

Abstract
Abstract The cerebellum plays crucial roles in controlling sensorimotor functions. The neural output from the cerebellar cortex is transmitted solely by Purkinje cells (PCs), whose impairment causes cerebellar ataxia. Spinocerebellar ataxia type 13 (SCA13) is an autosomal dominant disease, and SCA13 patients exhibit cerebellar atrophy and cerebellar symptoms. Recent studies have shown that missense mutations in the voltage-gated K+ channel Kv3.3 are responsible for SCA13. In the rodent brain, Kv3.3 mRNAs are expressed most strongly in PCs, suggesting that the mutations severely affect PCs in SCA13 patients. Nevertheless, how these mutations affect the function of Kv3.3 in PCs and, consequently, the morphology and neuronal excitability of PCs remains unclear. To address these questions, we used lentiviral vectors to express mutant mouse Kv3.3 (mKv3.3) channels harbouring an R424H missense mutation, which corresponds to the R423H mutation in the Kv3.3 channels of SCA13 patients, in mouse cerebellar cultures. The R424H mutant-expressing PCs showed decreased outward current density, broadened action potentials and elevated basal [Ca2+]i compared with PCs expressing wild-type mKv3.3 subunits or those expressing green fluorescent protein alone. Moreover, expression of R424H mutant subunits induced impaired dendrite development and cell death selectively in PCs, both of which were rescued by blocking P/Q-type Ca2+ channels in the culture conditions. We therefore concluded that expression of R424H mutant subunits in PCs markedly affects the function of endogenous Kv3 channels, neuronal excitability and, eventually, basal [Ca2+]i, leading to cell death. These results suggest that PCs in SCA13 patients also exhibit similar defects in PC excitability and induced cell death, which may explain the pathology of SCA13.
Key points.
The cerebellum plays crucial roles in controlling sensorimotor functions, and patients with spinocerebellar ataxia type 13 exhibit cerebellar atrophy and cerebellar symptoms.
The disease is an autosomal dominant disorder caused by missense mutations in the voltage-gated K+ channel Kv3.3, which is expressed intensely in the cerebellar Purkinje cells, the sole output neurons from the cerebellar cortex.
Here, we examined how these mutations cause the cerebellar disease by lentiviral expression of the mutant Kv3.3 in mouse cultured Purkinje cells.
Expression of the mutant Kv3.3 suppressed outward currents, broadened action potentials and elevated basal intracellular calcium concentration in Purkinje cells. Moreover, the mutant-expressing Purkinje cells showed impaired dendrites and extensive cell death, both of which were significantly rescued by blockade of P/Q-type Ca2+ channels.
These results suggest that Purkinje cells in the patients also exhibit similar abnormalities, which may account for the pathology of the disease.
Introduction
In most excitable cells, the high K+ permeability arises from delayed-rectifier K+ channels of the Kv class (Hille, 2001). One of the Kv subfamilies, known as Kv3, has generated particular interest because of its unique electrophysiological properties (Rudy & McBain, 2001). The Kv3 channels are high-voltage-activated K+ channels, and they exhibit fast activation and deactivation kinetics; therefore, Kv3 channels are activated during action potential depolarization and are indispensable for high-frequency firing in many neurons, such as fast-spiking cortical interneurons and cerebellar Purkinje cells (PCs; Erisir et al. 1999; McKay & Turner, 2004). Rodents and humans possess four Kv3 genes: Kv3.1–3.4. The Kv3 channels are composed of four pore-forming subunits and form heterotetrameric channels by combination of Kv3 members. In the rodent brain, Kv3.3 mRNA and protein are abundantly expressed in the cerebellum, in which the mRNA is most intensely expressed in PCs (Weiser et al. 1994; Chang et al. 2007). Interestingly, Kv3.3-deficient mice show normal PC morphology and no ataxic phenotype (Joho et al. 2006; Hurlock et al. 2008; Zagha et al. 2010).
The numerous diseases arising from channel dysfunction (channelopathies) illustrate the importance of ion channels to the organism. To date, missense mutations in more than 60 ion-channel genes have been associated with human disease (Ashcroft, 2006). Recently, missense mutations in the KV3.3 gene (also known as KCNC3), which encodes human Kv3.3 (hKv3.3) channels, were linked to autosomal dominant spinocerebellar ataxia type 13 (SCA13). Spinocerebellar ataxia type 13 is accompanied by cerebellar symptoms and by cerebellar atrophy, and three different mutations (R420H, R423H and F448L) have been identified, although the neurodegenerative changes in the post-mortem cerebellum have not been investigated (Waters et al. 2006; Figueroa et al. 2010). In Xenopus oocyte expression systems, coexpression of the R420H or R423H mutant subunits with wild-type (WT) hKv3.3 suppresses the current by a dominant-negative mechanism. Given the intense Kv3.3 expression in rodent PCs and the cerebellar atrophy in SCA13 patients, these mutations are expected to affect the neuronal excitability and morphology of PCs severely. Recently, Issa et al. reported that zebrafish expressing mutant zebrafish Kv3.3 subunits (homologous to the F448L mutant) in spinal motoneurons, which endogenously express Kv3.3, show defective axonal pathfinding (Issa et al., 2012). However, as they used a motoneuron-specific enhancer to drive expression, these zebrafish display no distinct cerebellar abnormality. Therefore, the pathology of SCA13 has not been elucidated, and other methodological approaches are needed.
In the present study, to investigate the effects of Kv3.3 mutations in PCs, we expressed mouse Kv3.3 (mKv3.3) channels harbouring the R424H missense mutation, which corresponds to the R423H mutation in hKv3.3, using a lentivirus system in mouse cerebellar cultures. Immunohistochemical analysis revealed that expression of R424H mutant subunits induced impaired dendrite development and cell death in PCs by 11 days in vitro (DIV) without significant alteration in granule cells. To examine the effects of R424H mutant subunits on the electrophysiological properties and free [Ca2+]i of PCs, we performed whole-cell patch-clamp recordings and calcium imaging from PCs at DIV 8–10. Action potential duration and basal [Ca2+]i were significantly increased in R424H mutant-expressing PCs compared with PCs expressing WT mKv3.3 or those expressing green fluoresent protein (GFP) alone. Furthermore, blockade of P/Q-type Ca2+ channels by ω-agatoxin IVA in the culture conditions rescued the dendritic maldevelopment and cell death in PCs caused by R424H mutant subunits.
Methods
Ethical approval
Newborn (i.e. within 24 h after birth) mice (ICR strain) of both sexes were used for cerebellar cultures. Xenopus oocytes were collected from anaesthetized Xenopus laevis. These animals were used according to the Guiding Principles for the Care and Use of Laboratory Animals approved by the guidelines of the National Institute of Health Sciences, Japan. All experiments also complied with The Journal of Physiology policy and UK regulations on animal studies (Drummond, 2009).
