

Abstract
Despite new and effective drug therapies, insulin resistance (IR), type 2 diabetes mellitus (T2D) and its complications remain major medical challenges. It is accepted that IR, often associated with over-nutrition and obesity, results from chronically elevated oxidant stress (OS) and chronic inflammation. Less acknowledged is that a major cause for this inflammation is excessive consumption of advanced glycation end products (AGEs) with the standard western diet. AGEs, which were largely thought as oxidative derivatives resulting from diabetic hyperglycemia, are increasingly seen as a potential risk for islet β-cell injury, peripheral IR and diabetes.
Here we discuss the relationships between exogenous AGEs, chronic inflammation, IR, and T2D. We propose that under chronic exogenous oxidant AGE pressure the depletion of innate defense mechanisms is an important factor, which raises susceptibility to inflammation, IR, T2D and its complications. Finally we review evidence on dietary AGE restriction as a non-pharmacologic intervention, which effectively lowers AGEs, restores innate defenses and improves IR, thus, offering new perspectives on diabetes etiology and therapy.
Keywords: AGE-Receptors, AGER1, SIRT1, Glycation, Oxidation, Diet, Inflammation, Innate Immunity
1. Introduction
As the incidence of type 2 diabetes (T2D) continues to increase [1] and its multifactorial etiology is still debated, new evidence points to lifestyle factors as critical predisposing factors [2, 3]. T2D as well as obesity are characterized by chronic inflammation and insulin resistance (IR), associated with chronically elevated oxidant stress (OS).
The importance of hyperglycemia in the pathogenesis of diabetic complications has been reinforced by clinical trials [4, 5] although more recent studies [6–8] point to additional risk factors. For instance, one way for hyperglycemia to cause cell injury is by fostering advanced glycation end products (AGEs). These are known to contribute to the complications of diabetes by raising intracellular oxidative stress (OS) [9–11]. Compelling epidemiological evidence suggests that elevated AGEs may be a significant risk factor for type 1 diabetes [12], for beta cell injury [13–16] and for peripheral IR [17, 18]. This evidence begs the question of the origin of the large concentrations of AGEs that could induce beta cell toxicity prior to diabetes onset. New studies have introduced an instructive view proposing that the abundance of pro-oxidant AGEs in the highly industrialized modern food environment could potentially account for the initiation and progression of pre-diabetes to diabetes [19].
2. High Systemic AGEs – Not Always From High Glucose
Among naturally occurring reducing sugars, glucose exhibits the slowest glycation rate, unlike fructose, glucose-6-phosphate or glyceraldehyde-3-phosphate, which are intracellularly present at small levels, but can form AGEs at a faster rate, especially under high temperatures, i.e. ex vivo [20]. The chemical transformation of amine-containing molecules by reducing sugars—whether on proteins, lipids or nucleotides [21]—results in AGEs or Maillard products. Since high OS triggers the formation of dicarbonyl derivatives or AGEs it follows that diabetes and conditions of chronic high OS will further accelerate this spontaneous process. Several well studied AGEs, such as carboxymethyllysine (CML), pentosidine, or derivatives of methylglyoxal (MG), i.e. MG-H1, are among the better-characterized AGE compounds, currently serving as AGE markers [19, 22].
It has now become evident that even in the absence of diabetes, AGEs can be introduced into the circulation together with nutrients processed by common methods such as dry heat [23] or during tobacco smoking [24]. Food processing, involving dry heat, ionization or irradiation, whether at the industrial or commercial levels, significantly accelerates the generation of new AGEs [25, 26]. Heat and dehydration are also common in home cooking. Such simple methods, though intended to improve safety, digestibility and transportability of foods, can amplify the formation of AGEs. For the food industry, AGEs in food are highly desirable, due to the profound effect of AGEs on food flavor, and hence on food consumption [27].
Human and animal studies demonstrated that about 10% of AGEs contained in a meal can be absorbed into the circulation, of which two thirds remain in the body for 72 hours [23,28], long enough to promote OS, more AGEs and potentially tissue injury. Common AGEs, such as CML, MG-derivatives and others are thought as capable of inducing inflammatory events [26]. Since the effects of both, exogenous and endogenous AGEs can be directly or indirectly blocked by anti-oxidants and anti-AGE agents [26], they have overlapping biological properties.
Among healthy subjects the daily intake of AGEs during regular meals is estimated to be excessive beyond a range compatible with low levels of inflammatory markers. We have defined a high- or low-AGE diet on whether the estimated dietary AGE intake is greater or lower than 15,000 AGE kU/day, which happens to be the median dietary AGE intake in our cohort of healthy community dwellers [29,30]. This is largely attributed to the fact that most favored methods of food preparation promote AGE formation. On a chronic basis, consumption of high AGE foods can cause a strain upon, and eventually a depletion of native anti-oxidant defenses, setting the stage for disease, i.e. diabetes. A premature compromise or else an inability to mount sufficient innate anti-oxidant defenses may account for the fact that high serum AGE levels in mothers closely correlated with those of their newborns. Moreover, high serum AGEs in mothers predict higher plasma insulin or HOMA levels, but lower adiponectin levels [31] during the first year of life, factors which may pre-condition infants, with or without genetic predisposition, to diabetes. In this context, crucial epidemiological evidence has emerged identifying high levels of circulating AGEs as a risk factor for developing T1D both, in ICA+ (islet cell autoantibodies) twin as well as in non-twin cohorts [12], which highlighted the evidently important question of the origin of high AGE levels prior to diabetes and at a young age.
Based on semiquantitative but well-validated immune assays new information on the AGE content of common foods [29, 30] have began to illustrate that dry heat-involving food processing methods (typically broiling, searing, and frying) significantly increase the content of protein, as well as lipid-related AGEs in foods, especially those of animal origin, unlike methods that utilize lower temperatures and more moisture or water (as in stewing, steaming, boiling). The same amount or type of nutrient, such as protein or fat, can dramatically influence the amount of glycoxidants delivered if prepared under dry-heat, the burden of which can lead to depleted defenses (Figure 1). At the same time, practical tools have been developed for assessing dietary AGE intake, or for adjusting diets to a lower AGE content, while maintaining optimal caloric and nutrient intake [29].
Figure 1.

Native and exogenous sources of oxidants, a most notable one being food derived AGEs, contribute to cellular and tissue injury in chronic diabetes.
