

Abstract

That the cationic polyelectrolyte poly(allylamine hydrochloride) (PAH) exerts a significant influence on CaCO3 precipitation challenges the idea that only anionic additives have this effect. Here, we show that in common with anionic polyelectrolytes such as poly(aspartic acid), PAH supports the growth of calcite thin films and abundant nanofibers. While investigating the formation of these structures, we also perform the first detailed structural analysis of the nanofibers by transmission electron microscopy (TEM) and selected area electron diffraction. The nanofibers are shown to be principally single crystal, with isolated domains of polycrystallinity, and the single crystal structure is even preserved in regions where the nanofibers dramatically change direction. The formation mechanism of the fibers, which are often hundreds of micrometers long, has been the subject of intense speculation. Our results suggest that they form by aggregation of amorphous particles, which are incorporated into the fibers uniquely at their tips, before crystallizing. Extrusion of polymer during crystallization may inhibit particle addition at the fiber walls and result in local variations in the fiber nanostructure. Finally, we investigate the influence of Mg2+ on CaCO3 precipitation in the presence of PAH, which gives thinner and smoother films, together with fibers with more polycrystalline, granular structures.
Keywords: calcite, crystallization, nanowire, PILP, bioinspired, TEM
Introduction
Soluble additives are widely used to control the precipitation of crystals from solution, generating particles with defined sizes, morphologies, polymorphs, orientations, or even mechanical properties.1,2 In selecting or designing suitable additive molecules for crystal-growth control, the macromolecules used by nature to control the formation of biominerals such as bones, teeth, and seashells have frequently been used as inspiration.3−5 From the earliest studies of the biomacromolecules extracted from CaCO3 biominerals, which are widely investigated due to their abundance, it is recognized that these are characteristically highly acidic, being rich in aspartic and glutamic acid.6−14 Strategies for the control of crystallization based on the use of small molecules and polymers functionalized with negatively charged groups have, therefore, been widely explored and have achieved considerable success.15 For example, negatively charged Langmuir monolayers16,17 and self assembled monolayers (SAMs)18,19 have been used to support the oriented growth of calcite, whereas negatively charged small molecules,20,21 polymers,22−26 and block copolymers27−29 have supported the formation of many remarkable morphologies including calcite microtrumpets, patterned CaCO3 thin films, porous particles, strontium carbonate flowers,30 and barium carbonate helices.31
Some of the most dramatic morphological effects of soluble additives are seen in the production of thin films and long fibers of crystals such as calcium carbonate, barium carbonate, and barium sulfate.31−34 This has principally been achieved using the polymeric additives poly(aspartic acid) (PAsp), poly(acrylic acid) (PAA),35−40 or carboxylate-rich copolymers,41−43 where these molecules bear many similarities to the highly acidic biomacromolecules implicated in the formation of biominerals. The activity of these additives has either been related to their ability to stabilize crystalline precursor particles and then to direct their oriented assembly31,34 or to stabilize a so-called PILP (polymer-induced liquid precursor)44 or amorphous precursor phase.38,40,41 Accumulation of PILP droplets on a substrate and their subsequent crystallization can generate thin films, whereas aggregation at active sites has been proposed to lead to fiber formation.32,33 Based on the knowledge that PILP formation is due to a phase separation effect,45 which occurs due to the interaction between the anionic polymer and the calcium cations, we recently demonstrated that the same principal could be applied to generate thin films and fibers of CaCO3 using poly(allyamine hydrochloride) (PAH) as an additive, where phase separation was now driven by the interaction between the positively charged amine groups and the anionic carbonate ions.46 Thus, PAH can have a significant effect on the precipitation of calcium carbonate, whereas most cationic small molecules are relatively ineffective in directing CaCO3 growth, with a number of exceptions.
In this article, we build on these initial results to further investigate the influence of PAH on calcium carbonate precipitation, comparing and contrasting with the effects seen for PAsp and PAA. The focus is placed on the effects of the solution conditions on thin film and fiber formation, and for the first time, extensive transmission electron microscopy (TEM) and electron diffraction (ED) were used to characterize the structures of the fibers, facilitated by their nanoscale widths. Our results show that PAH is particularly effective in promoting the growth of calcite fibers, which often reach lengths of over a couple of hundred micrometers, and that these fibers typically show single crystal type patterns under selected area electron diffraction (SAED). The TEM analysis, in turn, provided insight into the mechanisms of formation of the fibers, suggesting a particle-mediated process. Finally, the additional effects of magnesium ions on CaCO3 precipitation in the presence of PAH were also studied. As well as generating thinner and smoother CaCO3 films, addition of magnesium ions to the PAH/CaCO3 system results in extensive fiber formation at a significantly lower PAH concentration than in Mg2+-free solutions, although it happens at the expense of good single crystallinity.
Experimental Section
Calcium Carbonate Precipitation
Precipitation of calcium carbonate was carried out in the presence of poly(allylamine hydrochloride), [−CH2CH(CH2NH2·HCl)−]n (PAH) (Mw 15 KDa Aldrich) using the ammonium carbonate diffusion method47 under a wide range of polymer and calcium concentrations. Solutions were prepared as follows: PAH was added to a 10 mM or 1.5 mM CaCl2·2H2O solution to give concentrations of between 1 μg mL–1 and 2 mg mL–1, and 10 mL aliquots were placed in Petri dishes. Glass slides, which had been previously cleaned with Piranha solution (70 vol % sulphuric acid, 30 vol % hydrogen peroxide), were used as substrates and were placed upright in a Petri dish. The Petri dish was then covered with Parafilm which had been pierced four times with a needle, and it was placed in a sealed desiccator. A Petri dish containing 5 g of ammonium carbonate, covered with Parafilm that had been pierced four times with a needle, was also placed in the desiccator. Crystallization was then allowed to proceed over periods ranging from 3 h to 3 weeks. After crystallization, the glass slides were removed from the reaction solution, washed with ethanol, and blown dry with air. The influence of Mg2+ on the calcium carbonate precipitation was also investigated by adding a 150 mM solution of MgCl2·6H2O to the reaction solution to give a [Ca2+]:[Mg2+] ratio of 1:1, 1:3, and 1:5. Finally, control experiments were performed using identical procedures as described above, but now in the absence of PAH.