Molecular biology
The mouse cerebellum expresses the Kv3.3b gene, which is an alternatively spliced isoform of mKv3.3 (Goldman-Wohl et al. 1994). mKv3.3 cDNA that is nearly identical to Kv3.3b (Desai et al. 2008) was obtained as a kind gift from Dr Leonard K. Kaczmarek (Yale University, New Haven, CT, USA). The amino acid identity between hKv3.3 (Gene Accession number: AF055989) and mKv3.3 is 89%, and the sequence of the S4 transmembrane segment showed a complete match between the two species (Supplemental Fig. S1). To date, several reports have shown that there are three types of missense mutations (R420H, R423H and F448L) in hKv3.3 channels in distinct SCA13 pedigrees (Waters et al. 2006; Figueroa et al. 2010, 2011). In the present study, we focused on the R423H mutation. The S4 transmembrane segment of mKv3.3 has an arginine residue at position 424, which corresponds to the arginine at position 423 in hKv3.3. The arginine of mKv3.3 was replaced with histidine by overlap PCR using PrimeSTAR HS DNA Polymerase (Takara Bio, Shiga, Japan). The nucleotide exchanges were c.1271G>A and c.1272T>C. Wild-type and R424H mutant mKv3.3 cDNA were subcloned in pcDNA3 (Invitrogen, Carlsbad, CA, USA), and the mutation was confirmed by sequencing.
For lentiviral vector-based gene expression, the murine embryonic stem cell virus (MSCV) promoter, which drives PC-predominant expression of a transgene in cerebellum, was used (Hawley et al. 1994; Hanawa et al. 2004; Torashima et al. 2006; Takayama et al. 2008). A Kozak translation initiation sequence and 2A peptide sequence from porcine teschovirus-1 (P2A) for efficient cleavage of polyproteins were placed at the N- and C-termini of WT and R424H mutant mKv3.3 cDNA, respectively (Szymczak et al. 2004; Torashima et al. 2009). Then, stop codons were removed using the following PCR primer pair: a 5′ primer with an AgeI site and Kozak sequence (bold; 5′ primer, CGACCGGTGCCACCATGCTCAGTTCAGTGTGCGT) and a 3′ primer with an AgeI site and P2A (bold; 3′ primer, CGACCGGTGGCCCGGGGTTTTCTTCAACATCTCCTGCTTGCTTTAACAGAGAGAAGTTCGTGGCGCCGGAGCCGAGGATGGAGGGCAGGGTCG). A Gly-Ser-Gly linker (underlined in the 3′ primer) was also placed between the N-terminus of a WT (or R424H mutant) mKv3.3 sequence and P2A using the 3′ primer. This linker improves the cleavage efficacy of the P2A (Szymczak et al. 2004). The PCR products were subcloned in-frame into the AgeI site of pCL20c MSCV-GFP, which is present 8 bp upstream from the translation initiation site of GFP. Finally, lentiviral transfer vectors for the experiments (pCL20c MSCV-WT-P2A-GFP and pCL20c MSCV-R424H-P2A-GFP) were obtained, and the inserted sequences were verified by sequencing.
Lentiviral vector preparation
Vesicular stomatitis virus G protein (VSV-G) pseudotyped lentiviral vector particles were produced by transient transfection of HEK 293T cells with viral plasmids as described previously (Torashima et al. 2006). In brief, HEK 293T cells were transfected with a mixture of the following four plasmids: pCAGkGP1R, pCAG4RTR2, pCAG-VSV-G and the lentiviral transfer vector plasmid (pCL20c MSCV-GFP, pCL20c MSCV-WT-P2A-GFP or pCL20c MSCV-R424H-P2A-GFP). The medium containing viral particles was concentrated by ultracentrifugation and resuspended in 70 μl of Dulbecco’s phosphate-buffered saline (Wako Pure Chemical Industries, Osaka, Japan). The infectious titres of the virus were determined as follows: virus stocks were added to HEK 293T cells in the presence of Polybrene (6 μg ml−1; Sigma-Aldrich, St Louis, MO, USA). After 4 days, GFP-positive cells were counted using a Tali Image-Based Cytometer (Invitrogen), and the titres were adjusted to 1.0 × 1010 or 0.5 × 1010 transduction units/ml.
Cerebellar culture and lentivirus-mediated gene expression
Cerebellar cultures were prepared according to our previous protocol with some modifications (Hirai & Launey, 2000). After the mice were killed by decapitation, the cerebella of newborn mice were quickly removed and treated with 2 ml of a papain digestion solution containing 40 units of papain (Worthington Biochemical, Lakewood, NJ, USA), 2 mm l-cysteine hydrochloride and 1 mm EDTA in Ca2+–Mg2+-free Hank’s balanced salt solution (pH 7.0; Gibco, Grand Island, NY, USA; Tabata et al. 2000). Then, the cerebella were dissociated by trituration in Hank’s balanced salt solution containing 0.05% (w/v) DNase (Sigma-Aldrich) and 12 mm MgSO4. After centrifugation (180 ×g, 5 min), the cells were resuspended in DMEM/F12 (Gibco)-based medium containing 4.2 mm KCl, 1% (v/v) horse serum, 2% (v/v) B-27 (Gibco) and a mixture of a penicillin–streptomycin solution (1000 U ml−1 and 100 mg ml−1, respectively; Gibco) to a density of 10 × 106 cells ml−1 (Gimenez-Cassina et al. 2007). High (25 mm)-KCl-containing medium, which is often used for rat cerebellar cultures, was not used in this study because low (5 mm)-KCl-containing medium can improve the long-term viability of mouse cerebellar granule cells, and high-KCl medium maintains the gene expression patterns of granule cells in an immature condition (Mellor et al. 1998; Sato et al. 2005). A mixture of the cell suspension (20 μl) and the concentrated virus solution (1 μl) was plated onto plastic coverslips (Cell Desk LF1, Sumilon MS-92132; Sumitomo Bakelite, Tokyo, Japan) coated with poly-d-lysine (Sigma-Aldrich) and incubated for 10 h in a CO2 incubator for virus infection. The DMEM/F12-based medium (700 μl) was added to each dish and replaced by half once a week. Green fluorescent protein fluorescence was first observed at DIV 3, and its expression continued thereafter. In some experiments, ω-agatoxin IVA (0.2 μm; Peptide Institute, Tokyo, Japan), a P/Q-type Ca2+ channel blocker, was added to the culture medium every other day from DIV 2 (Mintz & Bean, 1993; Mikuni et al. 2013).