3. Are AGEs Diabetogenic Agents? Mice studies
Extensive evidence demonstrates that AGEs can cause tissue injury, directly linking them to long-term diabetic complications [2, 9–11]. Moreover, recent findings have shed new light suggesting that exogenous AGEs may actually cause reduced peripheral insulin responsiveness [12, 32, 33]. Studies of primary adipocytes from trans-generational mice exposed for life to specific AGEs, such as MG-albumin - added to a low AGE diet at levels matching those in a standard mouse chow-showed that sustained exposure to glycoxidants can alter insulin receptor and IRS-1 phosphorylation levels and pattern, leading to impaired glucose-uptake, or fat mobilization from adipose tissue [32]. Another noteworthy finding was that, by the fifth generation, MG-fed mice developed insulin resistance as early as 16–18 months of age, instead of 24–26 months of age seen in the regular chow-fed controls. The AGE-restricted cohort did not develop these changes until beyond the age of 36 months or a time interval corresponding to approximately 20 human years [32]. These findings have begun to shift prior dogmas and introduce the view that prolonged exposure to an oxidant force that normally does not exist in nature, over the span of several generations, can deplete innate immune defenses, thus miss-firing inflammatory responses or fostering metabolic defects, namely in insulin action. This view, while prompting further investigation into potential epigenetic changes, has begun to receive support in the clinical setting, as is discussed later [19, 22, 23].
Several well-controlled animal studies have suggested that AGEs are also involved in islet β-cell damage in both T1D and T2D. Restriction of external AGEs protected and maintained normal pancreatic islet morphology and function in diverse models of diabetes, whether of genetic i.e. non-obese diabetic (NOD) mice susceptible to autoimmune T1D or db/db+/+ mice prone to T2D [13, 14] (Figure 2), or non-genetic etiology, i.e. dietary fat-induced or age-related diabetes [15, 17]. Subsequent data assigned AGE-mediated β-cell toxicity to the inhibition of cytochrome-c oxidase and reduced ATP production, thus impairing insulin secretion [13, 34, 35]. AGEs can also promote immune cell (T-cell, macrophage) recruitment, activation and beta cell cytotoxicity, apoptosis or cell death. AGER1 normally suppresses the effects of AGEs and ROS, and may enhance SIRT1 expression and function in beta cells, positively regulating insulin secretion (Figure 3). However under chronic high-level AGE conditions, AGER1, SIRT1 and other defenses are downregulated, thus possibly contributing to beta cell dysfunction or destruction. An AGE inhibitor, aminoguanidine (AG) [34] protected islet beta cell function both in vivo and in isolated rat islet [34,35]. Other animal studies have expanded the mechanistic basis of these findings and together demonstrated that excessive AGEs have the potential of acting as diabetogenic agents in the proper context [36].
Figure 2.
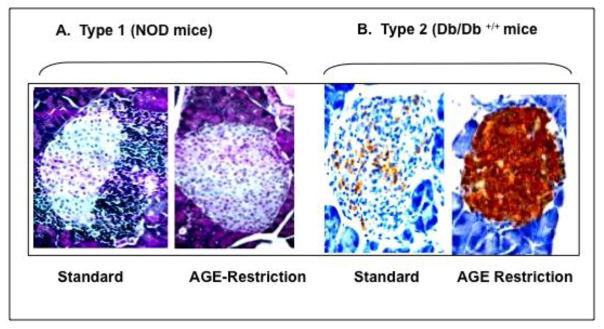
AGE restriction protects islet structure and function in mice genetically predisposed to type 1 (A) and type 2 diabetes (B). Non-obese diabetic (NOD) mice (A), and db/db +/+ mice (B) were exposed to either standard or Low-AGE diet for life. Pancreatic islets from mice on AGE-restriction did not display the mononuclear cell infiltration, typical of NOD mice fed a standard diet (AGE-rich) (H&E mag.x400,). Similarly, islets from age-matched db/db+/+ mice, after AGE-restriction, showed intact insulin production compared to those on regular diet. (insulin staining, mag.x400).
Figure 3.

Dietary AGEs can impair insulin secretion in pancreatic islet β-cells. Pro-oxidant AGEs can induce iNOS, and the generation of mitochondrial ROS, suppressing cytochrome-C levels and ATP generation, reducing insulin release. They can also suppress the deacetylase SIRT1, which regulates UCP2, thus impairing membrane depolarization and β-cell secretory function.
4. Diet-derived AGEs and Human Health
High AGE levels in T2D patients had been attributed to endogenous sources, namely hyperglycemia and OS [9] until it was noted that non-diabetic persons too could have “diabetic” levels of serum AGEs and OS, if they consumed a diet with a high AGE content [19, 22]. In pursuing this further, it was noted that unlike previous dogma, serum AGEs correlated with dietary AGE intake, as well as with established markers of OS and inflammation, such as hsCRP and TNFα, independent of age or diabetes [19, 22]. In self-declared normal controls from our population a significant association with HOMA, an indicator of IR, was also noted suggesting that the standard western diet could serve as a constant source of oxidants (Figures 4A,B). This, if supported by larger trials, may prove critical for those subjects with “non-obese” BMI who consume excessive AGEs as these may raise their risk for the metabolic syndrome or T2D.
Figure 4.


Traditional and non-traditional pathways to T1/T2D and complications. A, Over-nutrition promotes obesity and insulin resistance as well as increased OS, inflammation and β-cell injury leading to diabetes by unclear mechanisms. B, Modern diet, typically rich in animal food products, is also generally subject to heat-exposure. The resulting excess of oxidant AGEs include flavorful appetite-enhancing substances, which simultaneously promote food over-consumption and obesity, as well as insulin resistance, beta-cell injury and diabetes (type 1 or 2).
Other studies reported lowered levels of serum AGEs in diabetic patients after two months on a low-AGE diet [37]. This and other studies involving subjects with or without diabetes or with chronic kidney disease provide strong evidence that a low AGE diet program can be highly effective in reducing chronic OS and inflammation [22, 33, 37]. Together these studies bring into focus a new fact: the modern food environment can act as a significant source of AGEs which are capable of altering native defenses and of disturbing anti-oxidant balance in humans. Importantly, a dietary intervention, which utilizes a regimen of reduced AGE consumption, may represent a simple yet significant advance in the efficacy of anti-diabetic therapies [33, 37].
Another notable but unrecognized source of AGEs is cigarette smoking [24]. The processing or curing of tobacco involves AGE formation since the plant leaves are heat-dried in the presence of reducing sugars, added for purposes such as taste and smell. Subsequent combustion can lead to the inhalation of AGE derivatives and transfer into the circulation [24]. Levels of serum AGEs or LDL-apolipoprotein-B were found higher in chronic cigarette smokers than in nonsmokers. Also, AGE levels were higher in arterial wall samples or ocular lenses in smokers with diabetes compared to those from non-smoking diabetic persons [38]. Mechanistic and efficacy studies on smoking cessation have not been conducted from this perspective.
