB1.226/IpgC cross-linked product migrated with an apparent molecular mass near 40 kDa, which agrees with the expected molecular mass for an IpaB1.226/IpgC heterodimer (42.9 kDa) (Figure 4A). Similarly, the cross-linked, co-purified IpaB1.94/IpgC resulted in a complex that migrated at a molecular mass of 25 kDa, which is also consistent with heterodimer formation. The apparent molecular mass of these cross-linked complexes are consistent with the 1:1 stoichiometry observed using ITC and our previous report of a 1:1 stoichiometry for co-purified heterodimers of these IpgC:translocator complexes (whether IpaB or IpaC) (16). When recombinant IpgC was cross-linked and evaluated, a distinct band at 35 kDa was observed. This confirmed that IpgC alone does form a homodimer in solution (Fig. 4B), which is consistent with our previous reports as well as others (12, 13, 16).
Figure 4. Chemical cross-linking analysis of the IpaB N-terminal domain and IpgC.
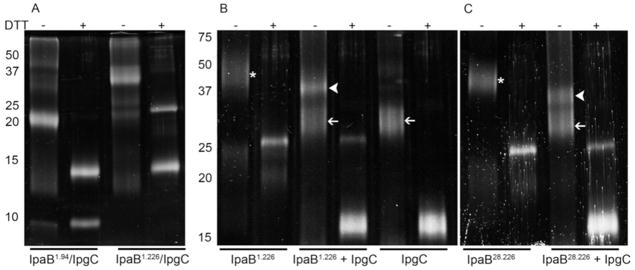
Aliquots of protein (10μM) were exposed to 168 uM of the cross-linking agent DSP at room temperature for 30 min. Cross-linking was quenched by the addition of a third volume of 3x SDS-PAGE sample buffer. The presence or absence of DTT in the sample buffer is indicated at the top of the gels. In panel A, IpaB1.94 and IpaB1.226 were co-expressed and co-purified with IpgC followed by cross-linking as described above. In panel B, IpgC, and IpaB1.226 were expressed and purified independently. Proteins were cross-linked at IpaB:IpgC molar ratios of 0:1, 1:2, and 1:0. In panel C, IpaB28.226 was tested at 0 or a two-fold molar excess of IpgC. White arrowheads show the position of IpaB/IpgC homodimer, thin white arrows show the position of IpgC homodimers and the asterisk (*) indicates the position of IpaB N-terminal domain homodimers. Addition of DTT cleaves the DSP cross-linker, resulting in the formation of modified monomeric proteins.
To complement the complexes that were formed in vivo during co-expression, we examined those that were formed by reconstituting the fragments in vitro, which cannot be accomplished with the multimerized full-length IpaB. Because IpaB1.94 could not be prepared in the absence of IpgC, we could only examine in vitro binding between IpgC and purified IpaB1.226 or IpaB28.226. In accordance with a previously reported 1:2 ternary complex (one IpaB peptide with two IpgC) (17), each of our IpaB N-terminal fragments were mixed with IpgC at a molar ratio of 1:2 (IpaB fragment:IpgC); however, this led to the formation of cross-linked complexes that migrated at two distinct molecular masses (Figure 4B and C). The smaller of these bands in each case corresponded to an IpgC homodimer (35 kDa), while the other to the expected mass of an IpaB fragment-IpgC heterodimer, 40 kDa for IpaB1.226-IpgC and 38 kDa for IpaB28.226-IpgC (Figure 4B and C). Interestingly, both IpaB N-terminal fragments appeared to form cross-linkable dimers in the absence of IpgC (Figure 4B and C). In all cases, the higher order complexes could be separated into their component proteins by adding DTT (Figure 4). It thus appears that when the IpaB N-terminal domain dimers are mixed with the IpgC dimers, they dissociate in favor of forming an IpaB fragment-IpgC heterodimer.