Histochemical examination
Cerebellar cultures were fixed in 4% (w/v) formaldehyde in PBS (pH 7.4) for 30 min at room temperature (RT) and incubated overnight at 4°C in PBS containing 0.3% Triton X-100, 0.12%λ-carrageenan, 1% goat serum and 0.02% sodium azide (PBS-XCG) with the following combination of primary antibodies: guinea-pig polyclonal anti-GFP antibody (1:1000 dilution, GFP-GP-Af1180-1; Frontier Institute, Hokkaido, Japan), rabbit polyclonal anti-calbindin D-28K antibody (1:2000 dilution, AB1778; Millipore, Billerica, MA, USA; calbindin is a marker protein of PCs) and mouse monoclonal anti-NeuN antibody (1:2000 dilution, MAB377; Millipore) in PBS-XCG. The samples were further incubated for 3 h at RT in PBS-XCG with the following secondary antibodies: AlexaFluor (AF) 488-conjugated goat anti-guinea-pig IgG antibody (A-11073; Invitrogen), AF 568-conjugated goat anti-rabbit IgG antibody (A-11011; Invitrogen) and AF 680-conjugated goat anti-mouse IgG antibody (A-21058; Invitrogen) in PBS-XCG. All secondary antibodies were used at a concentration of 5 μg ml−1. For examination of nuclear morphology, some cerebellar cultures were stained with Hoechst 33342 (1 μg ml−1 in PBS; Dojindo, Kumamoto, Japan) for 15 min at RT after the secondary antibody treatment.
Immunofluorescence was observed under a confocal microscope (A1R; Nikon, Tokyo, Japan) with the following appropriate filter sets: Hoechst 33342 (excitation, 403 nm; emission, 425–475 nm), AF 488 (excitation, 488 nm; emission, 500–550 nm), AF 568 (excitation, 561 nm; emission, 570–620 nm) and AF 680 (excitation, 639 nm; emission, 662–737 nm). Images were obtained with ×10 objective or ×60 water-immersion objective lenses. The confocal pinhole size was 1.0 airy unit. In some experiments, dendrites of PCs were traced from calbindin-positive areas using Neurolucida software (MBF Bioscience, Burlington, VT, USA). The extension of dendritic trees and dendrite complexity were measured by Sholl analysis in Neuroexplorer software (MBF Bioscience; Sholl, 1953; Sawada et al. 2010). Concentric spheres were centred on the cell body, and the radii were incremented by 10 μm. The number of branching points within each sphere was counted, and total dendritic length was measured. The cell densities of PCs and granule cells were calculated in each culture by averaging four values of cell density measured from a single image obtained around the centre of the culture with a ×10 objective lens. These measurements were performed using NIS-Elements AR 3.2 software (Nikon).
Data are provided as the means ± SD, and n is the number of experiments. Statistical significance was tested using the Mann–Whitney U test unless otherwise stated (significance, P < 0.05). GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA) and StatView 5 software (SAS Institute, Cary, NC, USA) were used for the analysis. In Figs 234 and 678 and Tables 1 and 2, statistical analysis was performed between cells expressing the R424H mutant and those expressing GFP alone or between cells expressing the R424H mutant and those expressing WT mKv3.3.
Figure 2. Lentivirus-mediated expression of R424H mutant subunits in cerebellar cultures decreases the density of Purkinje cells (PCs) but not of granule cells.

A–C, immunofluorescence images of cerebellar cultures infected with lentiviral vectors expressing green fluorescent protein (GFP) alone (Aa–c), WT subunits and GFP (Ba–c) or R424H mutant subunits and GFP (Ca–c; see Methods). Green fluorescent protein fluorescence was enhanced by immunostaining with guinea-pig anti-GFP antibody and AlexaFluor (AF) 488-conjugated goat anti-guinea-pig antibody. Purkinje cells were visualized by immunolabelling with rabbit anti-calbindin antibody (red signals in A–C). Ac′, Bc′ and Cc′, granule cells were selectively immunolabelled with mouse anti-NeuN mouse antibody (see Supplemental Fig. S3A and B). D, relative cell density of PCs plotted as a function of days in vitro (DIV). The density was normalized to the value of PCs expressing GFP alone at DIV 4. E, relative cell density of granule cells at DIV 14. The density was normalized to the mean cell density of granule cells in cultures lentivirally expressing GFP alone at DIV 14. F and G, R424H mutant-expressing PCs exhibiting chromatin condensation. F, representative fluorescence images of PCs at DIV 8. For nucleus detection, the PCs were stained with Hoechst 33342. Normal nuclei of PCs are indicated by arrows (GFP and WT), whereas a nucleus exhibiting chromatin condensation is marked with an arrowhead (R424H). G, summary of the percentages of PCs with chromatin condensation at DIV 8. The statistical analysis was conducted using Student’s unpaired t tests. In the following figures and tables, the statistical analysis was conducted between cells expressing the R424H mutant and those expressing GFP or between cells expressing R424H mutant and those expressing WT subunits. **P < 0.01 and ***P < 0.001.
Figure 3. R424H mutant-expressing PCs exhibit impaired dendritic development.
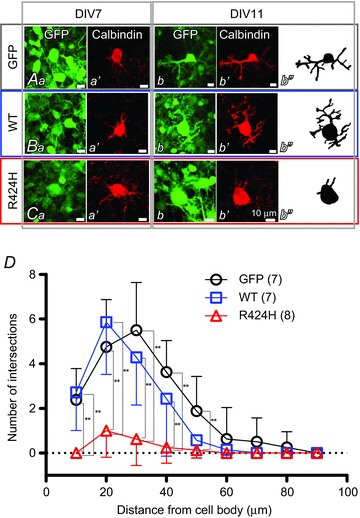
A–C, immunofluorescence images of PCs expressing GFP alone (Aa–b′), WT subunits and GFP (Ba–b′) and R424H mutant subunits and GFP (Ca–b′). Ab′′, Bb′′ and Cb′′, morphology of PCs expressing GFP (Ab′′), WT subunits and GFP (Bb′′) and R424H mutant subunits and GFP (Cb′′) are depicted for clarity. Each PC was traced using Neurolucida software. D, summary of dendrite complexity measured by Sholl analysis. Concentric spheres were centred on the cell body, and the radii were incremented by 10 μm. The number of branching points within each sphere was plotted as dendrite complexity (i.e. the number of intersections). **P < 0.01.
Figure 4. R424H mutant-expressing PCs exhibit suppressed peak outward current density.
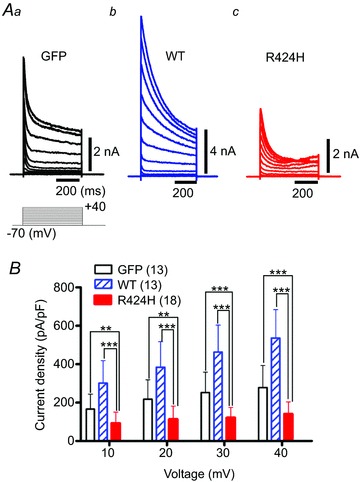
A, representative outward current traces recorded from PCs expressing GFP alone (Aa), WT subunits (Ab; note the vertical scale bar) and R424H mutant subunits (Ac). The currents were evoked by voltage steps from the −70 mV holding potential to voltages ranging from −60 to +40 mV in 10 mV increments. Leak currents were subtracted online by the P/4 protocol. The currents were recorded at DIV 8–10 in Hepes-buffered srtificial cerebrospinal fluid (ACSF) containing TTX, CdCl2, picrotoxin and 6,7-dinitroquinoxaline-2,3-dione (DNQX). B, summary of the peak outward current density, which was calculated by dividing the peak outward current by membrane capacitance. **P < 0.01 and ***P < 0.001.