5. AGE Balance: Keeping a Tide at Bay
AGE catabolism is dependent on tissue anti-oxidant reserves, macromolecular turnover and receptor-mediated AGE degradation, prior to renal elimination. Steady-state AGE levels reflect not only glycemia, but also the balance of oral intake, endogenous formation, and catabolism of AGEs. At least two types of cellular AGE receptors have been characterized: RAGE and AGER1. AGER1 binds, degrades AGEs and protects against oxidant injury (Figure 5). It has considerable anti-oxidant and anti-inflammatory properties based on studies on AGER1-transduced cells and transgenic mice [22, 39–42]. AGEs induce native AGER1. Prolonged supply of external AGEs, however, depletes AGER1. The ensuing surplus OS promotes inflammation via RAGE, TLR4, EGFR and other receptors regulating the activities of NF-κB, AP1, FOXO and other pathways. Chronically elevated AGEs, via high OS, are potent suppressors of NAD+, partly by reducing NAMPT leading to SIRT1 depletion. Decreased SIRT1 levels promote NF-κB p65 hyper-acetylation and enhanced transcription of inflammatory genes, such as TNFα, which contributes to insulin resistance. AGER1, by engaging AGEs, controls many of these effects. The protective effects of AGER1 may stem from its long extracellular tail with high-affinity AGE-binding domain that competitively interferes with other AGE cell surface interactions leading to ROS.
Figure 5.
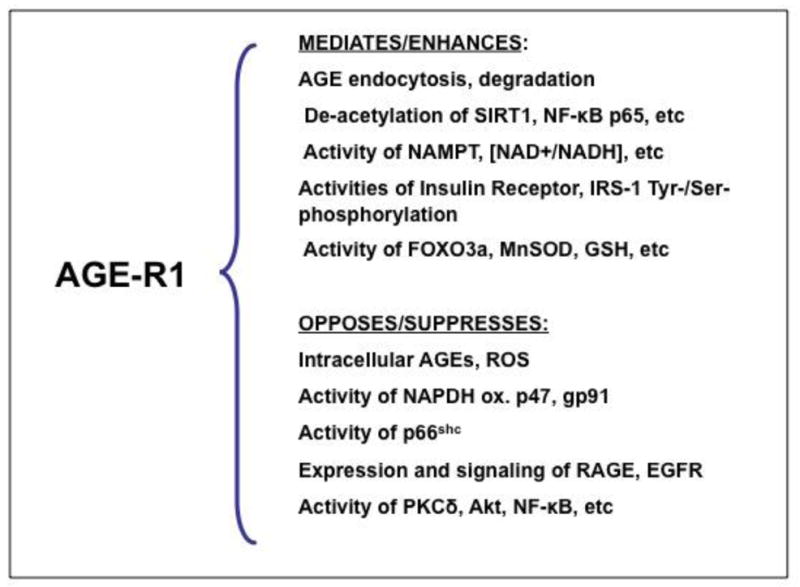
Partial list of known properties of AGER1, the best characterized anti-AGE receptor involved in the removal of AGEs, as well as in the maintenance of host defenses controlling their pro-inflammatory effects. Loss of function of AGER1 in chronic diabetes and other conditions of sustained oxidant AGE pressure is thought as one way by which depleted host defenses and anti-oxidant mechanisms can lead to the maladaptive inflammatory responses underlying diabetes and its complications.
The other type of multi-ligand receptor, RAGE is thought to promote and perpetuate cell activation and tissue injury via increased OS [43]44. The balance between these two receptors may be critical in the maintenance of oxidant homeostasis or progression to diabetes. Thus, AGER1 disrupts RAGE signaling [39] and promotes the expression and functions of SIRT1, a major deacetylase and a regulator of inflammation and the metabolic actions of insulin [32]. AGER1, like SIRT1 and other defense mechanisms, is suppressed in chronic diabetes. However both AGER1 and SIRT1 are restored after lowering the external oxidant burden by AGE restriction [32, 33]. Taken together, AGER1 expression levels correlate positively with the levels of other intracellular anti-oxidant mechanisms (SIRT1, NAMPT, SOD2, GSH) and negatively with pro-oxidant pathways (i.e, RAGE, NADPH oxidase, p66shc). Thus, assuming that AGER1 is important in the maintenance of normal AGE homeostasis, reduced AGER1 expression levels may signal a compromise in host defenses.
Degradation products of AGE-proteins resulting from the action of AGER1 and other receptor or non-receptor mechanisms give rise to AGE-peptides, which normally filter across the glomerular membrane. After filtration they can undergo variable degrees of tubular reabsorption or further catabolism by the proximal tubule, and excretion in the urine [45]. The active participation of the kidneys in the metabolism and excretion of AGEs is demonstrated by an inverse correlation between serum AGE levels and renal function estimated by glomerular filtration rate (GFR) [46]. A precise and quantitative analysis of the contribution of each of these processes is lacking in humans. It is, however, suggested in both animal and human studies, that even a modest degree of renal disease is associated with a markedly reduced excretion of oxidant AGEs [23, 46, 47].
6. Failing anti-AGE Defenses: a Road to High OS and Chronic Inflammation
Ambient levels of AGEs and OS regulate the expression of both AGER1 and RAGE receptors and their downstream pathways as needed to maintain the glycoxidative balance within cells. Thus, short-term elevations in AGEs can bring about an overexpression in AGER1, as well as in RAGE levels. On the other hand, reduced AGE levels, as seen after a low AGE diet in healthy subjects, downregulate AGER1 as well as RAGE levels [22]. Consistent with an active role of AGER1 in AGE turnover and elimination by the normal kidney, AGER1, but not RAGE levels correlate directly with urinary AGEs in healthy persons. Under chronic diabetes or chronic kidney disease, however, the situation is quite different. RAGE levels remain high, while AGER1 levels are uniformly suppressed even during full anti-diabetic therapy [18, 22, 33], indicating persistently high OS. However, when diabetic patients followed a low-AGE diet for 4 months, AGER1 levels were restored to normal, while RAGE levels were effectively suppressed [33]. This pattern further suggests that a functional depletion of AGER1 could be important in diabetic tissue injury as it could lead to intracellular AGE accumulation, ROS generation, and suppression of NAD+-dependent SIRT1, followed by a further increase in pro-inflammatory NF-κB activity. Thus a hyper-activation of RAGE and other pro-inflammatory genes could be the consequence of a failure of anti-AGE and anti-OS defenses, such as AGER1 and SIRT1, to fend off perpetual OS.