Characterization of the Calcium Carbonate Precipitates
The calcium carbonate crystals precipitated on the glass slides were investigated using a range of techniques including Raman microscopy, field emission gun scanning electron microscopy (FEG-SEM), transmission electron microscopy (TEM), and polarized optical microscopy. Optical microscopy was used to examine the morphologies of the precipitates, and observation between crossed polarizers provided information on their single crystal/polycrystalline/amorphous characters. To provide detailed morphological information, FEG-SEM was carried out using a LEO 1530 Gemini FEG-SEM operating at 3.00 kV with an in-lens detector. Samples were prepared by placing the slides on SEM stubs with adhesive carbon pads and then sputter-coating them with 10 nm Pt/Pd (80/20). Micro-Raman spectroscopy and electron diffraction allowed determination of the polymorphs of CaCO3 present. Raman measurements were performed using a Renishaw 2000 inVia-Raman microscope equipped with a 785 nm diode laser as excitation source. By focusing the laser onto the sample using a 50 (numerical aperture NA 0.75) objective, it was possible to determine the structure of individual particles. TEM was used to investigate the films and fibers at a spatial resolution of better than 2 Å, and samples were prepared by placing a carbon-coated, Formvar-covered Ni-grid in the reaction solution and removing it at different time points. Grids were then washed with ethanol and were left to dry. TEM was then performed using a 200 kV FEI Tecnai TF20 FEG-TEM and a 200 kV JEOL JEM2011 TEM. Diffraction patterns were obtained using selected area electron diffraction (SAED) and were analyzed using the JEMS software package.48 Kinematic simulation of electron diffraction using this software package provided information about relative lattice rotations as well as the position of the Laue circle, which visualizes the magnitude and direction of the lattice orientation changes. The angular spread of diffraction spots were determined using the angle tool in ImageJ software. The composition of the films and fibers were determined using atomic absorption spectroscopy (AAS) and thermogravimetric analysis (TGA), where crystals were removed from the glass substrate using a cover slide. Early stages of the reactions were investigated by isolating the precipitate formed in a 3 h reaction solution ([Ca2+] = 10 mM, [Mg2+] = 10 mM, [PAH] = 20 μg mL–1). The precipitate was subsequently washed with ethanol and reisolated by centrifugation followed by analysis with TGA and AAS. AAS was carried out using a Perkin-Elmer Atomic Absorption Spectrometer, and TGA was performed with a TA Instruments, SDT Q600 Simultaneous TGA/DSC operating with a 5 °C min–1 heating rate under air.
Results
Calcium carbonate was precipitated on exposure of a [Ca2+] = 10 mM solution, containing PAH at concentrations between 5 μg mL–1 and 2 mg mL–1, to ammonium carbonate vapor for 1 day. Although the precipitation of calcite rhombohedra in the presence of 5 μg mL–1 PAH indicated that the polymer had little effect at this concentration, an increase in the amount of PAH to 50 and 80 μg mL–1 led to the formation of calcite particles with rounded surfaces that were capped with small {104} faces (Figure 1a). Notably, unusual fibrous structures were also seen to originate from the surface of the calcite particles and propagate along the substrate (Figures 1a and b and Supporting Information Figure S1). These fibers often grew parallel to each other close to the parent crystal and then became highly convoluted as they extended further away; sudden changes of growth direction were also very common (Figure 1a). Examination at higher magnification using SEM suggested a nanoparticulate substructure and a few fibers were terminated with a bulge/bobble at their tip (arrowed, Figure 1b). TEM analysis of the wires also revealed a nanoparticulate substructure, and selected area diffraction of thinner fibers yielded single crystal type patterns (Figure 1c). No preferred growth direction was identified. Under these conditions, a small number of crystalline film domains were also observed (Supporting Information Figure S2a), which comprised a mixture of single crystalline calcite and polycrystalline vaterite as identified with Raman spectroscopy (Supporting Information Figure S2b).
Figure 1.
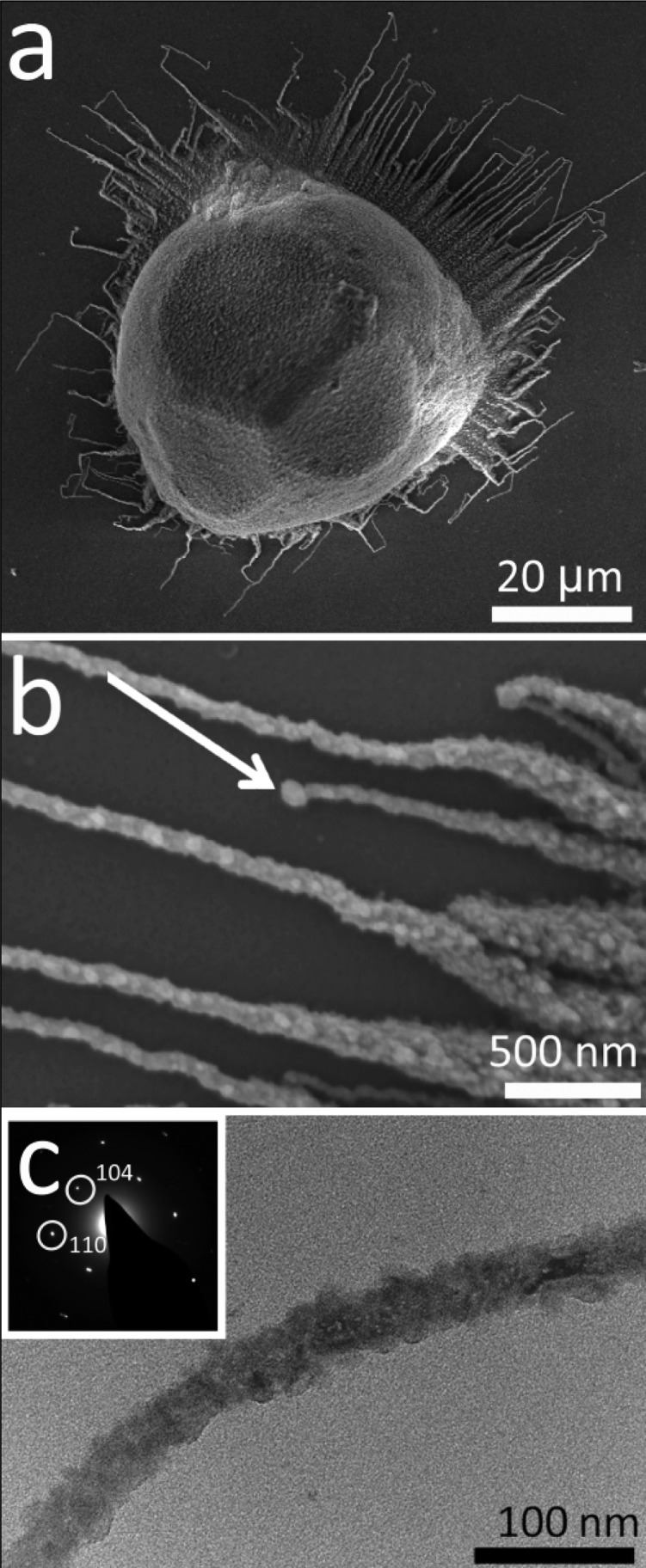
Calcium carbonate precipitated after 3 days from solutions of composition [Ca2+] = 10 mM and [PAH] = 80 μg mL–1. (a) Calcite rhombohedron with rounded faces and associated fibers. (b) A higher magnification image of fibers, where a “bobble” at the end of a fiber is arrowed. (c) TEM image and corresponding single crystal electron diffraction pattern of a calcite fiber.