Figure 6. R424H mutant-expressing PCs exhibit broadened action potential waveforms.
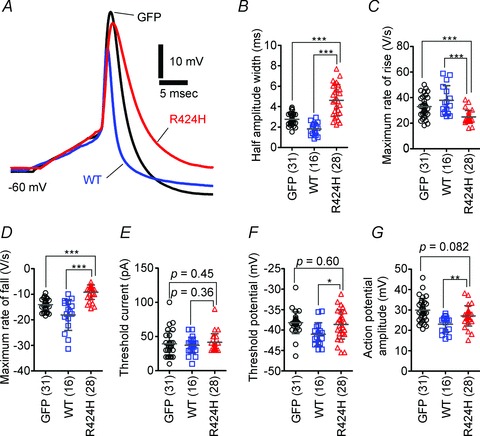
A, representative single action potential waveforms of PCs. The action potentials were evoked by short depolarizing current injection (10 ms duration). Action potentials were aligned by superimposing the rising phase of each trace. The resting membrane potentials were adjusted to −60 mV by current injection. B–G, comparison of action potential properties. B, half-amplitude width measured at the mid-point between the threshold and peak of the action potential. C and D, maximal rate of rise (C) and of fall (D) of action potentials. E, threshold current amplitude. F, threshold potential. G, action potential amplitude, as measured between the threshold and peak. *P < 0.05, **P < 0.01 and ***P < 0.001.
Figure 7. R424H mutant-expressing PCs exhibit altered firing patterns.

A–D, representative firing patterns of PCs expressing GFP alone (A), WT subunits (B) or R424H mutant subunits (C and D). Approximately half of the R424H mutant-expressing PCs fired at the onset of current injection (onset-type; C). The action potentials were evoked by long depolarizing current injection (200 ms duration). The resting membrane potentials were adjusted to −60 mV. E, comparison of firing patterns. The χ2 test was used for the statistical analyses. F, the firing frequency of the tonic-type cells is plotted as a function of injected current. *P < 0.05 and ***P < 0.001.
Figure 8. Significantly higher basal [Ca2+]i in PCs expressing R424H mutant.
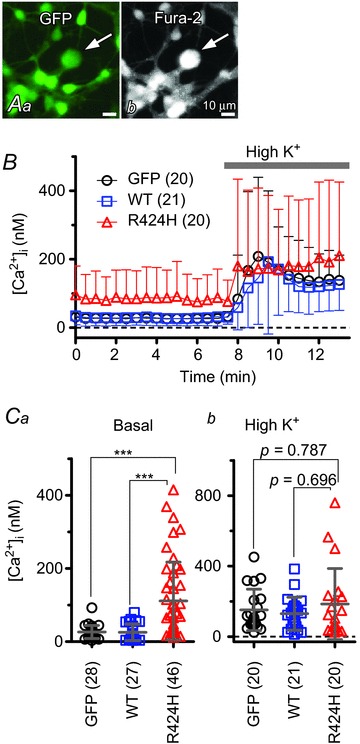
A, representative fluorescence images of a fura-2 AM-loaded cerebellar culture expressing GFP alone. Arrows indicate PCs. Aa, GFP fluorescence (excitation, 470–495 nm; emission, 510–550 nm). Ab, fura-2 fluorescence (excitation, 375–385 nm; emission, 470–550 nm). B, the time course of free [Ca2+]i in PCs. To depolarize PCs, high-K+ ACSF (High K+) was bath applied during the time indicated by the grey bar. C, summary of averaged [Ca2+]i in PCs. Basal [Ca2+]i was obtained as the average of a 7 min period from the beginning of the recordings (Ca), and elevated [Ca2+]i from a 5 min period during high-K+ ACSF perfusion (Cb). ***P < 0.001.
Table 1.
Basic electrophysiological properties of Purkinje cells
| Membrane capacitance (pF)a,d | Resting membrane potential (mV)a | Input resistance (MΩ)a,d | Percentage of cells showing spontaneous action potentialsb,e | Frequency of spontaneous action potentials (Hz)c,e | |
|---|---|---|---|---|---|
| GFP alone | 39.2 ± 24.3 | −58.3 ± 13.6 | 379 ± 278 | 80% | 0.43 ± 1.00 |
| (n= 35) | (n= 35) | (n= 35) | (12 of 15 cells) | (n= 15) | |
| WT mKv3.3 | 40.1 ± 16.1 | −59.9 ± 8.24 | 367 ± 181 | 75% | 0.25 ± 0.59 |
| (n= 17) | (n= 17) | (n= 17) | (6 of 8 cells) | (n= 8) | |
| R424H mutant | 22.3 ± 6.9*† | −59.5 ± 7.4 | 372 ± 169 | 64% | 0.12 ± 0.75 |
| (n= 28) | (n= 28) | (n= 28) | (9 of 14 cells) | (n= 14) |
Here and in Table 2, statistical analysis was conducted between cells expressing the R424H mutant and those expressing green fluorescent protein (GFP) alone or between cells expressing R424H mutant and those expressing wild-type (WT) mKv3.3. Data are given as the means ± SD, and n is the number of experiments. *P < 0.001 between GFP alone and R424H mutant. †P < 0.001 between WT mKv3.3 and R424H mutant. The statistical analysis indicated by superscript letters a, b and c was conducted using Student’s unpaired t test, the χ2 test and Mann–Whitney U test, respectively. dMembrane capacitance and input resistance were measured in voltage-clamp conditions. eSpontaneous action potentials were recorded at resting membrane potential for 300 s.
Table 2.