From the clinical and animal studies available, it may also be concluded that innate defenses and anti-oxidant mechanisms can be rapidly bolstered by the effective reduction of external oxidants, rather than by adding anti-oxidant supplements, which though perfectly functional, may be insufficient compared to the magnitude of their targets.
7. Diabetes, Diabetic Complications and the New Paradox
Because hyperglycemia has traditionally been thought to be the principal source of AGEs, intensive control of hyperglycemia was expected to also control the consequences of AGEs on diabetic vascular complications [4, 48]. Recently conducted large studies (ACCORD, ADVANCE, NADT) [6–8] have, however, failed to produce these long-anticipated results highlighting a need to identify new precipitating factors. For instance, measures to assess and modulate exogenously derived AGEs might have influenced these results. Furthermore, well-controlled cellular and animal studies suggest that a chronic exogenous overload of oxidant AGEs can incite β-cell injury [13] and thus the occurrence of T1D and T2D diabetes and their complications [49].
The contributing role of AGEs in human diabetes, but also in human non-diabetes related CVD or CKD is fairly well established [50–53]. A significant correlation was previously found between circulating AGE-apoB levels, vascular tissue AGEs and severity of atherosclerotic lesions in non-diabetic patients with coronary artery occlusive disease [49], and in diabetic subjects with aortic stiffness 53]. Serum pentosidine has also been shown to correlate positively with heart-brachial pulse wave velocity and with carotid intima-media thickness [54] in patients with T2D. In a random sample of Finnish T2D participants followed for 18 years serum levels of AGEs were associated with total and CVD mortality in women [55]. These studies reinforce the role of AGEs in diabetic complications, apart from that of hyperglycemia. When patients with either type 1 or 2 diabetes were placed on a low AGE diet for a brief period of 6 weeks, there was a significant reduction of markers of inflammation and endothelial dysfunction such as hsCRP, TNF and VCAM-1, in addition to markedly lower serum AGE levels [37].
The suggestion that food-derived AGEs could pose risk not only for diabetic vascular disease, but diabetes per se, has introduced a new paradox, whereby hyperglycemia could be the downstream effect of excessive AGEs, not a pre-requisite. Thus, in a study on T2D subjects, baseline serum AGEs were shown to correlate with fasting insulin, HOMA-IR and BMI and after a 4-month treatment with an AGE-restricted diet, a significant reduction in plasma insulin and leptin were associated with a marked rise in adiponectin [33], consistent with improved insulin sensitivity. There were also significant increases in AGER1 and SIRT1 levels tied to reduced mononuclear cell NF-κB activity and decreased TNFα levels, all consistent with suppressed inflammation in these subjects. These findings await confirmation in larger trials, but, since they were independent of any changes in standard medical therapy, they support the postulate that the externally derived pro-inflammatory AGEs are an important culprit [22, 33]. Furthermore, these abnormalities can be effectively - and economically - modulated by a modest decrease (~50%) of the amount of AGEs in the diet, without changes in nutrients or calories.
That dietary AGEs could also have a direct and immediate impact on tissues, such as the vasculature, was suggested in separate studies. An oral AGE challenge test produced an increase in serum AGEs, followed by a transient endothelial dysfunction, based on impaired flow mediated dilatation or FMD, and a marked rise in plasminogen activator inhibitor-1 or PAI-1 - in both diabetic and non-diabetic subjects [56]. The test beverage contained neither carbohydrates nor lipids, either of which could contribute to postprandial endothelial dysfunction. Finally, a single AGE-rich solid meal induced a more pronounced acute impairment of vascular function in diabetic subjects than did an otherwise nutritionally identical low-AGE meal [57]. The transient AGE effects shown by these studies underscored the possibility that repeated or sustained “stress” from the frequent intake of certain AGE-laden foods or beverages - apart from an excess of glucose or lipids - could set the stage for long-term vascular and other tissue injury.
8. Lifestyle Changes and Oral Agents that Can Prevent AGE Formation and AGE Gastrointestinal Absorption
After consuming a meal with a high AGE content, healthy adults show a rapid absorption of AGEs, with a rapidly rising peak in serum AGE level [23]. A close correlation was observed between AGEs consumed and AGEs in the circulation in a cross-section of healthy subjects [19]. Additional studies in healthy, diabetic or non-diabetic subjects with chronic renal insufficiency showed that lowering dietary AGE intake (by ~50%) could decrease circulating AGE levels and markers of inflammation and OS [22, 37, 58]. More importantly, an otherwise isocaloric but low in AGE diet seems able of improving hyper-insulinemia (by ~40%) in fully treated T2D patients, confirming that exogenous AGEs actively participate in the metabolic dysfunctional milieu of T2D. A marked recovery was noted in levels of AGER1, SIRT1 and adiponectin, three factors with anti-inflammatory properties, which were decreased at baseline. This return of innate defenses to normal suggests that this strategy carries promise as an efficient, low-cost treatment for those with diabetes mellitus or with prediabetes.
On the practical level, a low AGE intake can be easily achieved by using lower heat, higher humidity (e.g. instead of roasting, grilling or frying, use stewing, poaching, etc) and avoiding highly processed pre-packaged and fast foods [29, 30]. Patients with diabetes have found such a program to be easily incorporated into their personal and family life.
Various inhibitors of post-Amadori glycation intermediates (glyoxal, methylglyoxal, 3-deoxyglucosone) are described, including aminoguanidine [59–61], pyridoxamine, benfotiamine and ORB-9195 [62, 63]. Some of these, such as ORB-9195 may modulate the generation of glycation and lipoxidation derivatives [64, 65] and some may become available for clinical use in the future.
Several authors have suggested the use of antioxidants as anti-AGE agents, i.e. vitamin E [66], N-acetylcysteine [67], taurine [68], alpha lipoic acid [69], penicillamine [70], nicanartine [71] and others, despite the disappointing results of a range of studies with oral antioxidants. Since previous trials did not consider the large exogenous oxidant surplus entering the gastrointestinal tract and likely neutralizing them, their failure may not be surprising. More studies with new agents and expanded range of dosages will be needed to definitively establish their effectiveness [72, 73].