It is worth noting that the heterodimers formed between the IpaB fragments and IpgC are actually smaller than the respective IpaB fragment homodimers, yet FP measurements using probes at the IpaB N and C-termini gave rise to an increase in polarization. This is consistent with two different possibilities. First, these regions may be held more rigidly in the IpaB fragment-IpgC complex than in the IpaB N-terminal domain homodimer. Second, during the FP experiments this restriction overcomes the decrease in overall molecular volume caused by transition from an IpaB domain homodimer to an IpaB-IpgC heterodimer. Because the N- and C-termini could not be modeled in the crystal structure of IpaB28.226, they appear to be unstructured regions that perhaps gain some measure of structure upon chaperone binding. This phenomenon may very well account for the inherent instability of IpaB1.94 in the absence of IpgC. In contrast, the Trp residue at position 105 of IpaB (W105) is contained within the core structure of IpaB28.226 (see Figure 1). Thus, we performed FP experiments based on intrinsic Trp fluorescence. In this case, polarization decreased upon addition of IpgC with nearly maximal polarization decrease occurring when the two proteins are at an approximately equimolar concentration (Supplemental Figure S4). Taken together, these data are consistent with the IpaB N-terminal domain undergoing a significant conformational change upon chaperone binding.
Forster resonance energy transfer allows examination of relative probe positions within the IpaB N-terminal domain
To explore the possibility of a conformational change in IpaB1.226 or IpaB28.226 after IpgC binding, we used Förster resonance energy transfer (FRET) between the native W105 within the stable core of the IpaB fragment and an extrinsic Alexa350 acceptor linked at the less well-characterized termini of these proteins (IpaBN28.226 or IpaBN1.226 and IpaBC28.226, respectively). Unfortunately, the FRET-based distance determination for the IpaB N-terminal domain alone is complicated by the fact that IpaB28.226 and IpaB1.226 form homodimers in the absence of IpgC; this prevents a confident calculation of the distance between donor and acceptor in the absence of IpgC. However, the distance from W105 to A224 is approximately 18 Å in the crystal structure (see Figure 1, PDB code 3U0C), which is largely in agreement with the calculated distance based on FRET in the absence of chaperone (17 Å) for IpaBC28.226. In contrast, because we have demonstrated that this IpaB domain forms a heterodimer with IpgC (which lacks Trp residues), we were able to calculate distances from W105 to each of the IpaB domain termini when the chaperone was present (Table 4 and see Figure 5).
Table 4.
FRET-based determination of intermolecular distances for IpaB-IpgC complexes.
| Protein | No IpgC
|
1 μM IpgC
|
||
|---|---|---|---|---|
| FRETa efficiency (%) | Distance (Å)b | FRET efficiency (%) | Distance (Å)c | |
| IpaBN28.226 | 47.1 ± 3.2 | 21.4 | 37.9 ± 14.8 | 22.8 |
| IpaBC28.226 | 74.5 ± 5.9 | 17.6 | 46.1 ± 6.8 | 21.6 |
| IpaBN1.226 | 37.6 ± 3.3 | 22.8 | 6.5 ± 3.6 | 32.8 |
FRET was measured using the intrinsic probe W105 as the donor for IpaB28.226 or IpaB1.226 to either an N- or C-terminal Alexa350 acceptor probe as described in methods. FRET efficiencies and distances were calculated using equations 1 and 2, respectively.
This distance is measured with the realization that it may not precisely represent the distance separating the two probes because of homodimer formation.
This calculation refers to the condition in which IpaB/IpgC heterodimers are formed and should represent an accurate distance between the two probes.
Figure 5. Use of FRET to monitor intermolecular distances during the interaction of the IpaB N-terminal domain with IpgC.

Alexa350-IpaBN1.226 (D/A) or IpaBN1.226 (D) was excited at 295 nm and fluorescence spectra were collected from 300–400 nm in the presence and absence of 1μM IpgC. Open circles, IpaBN1.226 donor only without IpgC; open triangles, IpaBN1.226 donor only with IpgC; closed circles, IpaBN1.226 donor plus acceptor without IpgC; and closed triangles, IpaBN1.226 donor with acceptor and IpgC present. The data shown are representative of the data used to obtain the FRET efficiency values given in Table 4. The fluorescence spectra shown here are normalized relative to IpaBN1.226 donor only in the presence of IpgC.