Summary of spontaneous excitatory postsynaptic current (sEPSC) properties in Purkinje cells
| Percentage of cells showing sEPSCsa | Amplitude (pA)b | Frequency (Hz)b | 10–90% Rise time (ms)b | Decay time constant (ms)c | |
|---|---|---|---|---|---|
| GFP alone | 100% | 33.7 ± 9.3 | 0.62 ± 0.35 | 0.18 ± 0.04 | 0.84 ± 0.27 |
| (n= 15; 1502 events) | (15 of 15 cells) | (n= 15) | (n= 15) | (n= 15) | (n= 15) |
| WT mKv3.3 | 100% | 34.0 ± 12.0 | 0.73 ± 0.35 | 0.19 ± 0.03 | 0.87 ± 0.19 |
| (n= 10; 1041 events) | (10 of 10 cells) | (n= 10) | (n= 10) | (n= 10) | (n= 10) |
| R424H mutant | 23% | 23.1 ± 4.2 | 0.15 ± 0.10 | 0.20 ± 0.02 | 1.14 ± 0.21 |
| (n= 3; 103 events) | (3 of 13 cells)**†† | (n= 3) | (n= 3)*† | (n= 3) | (n= 3) |
All measurements were performed at a holding potential of −80 mV. The statistical analyses indicated by superscript letters a and b were conducted using Student’s unpaired t test and Fisher’s exact probability test, respectively. cThe sEPSC decay phases were fitted with a single exponential function. *P < 0.05, **P < 0.001 between GFP alone and R424H mutant. †P < 0.05, ††P < 0.001 between WT mKv3.3 and R424H mutant.
Expressions of heterologous proteins in Xenopus oocytes
The detailed procedures have been described previously (Kubo & Murata, 2001). Briefly, linearized WT or R424H mutant cDNA in pcDNA3 was used as a template to produce capped cRNA using T7 RNA polymerase (mMESSAGE mMACHINE; Ambion, Austin, TX, USA). Xenopus oocytes were collected from frogs anaesthetized in water containing 0.15% (w/v) tricaine. The isolated oocytes were treated with collagenase (2 mg ml−1; type 1; Sigma-Aldrich) and injected with 50 nl of nuclease-free water containing 10 ng of WT mKv3.3 cRNA, a mixture of 10 ng of WT and 10 ng of R424H mutant cRNAs, or 10 ng of R424H mutant cRNA. The oocytes were then incubated at 17°C in frog Ringer solution containing (mm): 88 NaCl, 1 KCl, 2.4 NaHCO3, 0.3 Ca(NO3)2, 0.41 CaCl2 and 0.82 MgSO4, pH 7.6, with 0.1% (v/v) penicillin–streptomycin solution (Sigma-Aldrich) for 2–3 days before recordings.
Electrophysiological recording from Xenopus oocytes
Potassium currents were recorded under a two-electrode voltage clamp using an OC-725C amplifier (Warner Instruments, Hamden, CT, USA) and Clampex 10.3 software (Molecular Devices, Sunnyvale, CA, USA). The signals were digitized at 10 kHz using a Digidata 1322A (Molecular Devices). The microelectrodes were filled with an electrode solution containing 3 m potassium acetate and 10 mm KCl. The oocytes were perfused with a bath solution containing (mm): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2 and 5 Hepes (pH adjusted to 7.2 with NaOH). All experiments were performed at 25°C. The data were analysed using Clampfit 10.3 software (Molecular Devices) and Igor Pro 6 software (Wavemetrics, Lake Oswego, OR, USA) with the added import functionality provided by the ReadPclamp XOP of the NeuroMatic software package (http://www.neuromatic.thinkrandom.com/). To calculate conductance–voltage relationships, outward currents were activated by applying 1000 ms voltage steps from a holding potential of −80 mV to potentials up to +70 mV in 10 mV increments. The equilibrium potential of K+ (EK=−104.0 mV) was calculated from the intracellular concentration of K+ (109.5 mm) in the Xenopus oocyte (Costa et al. 1989). To determine steady-state inactivation, cells were held at −80 mV before applying a 1000 ms prepulse to potentials between −80 and +60 mV in 10 mV increments, followed by a 250 ms test pulse to +20 mV. Steady-state inactivation (I/Imax) curves were fitted with the Boltzmann function, I/Imax= 1/[1 + exp(V−V½)/k], where k is a slope factor. To measure the rates of deactivation, outward currents were evoked by stepping from a holding potential of −80 mV to +50 mV for 5 ms and then stepping to potentials between −60 and +10 mV in 10 mV increments for 50 ms. The time constant of recovery (τrecove) from inactivation was determined by depolarizing the cells to +50 mV for 1000 ms from a holding potential of −80 mV. A step to −80 mV of variable duration was followed with test pulses to +50 mV for 200 ms in 500 ms increments. The data points were fitted with a single exponential function. The time constants of inactivation (τinacti) and deactivation were obtained by fitting current traces with a single exponential function on the inactivating and deactivating phases of the traces, respectively.
Whole-cell patch-clamp recordings from cultured PCs
In most recordings, cerebellar cultures were constantly perfused (2 ml min−1) with oxygenated artificial cerebrospinal fluid (ACSF) containing (mm): 120 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 17 d-glucose and 0.1 picrotoxin (Tocris Bioscience, Bristol, UK; a GABAA receptor antagonist), bubbled with 5% CO2–95% O2 at 26°C. The other recordings were performed in Hepes-buffered ACSF containing (mm): 140 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 10 d-glucose and 10 Hepes (pH adjusted to 7.4 with NaOH), bubbled with 100% O2. Outward currents were recorded in Hepes-buffered ACSF containing (mm): 0.2 CdCl2, 0.1 picrotoxin, 0.05 6,7-dinitroquinoxaline-2,3-dione (DNQX; Tocris Bioscience; a AMPA/kainate receptor antagonist,) and 0.001 TTX (Wako Pure Chemical Industries). Hepes buffer was used to avoid the precipitation of CdCO3.
Cells were visualized on the stage of an upright microscope (BX50WI; Olympus, Tokyo, Japan) using a ×40 water-immersion objective lens with Nomarski optics and a near-infrared CCD camera (C-3077-79; Hamamatsu Photonics, Hamamatsu, Japan). Green fluorescent protein-positive cells were visualized and selected using epifluorescence optics (Olympus). Patch pipettes were made from borosilicate glass capillaries to reduce pipette capacitance (GC150F-100; Harvard Apparatus, Holliston, MA, USA) and had a resistance of 1.5–2.5 MΩ when filled with a potassium gluconate-based internal solution containing (mm): 145 potassium gluconate, 5 KCl, 0.1 EGTA, 5 Mg-ATP, 5 disodium phosphocreatine, 0.3 Na2-GTP, 10 Hepes–KOH and 10 biocytin (Sigma-Aldrich), pH 7.3. The liquid junction potential (−10 mV) was corrected offline. Whole-cell patch-clamp recordings were conducted in GFP-positive PCs at DIV 8–10. Purkinje cells were identifiable by their large somatic size (Tabata et al. 2000), and their identity was confirmed by intracellular staining with biocytin. Patch-clamp recordings were acquired using a Multiclamp 700B amplifier with Clampex 10.3 software (Molecular Devices). Signals were filtered at 6–10 kHz and digitized at 10–50 kHz using a Digidata 1440A (Molecular Devices). In voltage-clamp conditions, series resistance was compensated electronically by 80–90%, and in current-clamp conditions, it was performed using the bridge balance and capacitance neutralization. Outward currents were activated with 500 ms voltage steps from −70 mV to voltages ranging from −60 to +40 mV in 10 mV increments. Leak currents were subtracted online by the P/4 protocol (Armstrong & Bezanilla, 1974). The recorded currents were normalized to cell capacitance, which was calculated from the transient current evoked by applying a small voltage step (−5 mV, 20 ms duration) from a holding potential of −70 mV in voltage-clamp conditions. Spontaneous excitatory postsynaptic currents (sEPSCs) were recorded at a holding potential of −80 mV for 250 s in the presence of picrotoxin and detected offline using the template search function in the Clampfit 10.3 software. Action potentials were evoked by depolarizing current pulses in current-clamp conditions (10 ms duration, from 0 to 200 pA in 10 pA increments; or 200 ms duration, from 0 to 200 pA in 20 pA increments). The resting membrane potentials were adjusted to −60 mV by current injection. Spontaneous firing was recorded at resting membrane potential for 300 s. The half-amplitude width of the action potential was measured at the mid-point between the threshold and peak. Action potential amplitude was measured between the threshold and peak.