In this context, a new study introduced sevelamer carbonate, an oral non-absorbable negatively charged polymer, known clinically for its phosphate-binding capacity [74]. The agent, in addition to phosphates, can bind AGEs in a pH-dependent manner, and thus possibly sequester AGEs in the gut. After 2 months, sevelamer carbonate, but not CaCO3, another phosphate-binder that does not bind AGEs, effectively lowered circulating AGEs, as well as markers of OS and inflammation in diabetic subjects with chronic kidney disease [74]. Of particular interest was that the agent restored AGER1 and SIRT-1 to normal levels. This alternative strategy, while currently under further clinical investigation, by confirming the importance of reducing absorption of oral AGEs, provides critical support to AGE-restriction and may offer an important adjunct treatment strategy.
Conclusions
A major shift over the past half century is the enrichment of the food environment with AGEs, palatable pro-oxidant substances, which can promote both overnutrition and oxidant overload. Sustained oxidant overload may overwhelm host defenses and lead to unopposed oxidant stress and chronic inflammation. These states can over time impair insulin production and/or sensitivity and lead to diabetes. The fact that AGEs contribute significantly to OS and inflammation in diabetes and diabetic complications is no longer debated. Hyperglycemia is a significant driving force for AGE formation, especially since it arrives upon a premise of pre-existing overt OS. It is becoming increasingly clear, however, that the common diet is a carrier of pre-formed AGEs, and thus, may be a key initiator or contributor to OS. This causal factor is of particular concern in subjects not only with diabetes but those with pre-diabetes. Thus, AGE overload as a cause of diabetes is an area of major clinical relevance. While many pharmacologic anti-AGE therapies are under development, their efficacy remains to be proven. Aiming to reduce both the exogenous or food-derived AGEs in addition to or beyond lowering endogenous hyperglycemia-derived AGEs seems to be a sound target for lasting benefits against diabetes and its complications. In the interim, screening for and advising those with high AGE markers or consumption may be a first move in this direction that is worth considering.
Acknowledgments
This work was supported by grants AG-23188 and AG-09453 (to H.V.) and from the National Institutes of Health, National Institute of Research Resources (Grant M01-RR-00071) to the General Clinical Research Center at Mount Sinai School of Medicine.
Footnotes
Conflict of Interest
Helen Vlassara has received grant support and support for travel to meetings for the study or otherwise from Sanofi for a co-investigator-initiated clinical investigation. She has patents (planned, pending or issued) from Cell Biolabs for development of monoclonal antibody, and receives royalties for monoclonal antibody. Her husband is principal investigator in investigator-initiated clinical trial, supported by Sanofi.
Jaime Uribarri is a co-author on a book on AGE-less Diet.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Amos AF, McCarty DJ, Zimmet P. The rising global burden of diabetes and its complications: estimates and projections to the year 2010. Diabet Med. 1997;14 (Suppl 5):S1–85. [PubMed] [Google Scholar]
- 2.Huebschmann AG, Regensteiner JG, Vlassara H, Reusch JE. Diabetes and advanced glycoxidation end products. Diabetes Care. 2006;29:1420–32. doi: 10.2337/dc05-2096. [DOI] [PubMed] [Google Scholar]
- 3.Ford ES, Giles WH, Mokdad AH. Increasing prevalence of the metabolic syndrome among U.S. Adults Diabetes Care. 2004;27:2444–9. doi: 10.2337/diacare.27.10.2444. [DOI] [PubMed] [Google Scholar]
- 4.The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–86. doi: 10.1056/NEJM199309303291401. No authors listed. [DOI] [PubMed] [Google Scholar]
- 5.Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854–65. No authors listed. [PubMed] [Google Scholar]
- 6.Skyler JS, et al. Intensive glycemic control and the prevention of cardiovascular events: implications of the ACCORD, ADVANCE, and VA Diabetes Trials: a position statement of the American Diabetes Association and a Scientific Statement of the American College of Cardiology Foundation and the American Heart Association. J Am Coll Cardiol. 2009;53:298–304. doi: 10.1016/j.jacc.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 7.Ginsberg HN, Elam MB, Lovato LC, Crouse JR, 3rd, Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein HC, Probstfield J, Grimm RH, Ismail-Beigi F, Bigger JT, Goff DC, Jr, Cushman WC, Simons-Morton DG, Byington RP ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Eng J Med. 2010;362:1563–74. doi: 10.1056/NEJMoa1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duckworth W, et al. Glucose control and vascular complications in veterans with type 2 diabetes. N Eng J Med. 2009;360:129–39. doi: 10.1056/NEJMoa0808431. [DOI] [PubMed] [Google Scholar]
- 9.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 10.Vlassara H, Palace MR. Diabetes and advanced glycation endproducts. J Intern Med. 2002;251:87–101. doi: 10.1046/j.1365-2796.2002.00932.x. [DOI] [PubMed] [Google Scholar]
- 11.Thorpe SR, Baynes JW. Role of the Maillard reaction in diabetes mellitus and diseases of aging. Drug Aging. 1996;9:69–77. doi: 10.2165/00002512-199609020-00001. [DOI] [PubMed] [Google Scholar]
- 12.Beyan H, Riese H, Hawa MI, Beretta G, Davidson HW, Hutton JC, Burger H, Schlosser M, Snieder H, Boehm BO, Leslie RD. Glycotoxin and autoantibodies are additive environmentally determined predictors of type 1 diabetes: a twin and population study. Diabetes. 2012;61:1192–8. doi: 10.2337/db11-0971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Z, Zhao C, Zhang XH, Zheng F, Cai W, Vlassara H, Ma ZA. Advanced glycation end products inhibit glucose-stimulated insulin secretion through nitric oxide-dependent inhibition of cytochrome c oxidase and adenosine triphosphate synthesis. Endocrinology. 2009;150:2569–76. doi: 10.1210/en.2008-1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peppa M, He C, Hattori M, McEvoy R, Zheng F, Vlassara H. Fetal or neonatal low-glycotoxin environment prevents autoimmune diabetes in NOD mice. Diabetes. 2003;52:1441–8. doi: 10.2337/diabetes.52.6.1441. [DOI] [PubMed] [Google Scholar]