The distance from W105 to Alexa350 in IpaBC28.226 was calculated to be 21.6 Å in the presence of an equimolar concentration of IpgC. This is an increase from the 18 Å predicted in the crystal structure, perhaps indicating that a modest conformational rearrangement has occurred upon association with IpgC. Since the crystal structure of the IpaB N-terminal domain does not include residues 1–73, the distances between W105 and Alexa350 at the N-termini of IpaB1.226 and IpaB28.226 could not be determined based on the static structure. Nevertheless, we were able to use FRET to calculate the distances from W105 to Alexa350 on IpaBN28.226 or IpaBN1.226 after the addition of IpgC. For the IpaBN1.226-IpgC heterodimer, the distance from W105 to the N-terminal Alexa350 acceptor was calculated at 32.8 Å. This represents a significant decrease in FRET efficiency relative to IpaBN1.226 alone (from 38% to about 7%). Meanwhile, the equivalent distance for IpaBN28.226 with IpgC was 22.8 Å, which was calculated from a FRET efficiency of 38% and which did not differ substantially from the FRET efficiency seen in the absence of IpgC (Table 4). It is important to state that these data cannot be used to specifically demonstrate a change in the IpaB N-terminal domain structure upon chaperone binding. However, they are nevertheless consistent with the portion of the IpaB N-terminal domain that is not visible in the published crystal structure assuming an elongated conformation upon association with IpgC.
Discussion
We previously used controlled digestion to identify a soluble, protease-resistant core of IpaB comprised of residues 28–226 (IpaB28.226). This region was used to determine the recently reported crystal structure of the IpaB N-terminal domain comprised of residues 74 to 224 to a resolution of 2.1 Å (18). The refined structure consisted of an extended coiled-coil possessing high structural homology to the extended portion of the pore-forming toxin colicin Ia (18, 25, 26). Purified IpaB has been shown to penetrate liposomes to cause the release of small molecules trapped inside, while the IpaB-IpgC chaperone complex is unable to do so (16). When co-expressed in E. coli, the N-terminal IpaB fragments IpaB1.226 and IpaB1.94 co-purify with IpgC, indicating that they contain important sequences that contribute to chaperone binding. Of these three fragments, it is interesting that IpaB28.226 associates with IpgC when the two are co-expressed, but the two separate during the extensive purification steps used here. Unlike full-length IpaB, however, IpaB1.226 and IpaB28.226 can be purified in the absence of IpgC and remain soluble in the absence of detergents. In this study we demonstrate three points: 1) the N-terminal IpaB fragments strongly interact with IpgC in vitro in a manner that supports a two chaperone binding site model; 2) the IpaB fragment:IpgC complex is a heterodimer whose formation is more favorable than maintenance of IpaB N-terminal domain and IpgC homodimers; and 3) upon association of IpgC, the N-terminal IpaB fragment probably undergoes a conformational change into what may be a more elongated structure.
ITC and FP data show that the binding affinity of IpgC for IpaB1.226 is stronger than that for IpgC binding to IpaB28.226 alone (Figures 2 and 3, Tables 2 and 3). It has been suggested that there are two chaperone binding domains (CBD) in IpaB with CBD 1 being close to the N terminus at residues 16–35 and CBD 2 located just downstream between residues 51–70 (12, 17). Thus, when the IpaB N-terminal fragment lacks CBD 1 as is the case for IpaB28.226, the binding affinity for IpgC is reduced relative to that seen when CBD 1 and 2 are present as in IpaB1.226 (Figures 3 and 4 and Table 3). These data firmly support a two-site model for chaperone binding by IpaB. Structural information remains lacking for this region of IpaB, which is consistent with the N-terminal fragment IpaB1.94 being rapidly degraded in the absence of IpgC. In contrast, IpaB1.94 is quite stable as part of the complex with IpgC, but is unstable and thus may be largely unstructured in the absence of chaperone. When all the data are considered, it would seem that residues within this region have a significant role in directing IpaB binding to IpgC. This could explain why the IpaB28.226/IpgC complex disassociates during the purification process while the IpaB1.226/IpgC complex remains intact.