Fluorescence imaging of calcium
To monitor the free [Ca2+]i of PCs, cerebellar cultures at DIV 8–10 were incubated with Hepes-buffered ACSF containing fura-2 AM (0.01 mm; Invitrogen) for 1 h at 37°C. The cells were visualized and perfused with Hepes-buffered ACSF at the same settings as those used for the patch-clamp recordings. For fura-2 excitation, the cultures were illuminated alternately at 340 and 380 nm wavelengths using a 100 W xenon lamp source, a fura-2 filter set (ET FURA2; Chroma Technology, Brattleboro, VT, USA), and a filter wheel (Ludl Electronic Products, Hawthorne, NY, USA). The fluorescence was filtered through a bandpass filter (470–550 nm) and captured using an EMCCD camera (iXon3 DU897; Andor Technology, Belfast, UK). The experiments were controlled by Andor iQ software (Andor Technology). The regions of interest were defined as the shape of the GFP-expressing Purkinje cell bodies. The [Ca2+]i was calculated according to the previously described method with a dissociation constant of 224 nm (Grynkiewicz et al. 1985). The maximal and minimal fluorescence ratios (Rmax and Rmin) were measured after addition of the calibration solutions. The Rmax was measured in Hepes-buffered ACSF containing 5 μm ionomycin (Sigma-Aldrich), and Rmin in Hepes-buffered, Ca2+-free ACSF containing 10 mm EGTA and 5 μm ionomycin. In some experiments, the cultures were perfused for 5 min with high-K+ ACSF, in which 12.5 mm NaCl was replaced by equimolar KCl (total [K+], 15 mm) to elevate [Ca2+]i. Basal [Ca2+]i was obtained as an average for a 7 min period from the beginning of the recordings, and elevated [Ca2+]i for a 5 min period during high-K+ ACSF perfusion.
Results
R424H mutant subunits exhibit a dominant-negative effect against WT mKv3.3 subunits without affecting WT subunit expression in heterologous expression systems
We examined whether the biophysical properties of hKv3.3 with the R423H mutation were conserved in mKv3.3 with the R424H mutation using the Xenopus oocyte expression system and two-electrode voltage-clamp recording (Figueroa et al. 2010; Minassian et al. 2012). In WT mKv3.3-expressing oocytes, depolarizing voltage steps from a holding potential of −80 mV evoked outward currents that became more apparent when the membrane was depolarized to potentials more positive than −10 mV (Supplemental Fig. S2Aa and S2C; a V1/2 of activation of 25.5 ± 3.1 mV, n= 19), and the currents showed inactivation (Supplemental Fig. S2Aa, n= 11) and fast deactivation (deactivation τ= 0.948 ± 0.15 ms at −40 mV, n= 11; trace not shown). These results well reflected the reported properties of Kv3.3 channels, i.e. fast activation, positively shifted voltage dependence, N-type inactivation and a fast deactivation rate (Rae & Shepard, 2000; Rudy & McBain, 2001; Desai et al. 2008). In R424H mutant-expressing oocytes, negligible currents were observed at potentials more positive than +20 mV (Supplemental Fig. S2Ac and S2B), and the peak amplitudes were not significantly larger than those recorded in water-injected oocytes (Supplemental Fig. S2B; not significant by Student’s unpaired t test), indicating that most of the currents were derived from endogenous channels present in Xenopus oocytes. When cRNA of WT and R424H mutant mKv3.3 was injected at a 1:1 ratio (WT+R424H), the V1/2 of activation was significantly left-shifted, by 9.05 mV, compared with oocytes expressing WT subunits alone (Supplemental Fig. S2C; WT+R424H, 15.6 ± 9.2 mV, n= 21; WT, 24.6 ± 6.1 mV, n= 19; P< 0.001 by Student’s unpaired t test). The value of k was decreased by 4.29 mV (WT + R424H, k= 9.82 ± 2.4 mV, n= 21; WT, k= 14.1 ± 3.1 mV, n= 19; P< 0.001 by Student’s unpaired t test), and the time constant of activation (τacti) at +40 mV was 2.03-fold slower (Supplemental Fig. S2Da and S2Db; P< 0.001 by Student’s unpaired t test). These results are consistent with previous reports of hKv3.3 with R423H mutation (Figueroa et al. 2010; Minassian et al. 2012) and indicate that the functional effects of this mutation are well conserved between humans and mice.
Coexpression of R424H mutant and WT subunits accelerates the inactivation kinetics and slows the recovery from inactivation compared with WT subunits alone
In order to reveal further the unknown biophysical properties of hKv3.3 with the R423H mutation, we investigated the inactivation kinetics, steady-state inactivation and recovery from inactivation of WT+R424H mutant channels. The τinacti of WT+R424H mutant channels was more than 2-fold faster than that of channels composed of WT subunits alone in the range of +10 to +70 mV depolarizing steps (Fig. 1A), and the V1/2 of inactivation was significantly left-shifted, by 9.67 mV, compared with that of WT subunits (Fig. 1B; WT+R424H, V1/2=−4.39 ± 7.09 mV, n= 12; WT, V1/2= 5.28 ± 5.33 mV, n= 11; P< 0.01 by Student’s unpaired t test). The τrecove from inactivation of WT+R424H was significantly slower than that of WT (Fig. 1C; WT+R424H, τrecove= 2.62 ± 0.60, n= 10; WT, τrecove= 1.78 ± 0.08, n= 7; P< 0.01 by Student’s unpaired t test). These results indicate that coexpression of WT and R424H mutant subunits accelerated the inactivation kinetics and slowed recovery from inactivation.
Figure 1. Coexpression of R424H mutant and wild-type (WT) subunits in Xenopus oocytes accelerates the inactivation kinetics and slows the recovery from inactivation compared with WT subunits alone.