On increasing the concentration of PAH to 200 μg mL–1, the rhombohedral shape of the particles was completely lost and only spherical particles from which fibers grew were observed (Supporting Information Figure S3). These fibers were much longer than those that formed at lower PAH concentrations, frequently resulting in lengths of 60 μm and aspect ratios of 200. A larger number of crystalline film domains were also obtained under these conditions. A further increase in the PAH concentration resulted in extensive crystalline thin film coverage of the glass substrate, and fibers were also occasionally observed but were often only a couple of micrometer long and associated with polycrystalline films.46 Finally, at concentrations of [PAH] = 2 mg mL–1, the polymer itself was deposited on the glass slide together with small CaCO3 crystals (Supporting Information Figure S4).
The influence of the Ca2+ concentration on calcium carbonate precipitation in the presence of PAH was also investigated. A reduction in the [Ca2+] to 1.5 mM ([PAH] = 0.5 mg mL–1) resulted in extensive fiber formation and virtually no film formation; only very small domains of crystalline films formed, which were always very rough and associated with fibers. The data, therefore, reveals an inverse relationship between the abundance of the thin films and fibers. Most of these fibers appeared to originate from a central core (Figure 2a) and achieved typical lengths and aspect ratios of 120 μm and 400, respectively, (Figure 2b inset). They varied significantly in diameter along their length, ranging from typical values of about 5 μm at their bases to a few nanometers at their tips. This was accompanied by a considerable variation in their morphologies, with both straight (Figure 2a) and bent and branched fibers being observed (Figure 2c). A number also abruptly changed direction, apparently at specific angles with respect to the long axis of the fiber (Figure 2d), and intergrowth was sometimes observed when they grew outward in close proximity to each other (Figure 2a inset and 2c). Subsequent variation in the PAH concentration at [Ca2+] = 1.5 mM revealed that fibers were present at a much wider range of polymer concentrations than were at higher calcium levels, such that distorted rhombohedra were formed at concentrations of [PAH] = 1 μg mL–1, whereas short fibers were first noted at [PAH] = 5 μg mL–1. Long fibers were generated at concentrations of [PAH] = 50 μg mL–1, and polymer-coated fibers were observed at high concentrations of [PAH] = 2 mg mL–1.
Figure 2.

SEM images of calcite fibers precipitated after 3 days from solutions containing [Ca2+] = 1.5 mM and [PAH] = 0.5 mg mL–1. (a) Fibers growing from a central core, where the inset shows two fibers merging and an internal structure based on nanosized particles. (b) Fibers with aspect ratios of up to 400, (c) a higher magnification image showing straight and convoluted fibers and the formation of branches (arrow) and (d) a fiber showing rapid changes in direction.
Though a number of articles have described the formation of CaCO3 fibers in the presence of polymers,32,41 the fibers formed have generally been too thick to enable detailed structural analysis by TEM. With their nanometer-scale thicknesses, the fibers formed in the presence of PAH are ideally suited to such a study. Fibers precipitated under the conditions [Ca2+] = 10 mM and [PAH] = 1 mg mL–1 were investigated using HR-TEM and selected area diffraction, where patterns were recorded along the fibers at separations of ≈100 nm. The majority of fibers show continuity in the crystal lattice and yield single crystal-type diffraction patterns (Figure 3a). No preferential crystallographic orientation was observed with respect to their long axes. Some areas are observed, however, where the fibers comprise well-defined 5–10 nm nanoparticles, and diffraction patterns indicate a number of crystal orientations (Figure 3b).
Figure 3.

TEM image and corresponding electron diffraction pattern (insets) of calcite fibers precipitated from reaction solutions containing (a) [Ca2+] = 10 mM and [PAH] = 1 mg mL–1 showing sets of lattice fringes (directions indicated by parallel lines) and (b) [Ca2+] = 10 mM and [PAH] = 1 mg mL–1 showing the nanoparticulate substructure.