Cross-linking experiments showed that IpgC and the IpaB N-terminal fragments form homodimers in solution and that incubating these two sets of homodimers with each other in vitro leads to the formation of IpaB fragment:IpgC heterodimers. It thus appears that the IpaB N-terminal domain and IpgC homodimers dissociate in favor of forming IpaB-IpgC heterodimers in solution. These results are consistent with the ITC data demonstrating a 1:1 stoichiometry. Furthermore, size exclusion chromatography elution profiles of IpaB1.226 and co-purifed IpaB1.226/IpgC demonstrate distinct molecular sizes which are consistent with stable homodimer and heterodimer formation, respectively (data not shown). While others have suggested that the association of IpaB:IpgC is 1:2 (12, 17), the current results are supported and complemented by our previous co-expression and co-purification of IpgC:IpaB or IpgC:IpaC complexes (16). Using the same cross-linking methodology as well as analytical ultracentrifugation, we demonstrated that IpgC complexes with full-length IpaB and IpaC exist as 1:1 heterodimers (16).
The FRET experiments presented here suggest that the extreme N terminus of IpaB adopts an extended conformation when it binds to IpgC. While such data are only suggestive that an actual conformational change occurs in the IpaB N-terminal domain upon association with IpgC, they are consistent with the unstructured region of the N terminus becoming elongated as a result of chaperone binding. When considered in the overall context of the findings presented here, however, there is good evidence that the N terminus of IpaB is unstructured and association with IpgC has a major impact on the conformation that it finally assumes. This is not unlike what has been shown for other T3SS effectors such as SptP (31). For example, the FP data indicate that N- and C-terminal probes become restrained in motion upon IpgC binding (based on an increase in mP value), despite the fact that conversion from an IpaB fragment homodimer to an IpaB-IpgC heterodimer reduces the overall size of the complex. This suggests that the termini become more rigid upon chaperone binding. In contrast, W105 displays reduced polarization when IpaB associates with IpgC, which could either be a reflection of the reduced size of the heterodimer or an increase in local flexibility upon association with IpgC.
The ITC data obtained here provide important insight into the thermodynamics of formation translocator/chaperone heterodimers from two individual homodimers. In addition to measuring the binding affinity (Kd) between chaperone and translocator, both enthalpy (ΔH) and entropy (ΔS) data suggest the forces that drive complex formation and the events involved in this process (32). For example, the thermodynamic parameters reported here reveal that both IpaB1.226 and IpaB28.226 bind to IpgC in an endothermic, entropy-driven manner (Table 3). This entropically favorable reaction directly correlates to the change in higher order structure (here shown as the dissociation of two homodimers to allow the formation of two separate heterodimers). While the change in enthalpy for the IpaB28.226-IpgC interaction (ΔH = 3.55 ± 0.11 kcal/mol) was lower than the measured value for the IpaB1.226-IpgC interaction (ΔH = 8.68 ± 0.14 kcal/mol), both were found to be endothermic and are likely to reflect disruption of the IpaB and IpgC homodimer contacts and displacement of ordered solvent from the proteins’ surface. This is further supported by the significant increases in entropy for both interactions (ΔS = 59.7 J/K and 37.2 J/K for IpaB1.226 and IpaB28.226, respectively). It is not surprising to find that the formation of the heterodimeric IpaB fragment/IpgC complex is thermodynamically spontaneous in vitro as the ATPase located at the T3SA cytoplasmic face is likely required for disruption of this stable complex in vivo (33).