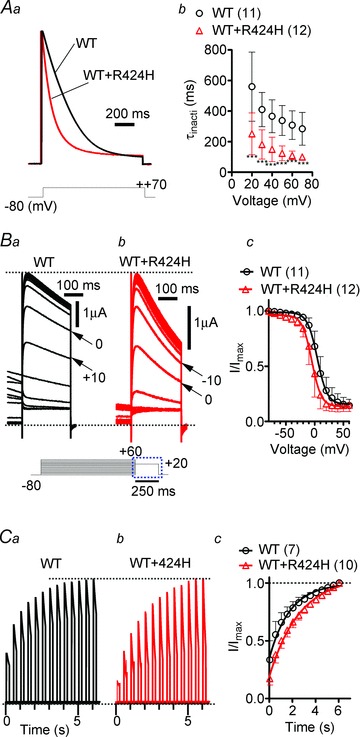
Aa, representative traces evoked by stepping from a −80 mV holding potential to +70 mV. The current traces are scaled to the same peak amplitude. Ab, the plots of the inactivation time constant (τinacti) were determined by fitting the falling phases of currents obtained in Supplemental Fig. S2A with a single exponential function. Ba and b, comparison of steady-state inactivation, which was obtained by changing the membrane potential from a prepulse potential ranging from −80 to +60 mV in 10 mV increments to a test voltage step of +20 mV to record tail currents. Ba and b shows traces of tail currents, and their corresponding voltage pulses are given under the traces surrounded by the dotted rectangle. Bc, the tail current amplitudes were normalized to the maximal current, and the resulting plots were fitted with the Boltzmann function, I/Imax= 1/[1 + exp(V−V1/2)/k], where k is a slope factor. C, recovery from inactivation in WT channels and WT+R424H mutant channels. Ca and b, the recovery time constant (τrecove) was determined by depolarizing the cells to +50 mV for 1000 ms from a holding potential of −80 mV. A step to −80 mV of variable duration was followed with test pulses to +50 mV for 200 ms in 500 ms increments. Cc, time course of the recovery from inactivation. The curves were fitted with a single exponential function to obtain τrecove. Here and in the following figures, error bars indicate standard deviation, the numbers in parentheses indicate the number of experiments, and statistical significance was tested using Mann-Whitney’s U test unless otherwise stated (significance, P < 0.05). ***P < 0.001.
Expression of R424H mutant subunits in cerebellar cultures induces PC death and impairs dendritic development
Spinocerebellar ataxia type 13 patients show cerebellar symptoms and cerebellar atrophy, suggesting shrinkage of the cerebellar cortex and degeneration of cerebellar neurons (Waters et al. 2006; Figueroa et al. 2010, 2011). In order to explore the effects of the R424H mutant on cell survival, dendritic development and electrophysiological properties in cerebellar neurons, mouse cerebellar cultures were infected at DIV 0 with lentiviruses expressing WT or R424H mutant subunits together with GFP. Given that SCA13 is an autosomal dominant disorder and Kv3 channels are formed by the assembly of four pore-forming subunits, hKv3.3 channels in SCA13 patients are considered to be heteromultimer channels consisting of WT and mutant subunits (MacKinnon, 1991; Figueroa et al. 2010; Minassian et al. 2012). When R424H mutant subunits were lentivirally expressed in cultured PCs, the subunits were expected to incorporate into endogenous mKv3.3 channels, mimicking the pathological condition.
Green fluorescent protein fluorescence was observed after DIV 3, and >90% of PCs were GFP positive (Fig. 2A–C). In the immunohistochemical experiments, PCs were immunostained with anti-calbindin antibody and granule cells with anti-NeuN antibody. Mullen et al. ()1992 reported that PCs are not stained with anti-NeuN antibody in vivo. In agreement with their report, PCs and GABAergic interneurons were NeuN negative in our culture conditions (Supplemental Fig. S3A and S3B). The mKv3.3 protein in WT-expressing PCs at DIV 10 was significantly overexpressed (by 8.3-fold) compared with that in PCs expressing GFP alone (Supplemental Fig. S3E). In contrast to the clear expression of mKv3.3 protein in PCs (Goldman-Wohl et al. 1994), mKv3.3 expression was not detected in cultured granule cells (Supplemental Fig. S3C), differing from a previous report using an in vivo preparation (Chang et al. 2007). Consistent with a previous report (Tabata et al. 2000), the relative densities of GFP- and WT-expressing PCs decreased in a day-dependent manner (Fig. 2A, B and D). Until DIV 7, the relative densities of PCs did not differ significantly between R424H mutant-expressing and control cultures (Fig. 2Aa, Ba, Ca and D; P= 0.346 between GFP and R424H; P= 0.222 between WT and R424H). At DIV 11, however, the density of PCs in R424H mutant-expressing cultures was significantly decreased (Fig. 2Ab, Bb, Cb and D; P < 0.01 between GFP and R424H; P < 0.01 between WT and R424H). At DIV 14, ∼40% of PCs still survived in GFP- or WT-expressing cultures (Fig. 2Ac, Bc, Cc and D), whereas there were few surviving PCs in R424H mutant-expressing cultures. Relative cell densities and the percentage of GFP-positive cells of granule cells at DIV 14 were also quantified, but there were no significant differences between R424H mutant-expressing and control cultures (Fig. 2Ac′, Bc′, Cc′ and E; cell densities, P= 0.0952 between GFP and R424H; P= 0.117 between WT and R424H; and percentage of GFP-positive cells, GFP, 65.9 ± 8.3%, n= 5; WT, 64.0 ± 7.4%, n= 5; R424H, 59.8 ± 8.1%, n= 5; P= 0.421 between GFP and R424H; P= 0.310 between WT and R424H), indicating that expression of R424H mutant subunits did not affect the survival of granule cells. The difference in cell survival between PCs and granule cells may be because cultured granule cells do not express endogenous mKv3.3 protein (Supplemental Fig. S3C), and R424H mutant subunits were thus unable to form multimeric channels with the endogenous mKv3.3 subunits in the cells (see Discussion). These results indicate that R424H mutant subunits induced PC death and worsened their survival in a day-dependent manner.
To examine the cell death-induced morphological defects in the nuclei of PCs, cerebellar cultures expressing GFP alone, WT mKv3.3 or the R424H mutant were stained with Hoechst 33342 at DIV 8 (Fig. 2F). Chromatin in nuclei of GFP- or WT-expressing PCs was stained moderately, with some small bright granules (Fig. 2F, arrows in GFP and WT panels), demonstrating normal nuclear morphology. In contrast, PCs expressing R424H mutant subunits showed clear chromatin condensation (Fig. 2F, arrowhead in R424H panel). The percentages of PCs exhibiting this chromatin condensation were significantly different between R424H mutant-expressing PCs and the control group (Fig. 2G; P < 0.001 by Student’s unpaired t test). These results suggest that the expression of R424H mutant subunits might induce apoptotic cell death in PCs.