A typical analysis of a bent fiber that changes its morphological orientation by more than 30° over a length of 0.6 μm is shown in Figure 4. The red (Laue) circles shown are the intersection of the Ewald sphere with the reciprocal lattice. If the lattice (crystal) is rotated off a low-index zone axis, the Laue circle is also shifted off-center, where the distance between the Laue circle center and the zero reflection is directly related to the tilt angle. The tilt angle for the sample can then be determined by comparison of the experimentally observed and simulated diffraction patterns, for which a crystal tilt is assumed. Interestingly, this analysis was used to demonstrate that the observed morphological distortion does not correspond to a distortion at the crystallographic level, with diffraction patterns recorded along the 0.6 μm length rotating by only 2.6° around an axis parallel to the electron beam. The changes in the intensity distribution in this set of diffraction patterns indicate lattice rotations around axes not parallel to the zone axis direction, although these are again small (≈5°, obtained from kinematic SAED pattern simulations) as compared to the physical change in orientation of the fiber. The angular spread of diffraction spots is found to be in the range of 8–20°, which is indicative of a high degree of alignment of the nanocrystalline domains over larger regions. However, dramatic changes of the nanocrystallite orientation are observed at some positions along fibers.
Figure 4.

Dark field TEM image and electron diffraction patterns (recorded at the indicated positions) of a calcite fiber precipitated from a solution of [Ca2+] = 10 mM and [PAH] = 1 mg mL–1. The red dots indicate simulated reflections belonging to the stated zone axis (ZA). The angles given in the diffraction patterns at positions 2, 3, and 4 refer to the amount of tilting of the lattice around an axis of rotation perpendicular to the electron beam. The magnitude and direction of the slight changes of the lattice orientation are illustrated by the red Laue circles.
A similar example of this behavior is shown by the fiber given in Supporting Information Figure S5, where the zone axis changes in orientation from close to [0 1 0] to [−2 8 −1] over a length of 300 nm, before switching to the original orientation again. This transition is related to a rotation around an axis parallel to the normal of the (−1 0 2) plane. The angle between these two zone axes is over 23°, showing that the crystal lattice changes its orientation dramatically. Interestingly, the growth direction of the fiber before and after the [−2 8 −1] section are identical, as evidenced by the absence of rotation in the diffraction pattern. Finally, a fiber was analyzed which showed sharp bends in its morphology (Figure 5). Here, the lattice is rotating; the (104) reflection and, thus, the orientation of the {104} planes remain unchanged while all other reflections change. Hence, the rotation is around an axis parallel to the normal of the (104) plane. Some change in the scattering intensity is observed between the points of rapid directional change, but these are much less than may be expected on the basis of the morphological distortion.
Figure 5.

Dark field TEM image and electron diffraction patterns (DPs, recorded at the indicated positions) of a calcite fiber precipitated from a solution of [Ca2+] = 10 mM and [PAH] = 1 mg mL–1. The red dots indicate simulated reflections belonging to the stated zone axis (ZA). The angles given in the DPs at positions 2–4 refer to the amount of rotation of the lattice around an axis of rotation perpendicular to the electron beam. The slight deviation of lattice orientation from the zone axis orientation is illustrated by the Laue circle in DP 4.
Further information on the mechanism of formation of the films and fibers was also obtained from time-dependent TEM studies, where Ni TEM grids immersed in the reaction solution were removed at intervals between 3 and 24 h. While small patches of amorphous film were identified at early reaction times, extensive coverage of the grid with an amorphous thin film occurred after approximately 8 h (Figure 6a). Sample extracted after 12 h, in contrast, had crystallized to calcite, and small protrusions were beginning to be observed (Figure 6b). With further incubation in solution to 14 h, the protrusions had evolved into small fibers (Figure 6c), demonstrating that fibers form after the films. Fully formed fibers were observed after 24 h (Figure 6d).
Figure 6.

TEM images and corresponding electron diffraction patterns of calcium carbonate films and fibers precipitated from a reaction solution containing [Ca2+] = 10 mM and [PAH] = 1 mg mL–1 after (a) 8 h, (b) 12 h, (c) 14 h, and (d) 24 h. The film was still amorphous after 8 h (a), whereas after 12 h, it had crystallized to calcite and small protrusions were observed (b). These protrusions developed further after 14 h (c) until fully developed calcite fibers are viewed after 24 h (d).
Previous studies with CaCO3 precipitation in the presence of poly(aspartic acid) (PAsp) and poly(acrylic acid) (PAA) have demonstrated that the formation of thin films (but not fibers) can be promoted through addition of Mg2+ ions to the reaction solution.49 Magnesium ions are a common component of biogenic ACC,50,51 and they also retard the crystallization of synthetic ACC such that thin films can form in the PAsp/CaCO3 and PAA/CaCO3 systems at lower polymer concentrations than in Mg2+-free solutions.49,52 Addition of Mg2+ to the PAH ([PAH] = 20 μg mL–1)/ CaCl2·2H2O reaction solution at a concentration of [Ca2+] = [Mg2+] = 10 mM led to the formation of ≈300 nm thick polycrystalline calcite films (Figure 7) that covered the entire glass substrate. No fibers were generated under these conditions. By comparison, [PAH] = 0.5–1.0 mg mL–1 was required to generate analogous films in the absence of magnesium. The films formed in the presence of Mg2+ were also much smoother and thinner than those formed with PAH alone46 (Figure 7b) and comprised only polycrystalline and amorphous domains; no single crystal domains were observed. Demonstrating the stabilizing effect of magnesium ions on ACC, these films were still only 30% crystalline after 3 days, as judged by examination with crossed polarizers in an optical microscope (Figure 7a) and had reached 80% crystallinity after 6 days.
Figure 7.

Polycrystalline calcite thin films precipitated after 3 days from a solution of composition [Ca2+] = 10 mM, [Mg2+] = 10 mM, and [PAH] = 20 μg mL–1. (a) An optical micrograph and (b) SEM image of a thin film deposited on a glass substrate, where the inset shows the film to be ≈300 nm thick. The films are only partially crystalline (≈30%) and show morphologically distinct areas (b).