- 15.Hofmann SM, Dong HJ, Li Z, Cai W, Altomonte J, Thung SN, Zeng F, Fisher EA, Vlassara H. Improved insulin sensitivity is associated with restricted intake of dietary glycoxidation products in the db/db mouse. Diabetes. 2002;51:2082–2089. doi: 10.2337/diabetes.51.7.2082. [DOI] [PubMed] [Google Scholar]
- 16.Sandu O, Song K, Cai W, Zheng F, Uribarri J, Vlassara H. Insulin resistance and type 2 diabetes in high-fat-fed mice are linked to high glycotoxin intake. Diabetes. 2005;54:2314–2319. doi: 10.2337/diabetes.54.8.2314. [DOI] [PubMed] [Google Scholar]
- 17.Cai W, He JC, Zhu L, Chen X, Zheng F, Striker GE, Vlasara H. Oral glycotoxins determine the effects of calorie restriction on oxidant stress, age-related diseases, and lifespan. Am J Pathol. 2008;173:327–336. doi: 10.2353/ajpath.2008.080152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cai W, He JC, Zhu L, Chen X, Wallenstein S, Striker GE, Vlassara H. Reduced oxidant stress and extended lifespan in mice exposed to a low glycotoxin diet: association with increased AGER1 expression. Am J Pathol. 2007;170:1893–902. doi: 10.2353/ajpath.2007.061281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uribarri J, Cai W, Peppa M, Goodman S, Ferrucci L, Striker G, Vlassara H. Circulating glycotoxins and dietary advanced glycation endproducts: Two links to inflammatory response, oxidative stress and aging. J Gerontol Med Sci. 2007;62:427–433. doi: 10.1093/gerona/62.4.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suarez G, Rajaram R, Oronsky AL, Gawinowicz MA. Nonenzymatic glycation of bovine serum albumin by fructose (fructation). Comparison with the Maillard reaction initiated by glucose. J boil Chem. 1989;264:3674–9. [PubMed] [Google Scholar]
- 21.Bucala R, Mitchell R, Arnold K, Innerarity T, Vlassara H, Cerami A. Identification of the major site of apolipoprotein B modification by advanced glycosylation end products blocking uptake by the low-density lipoprotein receptor. J Biol Chem. 1995;270:10828–32. doi: 10.1074/jbc.270.18.10828. [DOI] [PubMed] [Google Scholar]
- 22.Vlassara H, Cai W, Goodman S, Pyzik R, Yong A, Zhu L, Neade T, Beeri M, Silverman JM, Ferrucci L, Tansman L, Striker GE, Uribarri J. Protection against loss of innate defenses in adulthood by low AGE intake; role of a new anti-inflammatory AGE-receptor-1. J Clin End Met. 2009;94:4483–91. doi: 10.1210/jc.2009-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koschinsky T, He CJ, Mitsuhashi T, Bucala R, Liu C, Bueting C, Heitmann K, Vlassara H. Orally absorbed reactive advanced glycation end products (glycotoxins): an environmental risk factor in diabetic nephropathy. Proc Natl Acad Sci USA. 1997;94:6474–6479. doi: 10.1073/pnas.94.12.6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cerami C, Founds H, Nicholl I, Mitsuhashi T, Giordano D, Vanpatten S, Lee A, Al-Abed Y, Vlassara H, Bucala R, Cerami A. Tobacco smoke is a source of toxic reactive glycation products. Proc Natl Acad Sci U S A. 1997;94:13915–20. doi: 10.1073/pnas.94.25.13915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Brien J, Morrissey PA. Nutritional and toxicological aspects of the Maillard browning reaction in foods. Crit Rev Food Sci Nutr. 1989;28:211–48. doi: 10.1080/10408398909527499. [DOI] [PubMed] [Google Scholar]
- 26.Cai W, Gao Q-D, Zhu L, Peppa M, He C, Vlassara H. Oxidative stress-inducing carbonyl compounds from common foods: novel mediators of cellular dysfunction. Mol Med. 2002;8:337–46. [PMC free article] [PubMed] [Google Scholar]
- 27.Van Boekel MA. Formation of flavour compounds in the Maillard reaction. Biotechnol Adv. 2006;24:230–233. doi: 10.1016/j.biotechadv.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 28.He C, Sabol J, Mitsuhashi T, Vlassara H. Dietary glycotoxins: inhibition of reactive products by aminoguanidine facilitates renal clearance and reduces tissue sequestration. Diabetes. 1999;48:1308–1315. doi: 10.2337/diabetes.48.6.1308. [DOI] [PubMed] [Google Scholar]
- 29•.Uribarri J, Woodruff S, Goodman S, Cai W, Chen X, Pyzik R, Yong A, Striker GE, Vlassara H. Advanced glycatione end products in foods and a practical guide to their reduction in the diet. J Am Diet Assoc. 2010;110:911–916. doi: 10.1016/j.jada.2010.03.018. This review includes an extensive database with the AGE content of many foods allowing the practical implementation of an AGE-restricted diet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldberg T, Cai W, Peppa M, Dardaine V, Uribarri J, Vlassara H. Advanced glycoxidation end products in commonly consumed foods. J Am Diet Assoc. 2004;104:1287–1291. doi: 10.1016/j.jada.2004.05.214. [DOI] [PubMed] [Google Scholar]
- 31.Mericq V, Piccardo C, Cai W, Chen X, Zhu L, Striker GE, Vlassara H, Uribarri J. Maternally transmitted and food-derived glycotoxins: a factor preconditioning the young to diabetes? Diabetes care. 2010;33:2232–7. doi: 10.2337/dc10-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32••.Cai W, Ramdas M, Zhu L, Chen X, Striker GE, Vlassara H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc Natl Acad Sci USA. 2012;109:15888–93. doi: 10.1073/pnas.1205847109. This study in mice documented very well the mechanisms by which AGEs induce insulin resistance and demonstrated that successive generations of animals chronicall exposed to high AGE diets develop insulin resistance at a progressively earlier age. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33••.Uribarri J, Cai W, Ramdas M, Goodman S, Pyzik R, Chen X, Zhu L, Striker GE, Vlassara H. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: potential role of AGER1 and SIRT1. Diabetes Care. 2011;34:1610–6. doi: 10.2337/dc11-0091. This clinical trial documented for the first time that insulin resistance in humans can be reversed by an AGE-restricted diet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tajiri Y, Moller C, Grill V. Long-term effects of aminoguanidine on insulin release and biosynthesis: evidence that the formation of advanced glycosylation end products inhibits B cell function. Endocrinology. 1997;138:273–8. doi: 10.1210/endo.138.1.4851. [DOI] [PubMed] [Google Scholar]