There remains much to be learned about the roles of translocator proteins in T3SS function. The Shigella T3SA continues to be an important model system in understanding type III secretion and, in particular, for dissecting the steps that bridge T3SA assembly and T3SS induction. Prior to mobilization of IpaB to the needle tip complex, this protein is stored in the bacterial cytoplasm where it associates as a heterodimer with its cognate chaperone IpgC. This is a stable complex, which appears to require the active participation of an ATPase at the cytoplasmic face of the T3SA for separation. Once separated, IpaB adopts a conformation at the T3SA needle tip that is compatible with its new role as sensor of host cell (membrane) contact and, later, as an essential translocon pore component. This requires that new sets of protein-protein interactions form. On the other hand, IpgC remains in the bacterial cytoplasm where it forms a homodimer that may be shunted off to assume new roles in type III secretion. It is possible that the IpgC homodimer is now able to bind to the transcriptional regulator MxiE to promote the expression of late effector genes (34). Further exploration of the temporal interactions that occur for IpaB and IpgC is clearly warranted for better understanding the early interactions that ultimately allow Shigella invasion of human intestinal epithelial cells. This work and additional structural analysis of IpaB and IpgC is just one step in developing a complete understanding of the induction of type III secretion.
Supplementary Material
Acknowledgments
We gratefully acknowledge technical assistance from Kirk Pendleton and Daniel R. Picking.
Abbreviations and footnotes
- T3SS
type-III secretion system
- T3SA
type III secretion apparatus
- Ipg
Invasion plasmid gene
- Ipa
Invasion plasmid antigen
- Mxi
major exporter of Ipas
- CD
circular dichroism
- FM
fluorescein maleimide
- FP
fluorescence polarization
- mP
millipolarization units
- Alexa350
Alexa-Fluor 350 maleimide
- FRET
Förster resonance energy transfer
- ITC
isothermal titration calorimetry
- TCEP
(tris (2-carboxyethyl)phosphine)
- DSP
dithiobis[succinimidyl proprionate]
- TCEP
tris (2-carboxyethyl)phosphine
- DMSO
dimethylsulfoxide
- DMF
N′,N′-dimethylformamide
- TPR
tetratricopeptide repeat
- Tm
thermal unfolding midpoint
- IpaBN/C28.226
IpaB28.226 N or C-terminal Cys, respectively
- IpaBN/C1.226
IpaB1.226 N or C-terminal Cys, respectively
Footnotes
This work was supported by funding to WLP (NIH R01 AI067858) and WDP (Oklahoma Health Research Program Funding HR10-128S) and WDP/BVG (NIH R21 AI090149). MKP was supported by the Oklahoma State University Niblack Research Scholars Program.
The proteolysis data used to derive the protein fragments used in this paper (primarily IpaB28.226) and which were used for prior crystallographic analysis are summarized in Figure S1. A schematic of the functional organization of IpaB is provided in Figure S2. Full CD spectra and thermal folding profiles are included for each protein used in this study (Figure S3). Fluorescence polarization data acquired using the native Trp in the IpaB N-terminal domain is presented in Figure S4. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mueller C, Broz P, Cornelis G. The type III secretion system tip complex and translocon. Mol Microbiol. 2008;68:1085–1095. doi: 10.1111/j.1365-2958.2008.06237.x. [DOI] [PubMed] [Google Scholar]
- 2.Cornelis GR. The type III secretion injectisome, a complex nanomachine for intracellular ‘toxin’ delivery. Biol Chem. 2010;391:745–751. doi: 10.1515/BC.2010.079. [DOI] [PubMed] [Google Scholar]