Increase in the [Mg2+]:[Ca2+] ratio to 3–5 resulted in similar films being generated at PAH concentrations as low as 2 μg mL–1. The amorphous phase was also further stabilized under these conditions such that they were estimated to be ≈10% crystalline after 3 days under the conditions [Ca2+] = 10 mM, [Mg2+] = 30 mM, and [PAH] = 2 μg mL–1. The composition of the films was also investigated using atomic absorption spectroscopy (AAS) and thermogravimetric analysis (TGA). Data for a film grown under the conditions [Ca2+] = 10 mM, [Mg2+] = 10 mM, and [PAH] = 20 μg mL–1 showed that it comprised 2 wt % Mg2+ and 6.7 wt % of PAH after 3 h reaction time, whereas after 3 weeks, when the film was approximately 80% crystalline, the composition was 2 wt % Mg2+ and 3.3 wt % of PAH (Figure 8). Thus, the crystallization process was accompanied by preservation of the Mg2+ content but a loss of polymer from the film.
Figure 8.

Thermogravimetric analysis (TGA) data of precipitates formed in a solution of composition [Ca2+] = 10 mM, [Mg2+] = 10, mM and [PAH] = 20 μg mL–1 after (a) 3 h, when 6.7 wt % of PAH was present, and (b) 3 weeks, when 3.3 wt % of PAH was present.
Although no fibers were generated under these conditions, fiber formation was observed in the presence of Mg2+ when the metal ion concentrations were reduced to between [Ca2+] = [Mg2+] = 1.5–7.5 mM (Figure 9). The fibers formed under these conditions were again calcite and had similar morphologies to those formed in magnesium-free solutions. However, analysis by TEM and EDX showed that they now comprised both polycrystalline and single crystal domains and that they contained Mg2+ (Figure 9 and Supporting Information Figure S6).
Figure 9.

Images of CaCO3 thin films and fibers precipitated after 3 days from solutions of composition [Ca2+] = 1.5 mM, [Mg2+] = 1.5 mM, and [PAH] = 0.5 mg mL–1. (a) An optical image recorded under crossed polarisers and an SEM image (inset) of a calcite film from which fibers are growing. (b) A TEM image with corresponding electron diffraction pattern of a calcite fiber, and a corresponding EDX spectrum showing the presence of Mg2+ within the fibers.
Discussion
Our results show that although cationic at the pH values of the experiments, PAH has very similar effects on CaCO3 mineralization as the anionic PAA and PAsp, promoting the formation of thin films and fibers, although higher concentrations of PAH are typically required to give similar effects. In all cases, a phase separation occurs in the calcium carbonate growth solution driven by the association of these polymers with their oppositely charged counterions (Ca2+ for the anionic polymers and carbonate for the cationic PAH).45,46,53,54 Droplets rich in polymer, calcium, and carbonate ions therefore form, and subsequent calcium carbonate precipitation occurs at a rate dependent on the binding strength of the polymer and counterions.
A difference between these systems is, however, seen in the effect of Mg2+ ions. Though very smooth, polycrystalline calcite films are generated with both PAA/PAsp49 and PAH in the presence of Mg2+, compositional analysis showed that those formed with the PAH comprised only 2% Mg2+, whereas addition of PAA supported the formation of films containing up to 26% Mg2+. This is likely to derive from the fact that the anionic PAA/PAsp will bind strongly to Mg2+ ions, driving their incorporation in the PILP phase, whereas association of the cationic PAH with Mg2+ ions is weaker, only occurring due to carbonate ions present in the phase-separated material. Notably, fiber formation still occurs when Mg2+ ions are present in the PAH system, though no fibers form when magnesium ions are present in the PAA/PAsp system. The fibers formed in the former system are much more granular in structure and show more polycrystallinity than those in Mg2+-free solutions. This may derive from the well-known ability of Mg2+ ions to poison the growth of calcite crystallites, which then results in small crystal sizes.55,56
There has been much discussion about the mechanisms underlying organic additive-directed fiber-formation in inorganic systems. Microemulsions have proven extremely effective in promoting the growth of fibers,57−60 and a range of models have been proposed.61 It has been suggested that the assembly of crystalline precursor particles may be driven by preferential adsorption of the surfactant onto specific crystal faces,60 although nanowires can also form via the assembly of surfactant-coated amorphous particles. Considering the BaSO4 system, compelling early stage TEM analysis has shown that although precursor nanoparticles are present adjacent to all surfaces of the developing nanowires, propagation only occurs when the amorphous nanoparticles add to the ends.59 This provides clear evidence that a difference in the surfactant assembly on the sides as compared with the ends of the nanowires drives the directed assembly in this system.
A range of polymers have also been used to generate inorganic fibers, and again mechanisms based on the assembly of either crystalline or amorphous nanoparticles have been suggested.38,61 The formation of helical BaCO3 fibers was proposed to arise from the directed assembly of polymer stabilized crystalline precursor particles.31 Here, anionic block copolymers were proposed to preferentially adsorb onto specific crystal faces, and the subsequent electrostatic interaction between the nanoparticles causes 1D assembly. Looking at the BaSO4 system, a detailed investigation of fiber-formation in the presence of sodium polyacrylate demonstrated the presence of amorphous precursor nanoparticles.38,62 It has been suggested that these aggregate on the substrate prior to crystallizing. Subsequent growth then occurs by the directed assembly of crystalline nanoparticles, where this is driven by preferential adsorption of the polymer to specific faces only.
The mechanisms described above are based on the directed assembly of amorphous or crystalline nanoparticles. An alternative mechanism has also been proposed32 on the basis of the generation of PILP (polymer-induced liquid precursor) droplets during precipitation of CaCO3 in the presence of PAA or PAsp. Here, a SPS (solution precursor solid) mechanism was proposed, where this is analogous to the vapor–liquid–solid (VLS) and solution-liquid–solid (SLS) processes responsible for the catalytic formation of nanowires.63,64 VLS and SLS processes have been widely used to generate nanowires of semiconductors and carbon nanotubes, where these rely on delivery of the reagents from a fluid phase (gaseous, liquid, or supercritical). Catalytic particles play a key role in nanowire formation, reducing the activation energy for nucleation of the product material at the catalyst/nanowire interface, driving axial growth. Indeed SLS and VLS growth mechanisms are frequently identified by the presence of a catalytic particle at either the tip (float-growth mechanism) or base (root-growth mechanism) of the nanowire.63,64 In the SPS process, PILP droplets were suggested to provide a flux of reagents to the fiber, where in the absence of any additional catalytic particles, this process must be autocatalytic.