- 35.Tajiri Y, Grill V. Aminoguanidine exerts a beta-cell function-preserving effect in high glucose-cultured beta cells (INS-1) Int J Exp Diabetes Res. 2000;1:111–9. doi: 10.1155/EDR.2000.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coughlan MT, Yap FY, Tong DC, Andrikopoulos S, Gasser A, Thallas-Bonke V, Webster DE, Miyazaki J, Kay TW, Slattery RM, Kaye DM, Drew BG, Kingwell BA, Fourlanos S, Groop PH, Harrison LC, Knip M, Forbes JM. Advanced glycation end products are direct modulators of β-cell function. Diabetes. 2011;60:2523–32. doi: 10.2337/db10-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vlassara H, Cai W, Crandall J, Goldberg T, Oberstein R, Dardaine V, Peppa M, Rayfield EJ. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc Natl Acad Sci USA. 2002;99:15596–601. doi: 10.1073/pnas.242407999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicholl ID, Stiit AW, Moore JE, Ritchie AJ, Archer DB, Bucala R. Increased levels of advanced glycation endproducts in the lenses and blood vessels of cigarette smokers. Mol Med. 1998;4:594–601. [PMC free article] [PubMed] [Google Scholar]
- 39.Cai W, He C, Zhu L, Vlassara H. Advanced glycation end product (AGE) receptor 1 suppresses cell oxidant stress and activation signaling via EGF receptor. PNAS USA. 2006;103:13801–806. doi: 10.1073/pnas.0600362103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cai W, Torreggiani M, Zhu L, Chen X, He JC, Striker GE, Vlassara H. AGER1 regulates endothelial cell NADPH oxidase-dependent oxidant stress via PKC-delta: implications for vascular disease. Am J Physiol Cell Physiol. 2010;298:C624–32. doi: 10.1152/ajpcell.00463.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu H, He JC, Cai W, Liu H, Zhu L, Vlassara H. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc Natl Acad Sci USA. 2004;101:11767–72. doi: 10.1073/pnas.0401588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Torreggiani M, Liu H, Wu J, Zheng F, Cai W, Striker G, Vlassara H. Advanced glycation end product receptor-1 transgenic mice are resistant to inflammation, oxidative stress, and post-injury intimal hyperplasia. Am J Pathol. 2009;175:1722–32. doi: 10.2353/ajpath.2009.090138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP. Understanding RAGE, the receptor for advanced glycatione end products. J Mol Med (Berl) 2005;83:876–86. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- 44.Lukic IK, Humpert PM, Nawroth PP, Bierhaus A. The RAGE pathway: activation and perpetuation in the pathogenesis of diabetic neuropathy. Ann N Y Acad Sci. 2008 Apr;1126:76–80. doi: 10.1196/annals.1433.059. [DOI] [PubMed] [Google Scholar]
- 45.Saito A, Nagai R, Tanuma A, Hama H, Cho K, Takeda T, Yoshida Y, Toda T, Shimizu F, Horiuchi S, Gejyo F. Role of megalin in endocytosis of advanced glycation end products: implications for a novel protein binding to both megalin and advanced glycation end products. J Am Soc Nephrol. 2003;14:1123–31. doi: 10.1097/01.asn.0000062962.51879.f8. [DOI] [PubMed] [Google Scholar]
- 46.Vlassara H, Torreggiani M, Post JB, Zheng F, Uribarri J, Striker GE. Role of oxidants/inflammation in declining renal function in chronic kidney diseases and normal aging. Kidney Int Suppl. 2009;114:S3–S11. doi: 10.1038/ki.2009.401. [DOI] [PubMed] [Google Scholar]
- 47.Makita Z, Radoff S, Rayfield EJ, Yang Z, Skolnik E, Delaney V, Friedman EA, Cerami A, Vlassara H. Advanced glycosylation end products in patients with diabetic nephropathy. N Engl J Med. 1991;325:836–42. doi: 10.1056/NEJM199109193251202. [DOI] [PubMed] [Google Scholar]
- 48.Odetti P, Traverso N, Cosso L, Noberasco G, Pronzato MA, Marinari UM. Good glycaemic control reduces oxidation and glycation end-products in collagen of diabetic rats. Diabetologia. 1996;39:1440–7. doi: 10.1007/s001250050596. [DOI] [PubMed] [Google Scholar]
- 49.Vlassara H, Striker GE. Glycotoxins in the diet promote diabetes and diabetic complications. Curr Diab Rep. 2007;7:235–41. doi: 10.1007/s11892-007-0037-z. [DOI] [PubMed] [Google Scholar]
- 50.Schleicher ED, Wagner E, Nerlich AG. Increased accumulation of the glycoxidation product N(epsilon)-(carboxymethyl)lysine in human tissues in diabetes and aging. J Clin Invest. 1997;99:457–68. doi: 10.1172/JCI119180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoshida S, Yamada K, Hamaguchi K, Nishimura M, Hatakeyama E, Tsuchida H, Sakamoto K, Kashiwabara H, Yokoyama T, Ikeda K, Horiuchi S. Immunohistochemical study of human advanced glycation end-products (AGE) and growth factors in cardiac tissues of patients on maintenance dialysis and with kidney transplantation. Clin Nephrol. 1998;49:273–80. [PubMed] [Google Scholar]
- 52.Sakata N, Imanaga Y, Meng J, Tachikawa Y, Takebayashi S, Nagai R, Horiuchi S. Increased advanced glycation end products in atherosclerotic lesions of patients with end-stage renal disease. Atherosclerosis. 1999;142:67–77. doi: 10.1016/s0021-9150(98)00192-0. [DOI] [PubMed] [Google Scholar]