- 3.Schroeder GN, Hilbi H. Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin Microbiol Rev. 2008;21:134–156. doi: 10.1128/CMR.00032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Espina M, Olive AJ, Kenjale R, Moore DS, Ausar SF, Kaminski RW, Oaks EV, Middaugh CR, Picking WD, Picking WL. IpaD Localizes to the Tip of the Type III Secretion System Needle of Shigella flexneri. Infect Immun. 2006;74:4391–4400. doi: 10.1128/IAI.00440-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darboe N, Kenjale R, Picking WL, Picking WD, Middaugh CR. Physical characterization of MxiH and PrgI, the needle component of the type III secretion apparatus from Shigella and Salmonella. Protein Sci. 2006;15:543–552. doi: 10.1110/ps.051733506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blocker A, Jouihri N, Larquet E, Gounon P, Ebel F, Parsot C, Sansonetti P, Allaoui A. Structure and composition of the Shigella flexneri “needle complex”, a part of its type III secreton. Mol Microbiol. 2001;39:652–663. doi: 10.1046/j.1365-2958.2001.02200.x. [DOI] [PubMed] [Google Scholar]
- 7.Veenendaal AK, Hodgkinson JL, Schwarzer L, Stabat D, Zenk SF, Blocker AJ. The type III secretion system needle tip complex mediates host cell sensing and translocon insertion. Mol Microbiol. 2007;63:1719–1730. doi: 10.1111/j.1365-2958.2007.05620.x. [DOI] [PubMed] [Google Scholar]
- 8.Epler CR, Dickenson NE, Bullitt E, Picking WL. Ultrastructural Analysis of IpaD at the Tip of the Nascent MxiH Type III Secretion Apparatus of Shigella flexneri. Journal of Molecular Biology. doi: 10.1016/j.jmb.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olive AJ, Kenjale R, Espina M, Moore DS, Picking WL, Picking WD. Bile salts stimulate recruitment of IpaB to the Shigella flexneri surface, where it colocalizes with IpaD at the tip of the type III secretion needle. Infect Immun. 2007;75:2626–2629. doi: 10.1128/IAI.01599-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Epler CR, Dickenson NE, Olive AJ, Picking WL, Picking WD. Liposomes recruit IpaC to the Shigella flexneri type III secretion apparatus needle as a final step in secretion induction. Infect Immun. 2009;77:2754–2761. doi: 10.1128/IAI.00190-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parsot C, Hamiaux C, Page AL. The various and varying roles of specific chaperones in type III secretion systems. Curr Opin Microbiol. 2003;6:7–14. doi: 10.1016/s1369-5274(02)00002-4. [DOI] [PubMed] [Google Scholar]
- 12.Lunelli M, Lokareddy RK, Zychlinsky A, Kolbe M. IpaB-IpgC interaction defines binding motif for type III secretion translocator. Proc Natl Acad Sci U S A. 2009;106:9661–9666. doi: 10.1073/pnas.0812900106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barta ML, Zhang L, Picking WL, Geisbrecht BV. Evidence for alternative quaternary structure in a bacterial Type III secretion system chaperone. BMC Struct Biol. 2010;10:21. doi: 10.1186/1472-6807-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Büttner C, Sorg I, Cornelis G, Heinz D, Niemann H. Structure of the Yersinia enterocolitica type III secretion translocator chaperone SycD. J Mol Biol. 2008;375:997–1012. doi: 10.1016/j.jmb.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 15.Job V, Mattei P, Lemaire D, Attree I, Dessen A. Structural basis of chaperone recognition of type III secretion system minor translocator proteins. J Biol Chem. 2010 doi: 10.1074/jbc.M110.111278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Birket SE, Harrington AT, Espina M, Smith ND, Terry CM, Darboe N, Markham AP, Middaugh CR, Picking WL, Picking WD. Preparation and characterization of translocator/chaperone complexes and their component proteins from Shigella flexneri. Biochemistry. 2007;46:8128–8137. doi: 10.1021/bi700099c. [DOI] [PubMed] [Google Scholar]