The absence of catalytic particles during CaCO3 nanowire formation casts some doubt on the proposed SPS mechanism. In subsequent work by the same authors investigating the formation of SrCO3 and BaCO3 fibers in the presence of PAA, a combination of aggregation-based and SPS mechanisms was proposed.33 There, PILP droplets coalesced on a substrate to form a thin film, and points of high energy were suggested to provide seeds for fiber growth. PILP droplets could then continue to preferentially adsorb at these sites, leading to one-dimensional growth. Under conditions where the tip remains as a liquid-like droplet, it could follow the SPS mechanism and lead to more homogeneous fibers, whereas rapid solidification of the tip could lead to fibers with more granular textures. Alternative studies have investigated fiber formation in the presence of PAA40 and a carboxylated block copolymer41 and also suggested that this proceeds via the attachment of amorphous/PILP precursor particles to developing fibers, although these did not invoke an SPS mechanism.
The results presented here and in our previous study with PAH show that amorphous/PILP particles coalesce on the substrate to form a thin film, which then crystallizes with time. Fibers grow from this film at later stages of the reaction, again fed by a supply of amorphous particles. As shown by our early stage studies (Figure 6), the fibers form at active sites on the substrate and initially grow as wide-base spines. This may be driven by the intrinsic concentration gradients generated around them, where the tip experiences a higher concentration of reagents than the base.65 The growth then continues to give high aspect ratio, narrow fibers. As the precursor particles are amorphous and, therefore, isotropic in morphology and physicochemical properties, simple aggregation would not be expected to lead to a high anisotropic morphology. Instead, the strong anisotropy of the fiber morphology must derive from the fiber itself, such that the precursor particles can combine with the fiber at its tip, but not at the sides. Such a mechanism was observed for the formation of BaSO4 nanowires in microemulsions59 and may derive from differences in the polymer coating between the walls and tip.
Our detailed electron microscopy studies also provide insight into the possible mechanisms of formation of the fibers. Fiber surfaces vary from rough to smooth, depending on their size and the reaction conditions, where the rough surfaces suggest a particle-mediated growth mechanism. Although fibers primarily diffract as single crystals (Figure 3a), detailed analysis using selected area electron diffraction reveals imperfections. Further, the platelet-type substructure observed by dark-field TEM (Figure 4) indicates a competition between crystal growth and defect formation. The dramatic bending and distortions exhibited by the calcite nanofibers studied here is also indicative of the presence of structural and surface defects, where these can not only affect crystal growth rates but also have a significant impact on the overall morphology of a nanostructure. Indeed, stacking faults were shown to be the primary source of the bending and buckling observed in nanowires of II–VI and GaN semiconductors,66−68 where in common with our calcite nanofibers, these morphological changes occur without a change in the orientation of the crystal lattice. The beam sensitivity of the calcite crystals and the overall convoluted structure of the fibers rendered it impossible to study the atomic structure of the observed defects. However, the diffraction pattern analyses clearly support the suggestion that the changes in fiber orientation are most likely due to the introduction of planar defects such as stacking faults.
Finally, some regions of polycrystallinity are often observed in the calcite nanofibers, where these frequently occur in combination with a well-defined nanoparticulate substructure (Figure 3b). At 5–10 nm in size, these calcite nanoparticles are far smaller than any that can be isolated from solution precipitation, suggesting that they may develop during crystallization of an amorphous phase. That the original orientation of a fiber is often regained after a zone of polycrystallinity also demonstrates transfer of structural information through this area. These variations in structure may occur due to local differences in the amounts of polymer extruded from the mineral during crystallization, which may even generate local areas of amorphous calcium carbonate that are too rich in polymer to crystallize.69
Conclusions
These experiments demonstrate that poly(allylamine hydrochloride) (PAH) is extremely effective in directing the formation of CaCO3 thin films and fibers, where the extent of fiber formation depends on the reaction conditions. That such dramatic changes in crystal morphologies and structures can be achieved with a positively charged additive runs counter to the wealth of literature that focuses on the use of negatively charged additives to control CaCO3 crystallization.1,46 However, it is stressed that the effect of PAH is due to a carbonate ion induced phase separation of PAH, rather than direct interaction with the growing crystal. Analysis of the structures of the fibers and their formation mechanisms suggests that they form by a particle-mediated aggregation mechanism, where amorphous, polymer-stabilized particles add to the tips of developing fibers and subsequently crystallize. The integration of new material uniquely at the fiber tips indicates a significant difference in structure between the fiber walls and tips and may derive from a polymer coating on the fiber walls. These suggestions are also supported by structural analyses of the fibers, which show that although these often exhibit highly convoluted morphologies, the physical distortions are not paralleled by changes in the lattice. Finally, we demonstrate that magnesium ions can be used to further tune the formation of CaCO3 in the presence of PAH, generating smoother and thinner thin films and fibers with a more granular and polycrystalline texture. In summary, although templating methods can be used to generate single crystal CaCO3 fibers70−72—such that the morphology and size is precisely defined by the template—the use of additives offers many advantages such as experimental simplicity and facile scale-up. The ultimate exploitation of these to control structure and morphology in a predictable fashion, therefore, requires greater understanding of the mechanisms by which additives such as PAH control crystal nucleation and growth.
Acknowledgments
This work was supported by an Engineering and Physical Sciences Research Council (EPSRC) Leadership Fellowship (F.C.M. and Y.-Y.K., EP/H005374/1) and an EPSRC Programme Grant (A.V., R.K., and F.C.M., EP/I001514/1) which funds the Materials Interface with Biology (MIB) consortium. H.L. is most grateful to the DAAD for funding his visit to the U. K.