- 53.Stitt AW, He C, Friedman S, Scher L, Rossi P, Ong L, Founds H, Li YM, Bucala R, Vlassara H. Elevated AGE-modified ApoB in sera of euglycemic, normolipidemic patients with atherosclerosis: relationship to tissue AGEs. Mol Med. 1997;3:617–27. [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshida N, Okumura K, Aso Y. High serum pentosidien concentrations are associated with increased arterial stiffness and thickness in patients with type 2 diabetes. Metabolism. 2005;54:345–50. doi: 10.1016/j.metabol.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 55.Kilhovd BK, Juutilainen A, Lehto S, Ronnemaa T, Torjesen PA, Hanssen KF, laakso M. Increased serum levels of advanced glycation endproducts predict total, cardiovascular and coronary mortality in women with type 2 diabetes: a population-based 18 year follow-up study. Diabetologia. 2007;50:1409–17. doi: 10.1007/s00125-007-0687-z. [DOI] [PubMed] [Google Scholar]
- 56.Uribarri J, Stirban A, Sander D, Cai W, Negrean M, Buenting CE, Koschinsky T, Vlassara H. Single oral challenge by advanced glycation end products acutely impairs endothelial function in diabetic and nondiabetic subjects. Diabetes Care. 2007;30:2579–82. doi: 10.2337/dc07-0320. [DOI] [PubMed] [Google Scholar]
- 57.Negrean M, Stirban A, Stratmann B, Gawlowski T, Horstmann T, Götting C, Kleesiek K, Mueller-Roesel M, Koschinsky T, Uribarri J, Vlassara H, Tschoepe D. Effects of low- and high-advanced glycation endproduct meals on macro- and microvascular endothelial function and oxidative stress in patients with type 2 diabetes mellitus. Am J Clin Nutr. 2007;85:1236–43. doi: 10.1093/ajcn/85.5.1236. [DOI] [PubMed] [Google Scholar]
- 58.Uribarri J, Peppa M, Godlberg T, Cai W, Lu M, He C, Vlassara H. Restriction of dietary glycotoxins markedly reduces elevated advanced glycation end products in renal failure patients. J Am Soc Nephrol. 2003;14:728–731. doi: 10.1097/01.asn.0000051593.41395.b9. [DOI] [PubMed] [Google Scholar]
- 59.Soulis T, Cooper ME, Vranes D, Bucala R, Jerums G. Effects of aminoguanidine in preventing experimental diabetic nephropathy are related to the duration of treatment. Kidney Int. 1996;50:627–34. doi: 10.1038/ki.1996.358. [DOI] [PubMed] [Google Scholar]
- 60.Panagiotopoulos S, O’Brien RC, Bucala R, Cooper ME, Jerums G. Aminoguanidine has an anti-atherogenic effect in the cholesterol-fed rabbit. Atherosclerosis. 1998;136:125–31. doi: 10.1016/s0021-9150(97)00192-5. [DOI] [PubMed] [Google Scholar]
- 61.Kelly DJ, Gilbert RE, Cox AJ, Soulis T, Jerums G, Cooper ME. Aminoguanidine ameliorates overexpression of prosclerotic growth factors and collagen deposition in experimental diabetic nephropathy. J Am Soc Nephrol. 2001;12:2098–107. doi: 10.1681/ASN.V12102098. [DOI] [PubMed] [Google Scholar]
- 62.Voziyan X, PA, Hudson BG. Pyridoxamine: the many virtues of a maillard reaction inhibitor. Ann N Y Acad Sci. 2005;1043:807–16. doi: 10.1196/annals.1333.093. [DOI] [PubMed] [Google Scholar]
- 63.Williams ME, Bolton WK, Khalifah RG, Degenhardt TP, Schotzinger RJ, McGill JB. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am J Nephrol. 2007;27:605–14. doi: 10.1159/000108104. [DOI] [PubMed] [Google Scholar]
- 64.Nakamura S, Makita Z, Ishikawa S, Yasumura K, Fujii W, Yanagisawa K, Kawata T, Koike T. Progression of nephropathy in spontaneous diabetic rats is prevented by OPB-9195, a novel inhibitor of advanced glycation. Diabetes. 1997;46:895–9. doi: 10.2337/diab.46.5.895. [DOI] [PubMed] [Google Scholar]
- 65.Wada R, Nishizawa Y, Yagihashi N, Takeuchi M, Ishikawa Y, Yasumura K, Nakano M, Yagihashi S. Effects of OPB-9195, anti-glycation agent, on experimental diabetic neuropathy. Eur J Clin Invest. 2001;31:513–20. doi: 10.1046/j.1365-2362.2001.00826.x. [DOI] [PubMed] [Google Scholar]
- 66.Odetti P, Robaudo C, Valentini S, Gurreri G, Garibaldi S, Angeletti S, Deferrari G. Effect of a new vitamin E-coated membrane on glycoxidation during hemodialysis. Contrib Nephrol. 1999;127:192–9. doi: 10.1159/000060001. [DOI] [PubMed] [Google Scholar]
- 67.Nakayama M, Izumi G, Nemoto Y, Shibata K, Hasegawa T, Numata m, Wang K, Kawaguchi Y, Hosoya T. Suppression of N(epsilon)-(carboxymethyl)lysine generation by the antioxidant N-acetylcysteine. Perit Dial Int. 1999;19:207–10. [PubMed] [Google Scholar]
- 68.Trachtman H, Futterweit S, Prenner J, Hanon S. Antioxidants reverse the antiproliferative effect of high glucose and advanced glycosylation end products in cultured rat mesangial cells. Biochem Biophys Res Commun. 1994;199:346–52. doi: 10.1006/bbrc.1994.1235. [DOI] [PubMed] [Google Scholar]
- 69.Kunt T, Forst T, Wihelm A, Rtitschler H, Pfuentzner A, Harzer O, Engelbach M, Zschaebitz A, Stofft E, Beyer J. Alpha-lipoic acid reduces expression of vascular cell adhesion molecule-1 and endothelial adhesion of human monocytes after stimulation with advanced glycation end products. Clin Sci (Lond) 1999;96:75–82. [PubMed] [Google Scholar]
- 70.Jakus V, Hrnclarova M, Carsky J, Krahulec B, Rietbrock N. Inhibition of nonenzymatic protein glycation and lipid peroxidation by drugs with antioxidant activity. Life Sci. 1999;65:1991–3. doi: 10.1016/s0024-3205(99)00462-2. [DOI] [PubMed] [Google Scholar]
- 71.Hammes HP, Bartmann A, Engel L, Wulfroth P. Antioxidant treatment of experimental diabetic retinopathy in rats with nicanartine. Diabetologia. 1997;40:629–34. doi: 10.1007/s001250050726. [DOI] [PubMed] [Google Scholar]
- 72.Vlassara H. AGEs as a preventable cause of diabetes and its complications. Diabetological Chronicles. 2011;24(3):155–160. [Google Scholar]
- 73.Vlassara H, Striker G. AGE restriction in diabetes mellitus: a paradigm shift. Nature Reviews Endocrinology. 2011;7:526–39. doi: 10.1038/nrendo.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74•.Vlassara H, Uribarri J, Cai W, Goodman S, Pyzik R, Post J, Grosjean F, Woodward M, Striker GE. Effects of sevelamer on HbA1c, inflammation, and advanced glycation end products indiabetic kidney disease. Clin J Am Soc Nephrol. 2012;7:934–42. doi: 10.2215/CJN.12891211. This clinical trial in patients with diabetic nephropathy demonstrates that an oral resin, sevelamer carbonate, used clinically to control hyperphosphatemia is also effective in reducing circulating AGEs and markers of inflammation and oxidative stress in this population. [DOI] [PMC free article] [PubMed] [Google Scholar]