- 17.Lokareddy RK, Lunelli M, Eilers B, Wolter V, Kolbe M. Combination of two separate binding domains defines stoichiometry between type III secretion system chaperone IpgC and translocator protein IpaB. J Biol Chem. 2010;285:39965–39975. doi: 10.1074/jbc.M110.135616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barta ML, Dickenson NE, Patil M, Keightley A, Wyckoff GJ, Picking WD, Picking WL, Geisbrecht BV. The Structures of Coiled-Coil Domains from Type Three Secretion System Translocators Reveal Homology to Pore-Forming Toxins. Journal of Molecular Biology. 2012 doi: 10.1016/j.jmb.2012.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geisbrecht B, Bouyain S, Pop M. An optimized system for expression and purification of secreted bacterial proteins. Protein Expr Purif. 2006;46:23–32. doi: 10.1016/j.pep.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Garcia BL, Summers BJ, Lin Z, Ramyar KX, Ricklin D, Kamath DV, Fu ZQ, Lambris JD, Geisbrecht BV. Diversity in the C3B contact residues and tertiary structures of the staphylococcal complement inhibitor (SCIN) protein family. J Biol Chem. 2011 doi: 10.1074/jbc.M111.298984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geisbrecht BV, Bouyain S, Pop M. An optimized system for expression and purification of secreted bacterial proteins. Protein Expr Purif. 2006;46:23–32. doi: 10.1016/j.pep.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 22.Martinez-Becerra FJ, Kissmann JM, Diaz-McNair J, Choudhari SP, Quick AM, Mellado-Sanchez G, Clements JD, Pasetti MF, Picking WL. A broadly protective Shigella vaccine based on Type III secretion apparatus proteins. Infect Immun. 2011 doi: 10.1128/IAI.06174-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lakowicz JR. Principles of Fluorescence Spectroscopy. Plenum Press; New York: 1983. [Google Scholar]
- 24.Oyston PC, Mellado-Sanchez G, Pasetti MF, Nataro JP, Titball RW, Atkins HS. A Yersinia pestis guaBA mutant is attenuated in virulence and provides protection against plague in a mouse model of infection. Microb Pathog. doi: 10.1016/j.micpath.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiener M, Freymann D, Ghosh P, Stroud RM. Crystal structure of colicin Ia. Nature. 1997;385:461–464. doi: 10.1038/385461a0. [DOI] [PubMed] [Google Scholar]
- 26.Soelaiman S, Jakes K, Wu N, Li C, Shoham M. Crystal structure of colicin E3: implications for cell entry and ribosome inactivation. Mol Cell. 2001;8:1053–1062. doi: 10.1016/s1097-2765(01)00396-3. [DOI] [PubMed] [Google Scholar]
- 27.McGhie EJ, Hume PJ, Hayward RD, Torres J, Koronakis V. Topology of the Salmonella invasion protein SipB in a model bilayer. Mol Microbiol. 2002;44:1309–1321. doi: 10.1046/j.1365-2958.2002.02958.x. [DOI] [PubMed] [Google Scholar]
- 28.Hume PJ, McGhie EJ, Hayward RD, Koronakis V. The purified Shigella IpaB and Salmonella SipB translocators share biochemical properties and membrane topology. Mol Microbiol. 2003;49:425–439. doi: 10.1046/j.1365-2958.2003.03559.x. [DOI] [PubMed] [Google Scholar]
- 29.Baudry B, Kaczorek M, Sansonetti PJ. Nucleotide sequence of the invasion plasmid antigen B and C genes (ipaB and ipaC) of Shigella flexneri. Microb Pathog. 1988;4:345–357. doi: 10.1016/0882-4010(88)90062-9. [DOI] [PubMed] [Google Scholar]
- 30.Page A, Fromont-Racine M, Sansonetti P, Legrain P, Parsot C. Characterization of the interaction partners of secreted proteins and chaperones of Shigella flexneri. Mol Microbiol. 2001;42:1133–1145. doi: 10.1046/j.1365-2958.2001.02715.x. [DOI] [PubMed] [Google Scholar]
- 31.Stebbins CE, Galan JE. Maintenance of an unfolded polypeptide by a cognate chaperone in bacterial type III secretion. Nature. 2001;414:77–81. doi: 10.1038/35102073. [DOI] [PubMed] [Google Scholar]
- 32.Ramirez K, Ditamo Y, Rodriguez L, Picking WL, van Roosmalen ML, Leenhouts K, Pasetti MF. Neonatal mucosal immunization with a non-living, non-genetically modified Lactococcus lactis vaccine carrier induces systemic and local Th1-type immunity and protects against lethal bacterial infection. Mucosal immunology. 3:159–171. doi: 10.1038/mi.2009.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akeda Y, Galan JE. Chaperone release and unfolding of substrates in type III secretion. Nature. 2005;437:911–915. doi: 10.1038/nature03992. [DOI] [PubMed] [Google Scholar]
- 34.Mavris M, Page AL, Tournebize R, Demers B, Sansonetti P, Parsot C. Regulation of transcription by the activity of the Shigella flexneri type III secretion apparatus. Mol Microbiol. 2002;43:1543–1553. doi: 10.1046/j.1365-2958.2002.02836.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.