Supporting Information Available
Further characterization of thin films and fibers. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Meldrum F. C.; Colfen H. Chem. Rev. 2008, 108, 4332–4432. [DOI] [PubMed] [Google Scholar]
- Song R. Q.; Colfen H. CrystEngComm 2011, 13, 1249–1276. [Google Scholar]
- Weiner S.; Addadi L. J. Mater. Chem. 1997, 7, 689–702. [Google Scholar]
- Sommerdijk N.; de With G. Chem. Rev. 2008, 108, 4499–4550. [DOI] [PubMed] [Google Scholar]
- Kim Y. Y.; Ganesan K.; Yang P. C.; Kulak A. N.; Borukhin S.; Pechook S.; Ribeiro L.; Kroger R.; Eichhorn S. J.; Armes S. P.; Pokroy B.; Meldrum F. C. Nat. Mater. 2011, 10, 890–896. [DOI] [PubMed] [Google Scholar]
- Piez K. A. Science 1961, 134, 841–&. [DOI] [PubMed] [Google Scholar]
- Simkiss K. Comp. Biochem. Phys. 1965, 16, 427–&. [DOI] [PubMed] [Google Scholar]
- Weiner S. Calcif. Tissue Int. 1979, 29, 163–167. [DOI] [PubMed] [Google Scholar]
- Samata T.; Hayashi N.; Kono M.; Hasegawa K.; Horita C.; Akera S. FEBS Lett. 1999, 462, 225–229. [DOI] [PubMed] [Google Scholar]
- Gotliv B. A.; Addadi L.; Weiner S. ChemBioChem 2003, 4, 522–529. [DOI] [PubMed] [Google Scholar]
- Michenfelder M.; Fu G.; Lawrence C.; Weaver J. C.; Wustman B. A.; Taranto L.; Evans J. S.; Morsel D. E. Biopolymers 2003, 70, 522–533. [DOI] [PubMed] [Google Scholar]
- Endo H.; Takagi Y.; Ozaki N.; Kogure T.; Watanabe T. Biochem. J. 2004, 384, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J. S. Chem. Rev. 2008, 108, 4455–4462. [DOI] [PubMed] [Google Scholar]
- Sato A.; Nagasaka S.; Furihata K.; Nagata S.; Arai I.; Saruwatari K.; Kogure T.; Sakuda S.; Nagasawa H. Nat. Chem. Biol. 2011, 7, 197–199. [DOI] [PubMed] [Google Scholar]
- Naka K.; Chujo Y. Chem. Mater. 2001, 13, 3245–3259. [Google Scholar]
- Heywood B. R.; Mann S. Adv. Mater. 1994, 6, 9–20. [Google Scholar]
- Loste E.; Diaz-Marti E.; Zarbakhsh A.; Meldrum F. C. Langmuir 2003, 19, 2830–2837. [Google Scholar]
- Aizenberg J.; Black A. J.; Whitesides G. H. J. Am. Chem. Soc. 1999, 121, 4500–4509. [Google Scholar]
- Travaille A. M.; Kaptijn L.; Verwer P.; Hulsken B.; Elemans J.; Nolte R. J. M.; van Kempen H. J. Am. Chem. Soc. 2003, 125, 11571–11577. [DOI] [PubMed] [Google Scholar]
- Didymus J. M.; Oliver P.; Mann S. J. Chem. Soc. Faraday Trans. 1993, 89(15), 2891–2900. [Google Scholar]
- Mukkamala B. S. Chem. Commun. 2004, 918–919. [Google Scholar]
- Volkmer D.; Fricke M.; Huber T.; Sewald N. Chem. Commun. 2004, 1872–1873. [DOI] [PubMed] [Google Scholar]
- Wang T. X.; Colfen H.; Antonietti M. J. Am. Chem. Soc. 2005, 127, 3246–3247. [DOI] [PubMed] [Google Scholar]
- Elhadj S.; Salter E. A.; Wierzbicki A.; De Yoreo J. J.; Han N.; Dove P. M. Cryst. Growth Des. 2006, 6, 197–201. [Google Scholar]
- Sugawara A.; Ishii T.; Kato T. Angew. Chem., Int. Ed. 2003, 42, 5299–5303. [DOI] [PubMed] [Google Scholar]
- Dmitrovic V.; Habraken G. J. M.; Hendrix M.; Habraken W.; Heise A.; de With G.; Sommerdijk N. Polymers 2012, 4, 1195–1210. [Google Scholar]
- Yu S. H.; Cölfen H. J. Mater. Chem. 2004, 14, 2124–2147. [Google Scholar]
- Kulak A. N.; Iddon P.; Li Y. T.; Armes S. P.; Colfen H.; Paris O.; Wilson R. M.; Meldrum F. C. J. Am. Chem. Soc. 2007, 129, 3729–3736. [DOI] [PubMed] [Google Scholar]
- Guo X. H.; Liu L.; Wang W.; Zhang J.; Wang Y. Y.; Yu S. H. CrystEngComm 2011, 13, 2054–2061. [Google Scholar]
- Terada T.; Yamabi S.; Imai H. J. Cryst. Growth 2003, 253, 435–444. [Google Scholar]
- Yu S. H.; Colfen H.; Tauer K.; Antonietti M. Nat. Mater. 2005, 4, 51–U5. [DOI] [PubMed] [Google Scholar]
- Olszta M. J.; Gajjeraman S.; Kaufman M.; Gower L. B. Chem. Mater. 2004, 16, 2355–2362. [Google Scholar]
- Homeijer S. J.; Barrett R. A.; Gower L. B. Cryst. Growth Des. 2010, 10, 1040–1052. [Google Scholar]
- Yu S. H.; Antonietti M.; Colfen H.; Hartmann J. Nano Lett. 2003, 3, 379–382. [Google Scholar]
- Balz M.; Therese H. A.; Li J. X.; Gutmann J. S.; Kappl M.; Nasdala L.; Hofmeister W.; Butt H. J.; Tremel W. Adv. Funct. Mater. 2005, 15, 683–688. [Google Scholar]
- Gower L. B. Chem. Rev. 2008, 108, 4551–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Ito T.; Yanagihara Y.; Oaki Y.; Nishimura T.; Kato T. CrystEngComm 2010, 12, 2021–2024. [Google Scholar]
- Wang T. X.; Reinecke A.; Colfen H. Langmuir 2006, 22, 8986–8994. [DOI] [PubMed] [Google Scholar]
- Zhu J. H.; Song J. M.; Yu S. H.; Zhang W. Q.; Shi J. X. CrystEngComm 2009, 11, 539–541. [Google Scholar]
- Long X.; Ma Y.; Cho K.-R.; Li D.; De Yoreo J. J.; Qi L. Cryst. Growth Des. 2013, dx.doi.org/10.1021/cg4010399. [Google Scholar]
- Kim Y. Y.; Kulak A. N.; Li Y. T.; Batten T.; Kuball M.; Armes S. P.; Meldrum F. C. J. Mater. Chem. 2009, 19, 387–398. [Google Scholar]
- Schenk A. S.; Zope H.; Kim Y. Y.; Kros A.; Sommerdijk N.; Meldrum F. C. Faraday Discuss. 2012, 159, 327–344. [Google Scholar]
- Moore L.; Hopwood J. D.; Davey R. J. J. Cryst. Growth 2004, 261, 93–98. [Google Scholar]
- Gower L. B.; Odom D. J. J. Cryst. Growth 2000, 210, 719–734. [Google Scholar]
- Bewernitz M. A.; Gebauer D.; Long J.; Colfen H.; Gower L. B. Faraday Discuss. 2012, 159, 291–312. [Google Scholar]
- Cantaert B.; Kim Y. Y.; Ludwig H.; Nudelman F.; Sommerdijk N.; Meldrum F. C. Adv. Funct. Mater. 2012, 22, 907–915. [Google Scholar]
- Ihli J.; Bots P.; Kulak A.; Benning L. G.; Meldrum F. C. Adv. Funct. Mater. 2013, 23, 1965–1973. [Google Scholar]
- Stadelmann P.Electron Microscopy Software - Java Version (JEMS); CIME—EPFL: Lausanne, Switzerland, 1999–2011. [Google Scholar]
- Cheng X. G.; Varona P. L.; Olszta M. J.; Gower L. B. J. Cryst. Growth 2007, 307, 395–404. [Google Scholar]
- Raz S.; Weiner S.; Addadi L. Adv. Mater. 2000, 12, 38–+. [Google Scholar]
- Loste E.; Wilson R. M.; Seshadri R.; Meldrum F. C. J. Cryst. Growth 2003, 254, 206–218. [Google Scholar]
- DiMasi E.; Kwak S. Y.; Amos F. F.; Olszta M. J.; Lush D.; Gower L. B. Phys. Rev. Lett. 2006, 97, 4. [DOI] [PubMed] [Google Scholar]
- Axelos M. A. V.; Mestdagh M. M.; Francois J. Macromolecules 1994, 27, 6594–6602. [Google Scholar]
- Brunner E.; Lutz K.; Sumper M. Phys. Chem. Chem. Phys. 2004, 6, 854–857. [DOI] [PubMed] [Google Scholar]
- Lippman F. Sedimentary carbonate minerals. 1973. [Google Scholar]
- Ihli J.; Kim Y. Y.; Noel E. H.; Meldrum F. C. Adv. Funct. Mater. 2013, 23, 1575–1585. [Google Scholar]
- Kuang D. B.; Xu A. W.; Fang Y. P.; Ou H. D.; Liu H. Q. J. Cryst. Growth 2002, 244, 379–383. [Google Scholar]
- Rees G. D.; Evans-Gowing R.; Hammond S. J.; Robinson B. H. Langmuir 1999, 15, 1993–2002. [Google Scholar]
- Li M.; Mann S. Langmuir 2000, 16, 7088–7094. [Google Scholar]
- Li M.; Schnablegger H.; Mann S. Nature 1999, 402, 393–395. [Google Scholar]
- Colfen H.; Mann S. Angew. Chem., Int. Ed. 2003, 42, 2350–2365. [DOI] [PubMed] [Google Scholar]
- Wang T. X.; Colfen H. Langmuir 2006, 22, 8975–8985. [DOI] [PubMed] [Google Scholar]
- Kolasinski K. Curr. Opin. Solid State Mater. Sci. 2006, 10, 182–191. [Google Scholar]
- Wang F. D.; Dong A. G.; Sun J. W.; Tang R.; Yu H.; Buhro W. E. Inorg. Chem. 2006, 45, 7511–7521. [DOI] [PubMed] [Google Scholar]
- Miura T.; Kotachi A.; Oaki Y.; Imai H. Cryst. Growth Des. 2006, 6, 612–615. [Google Scholar]
- Hughes S. M.; Alivisatos A. P. Nano Lett. 2013, 13, 106–110. [DOI] [PubMed] [Google Scholar]
- Selvig E.; Hadzialic S.; Skauli T.; Steen H.; Hansen V.; Trosdahl-Iversen L.; van Rheenen A. D.; Lorentzen T.; Haakenaasen R. Phys. Scr. 2006, T126, 115–120. [Google Scholar]
- Cherns D.; Meshi L.; Griffiths I.; Khongphetsak S.; Novikov S. V.; Farley N. R. S.; Campion R. O.; Foxon C. T. Appl. Phys. Lett. 2008, 93, 111911. [Google Scholar]
- Seto J.; Ma Y. R.; Davis S. A.; Meldrum F.; Gourrier A.; Kim Y. Y.; Schilde U.; Sztucki M.; Burghammer M.; Maltsev S.; Jager C.; Colfen H. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 3699–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loste E.; Meldrum F. C. Chem. Commun. 2001, 901–902. [Google Scholar]
- Loste E.; Park R. J.; Warren J.; Meldrum F. C. Adv. Func. Mater. 2004, 14, 1211–1220. [Google Scholar]
- Kim Y. Y.; Hetherington N. B. J.; Noel E. H.; Kroger R.; Charnock J. M.; Christenson H. K.; Meldrum F. C. Angew. Chem., Int. Ed. 2011, 50, 12572–12577. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.