At DIV 7, several neurites were observed in GFP- and WT-expressing PCs, which had morphologies similar to those of R424H mutant-expressing PCs at this stage (Fig. 3Aa′, Ba′ and Ca′). However, at DIV 11, GFP- and WT-expressing PCs had elongated immature dendrites, whereas R424H mutant-expressing PCs did not show dendritic extension (Fig. 3Ab′, Bb′ and Cb′). Sholl analyses of the dendritic arbors revealed that the number of dendritic intersections in R424H mutant-expressing PCs was significantly smaller than that in GFP-expressing (Fig. 3D, at distances of 10–50 μm from the cell body; P < 0.01) or WT-expressing PCs (Fig. 3D, at distances of 10–40 μm; P < 0.01). These results clearly demonstrate that in addition to the induction of cell death, expression of R424H mutant subunits in PCs decreases the survival rate and impairs dendritic development.
R424H mutant-expressing PCs exhibit lower outward current density
The results using Xenopus oocytes showed that expression of R424H mutant subunits significantly modulated WT mKv3.3 channel function (Fig. 1 and Supplemental Fig. S2). Thus, similar effects would be predicted in PCs lentivirally expressing R424H mutant subunits. In order to determine how these electrophysiological properties were affected, whole-cell patch-clamp recordings were performed using cultured PCs expressing GFP only, WT subunits or R424H mutant subunits at DIV 8–10.
Purkinje cells could be identified by their large cell bodies (cell body diameters of PCs at DIV 10, 17.2 ± 2.6 μm, n= 78; those of other neurons, 7.26 ± 1.7 μm, n= 226), and the recordings were confirmed by a combination of intracellular staining and immunocytochemical staining (Supplemental Fig. S4A). The cell capacitance of R424H mutant-expressing PCs was significantly lower than that of GFP- or WT-expressing PCs (Table 1; P < 0.001 in R424H versus GFP and in R424H versus WT), reflecting the impairment of dendritic development (Fig. 3D). However, the resting membrane potential and input resistance of R424H mutant-expressing PCs showed no difference when compared with PCs expressing GFP alone or those expressing WT subunits (Table 1; resting membrane potential, R424H versus GFP, P= 0.533; R424H versus WT, P= 0.874; and input resistance, R424H versus GFP, P= 0.882; R424H versus WT, P= 0.913; analyses by Student’s unpaired t test). Spontaneous action potentials were also observed in some PCs in all groups (Table 1 and Supplemental Fig. S4B and C). The percentages of PCs generating spontaneous firing and the frequency of the firing were comparable among the three groups (Table 1).
Outward currents were recorded in Hepes-buffered ACSF containing TTX, CdCl2, picrotoxin and DNQX (see Methods). Representative current traces recorded from GFP-, WT- and R424H mutant-expressing PCs are illustrated in Fig. 4A. Depolarizing voltage pulses (more positive than −10 mV) evoked outward currents with a transient peak in GFP-, WT- and R424H mutant-expressing PCs (Fig. 4A). In WT-expressing PCs, peak amplitudes of the transient currents were larger than those in GFP-expressing PCs, indicating that lentivirally expressed mKv3.3 formed functional channels (Fig. 4Ab). In contrast, the peak amplitudes in R424H mutant-expressing PCs were smaller than those in GFP-expressing PCs (Fig. 4Aa and Ac). As the membrane capacitance of R424H mutant-expressing PCs was significantly smaller than that of GFP- and WT-expressing PCs (Table 1), the peak current amplitudes at voltages between +10 and +40 mV were normalized to membrane capacitances (current densities, in picoamperes per picofarad; Fig. 4B). The current densities in R424H mutant-expressing PCs were ∼2-fold smaller than those in GFP-expressing PCs at voltages between +10 and +40 mV (Fig. 4B), confirming that the expression of the R424H mutant subunits in cultured PCs suppressed outward currents in a dominant-negative manner.
Expression of R424H mutant subunits reduces sEPSCs in PCs
In standard culture conditions, PCs receive excitatory synaptic inputs from granule cells via the dendrites (Hirano et al. 1986; Hirano & Kasono, 1993). In order to examine how R424H mutant-induced impairment of dendritic development affects the synaptic inputs to PCs, sEPSCs were recorded from PCs in the presence of picrotoxin at a holding potential of −80 mV (Fig. 5). Examples of sEPSCs in GFP- or WT-expressing PCs appear as downward deflections in the current traces (Fig. 5Aa and Ba). These currents were abolished by application of 20 μm DNQX (traces not shown) and were thus identified as being mediated by AMPA/kainate receptors. Ensemble averages of the events in individual cells are also shown in Fig. 5Ab and Bb. Although sEPSCs in both groups were observed in all cells tested (Table 2), most R424H mutant-expressing PCs (10 of 13 cells) did not show sEPSCs during the 250 s recording period (Fig. 5C and Table 2; R424H and GFP, P < 0.001; R424H and WT, P < 0.001 by Fisher’s exact probability test). Even in PCs showing sEPSCs, the frequency was significantly lower than in GFP- or WT-expressing PCs (Table 2). The R424H mutant-expressing PCs might receive few, if any, excitatory synaptic contacts onto their somata and dendrites.
Figure 5. Absence of spontaneous excitatory postsynaptic currents (sEPSCs) in R424H mutant-expressing PCs.
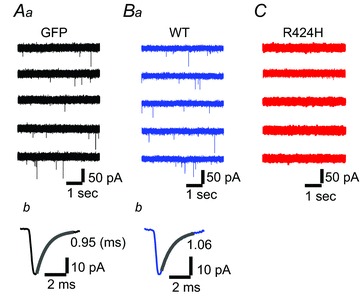
Aa, Ba and C, representative current traces recorded from PCs expressing GFP alone (Aa), WT subunits (Ba) or R424H mutant subunits (C). The sEPSCs appear as downward deflections in Aa and Ba. The PCs were held at −80 mV in the presence of picrotoxin. Ab and Bb, averaged sEPSCs from the same cell as in Aa and Ba, with superimposed single exponential fit. The decay time constant is indicated beside each trace.
R424H mutant-expressing PCs exhibit broadened action potentials and altered firing patterns
Expression of R424H mutant subunits in PCs suppressed outward current density, suggesting the alteration of the action potential waveform and firing properties in R424H mutant-expressing PCs. To examine these possibilities, single action potentials (Fig. 6) and repetitive firings (Fig. 7) were evoked in current-clamp conditions. When a single action potential was evoked by short current injection (10 ms duration), R424H mutant-expressing PCs showed a broadened action potential waveform (Fig. 6A). The half-amplitude widths of R424H mutant-expressing PCs were 1.7-fold larger than those of GFP-expressing PCs (Fig. 6B; P < 0.001). The maximal rate of rise and maximal rate of fall in R424H mutant-expressing PCs were 0.75- and 0.65-fold of those in GFP-expressing PCs, respectively (Fig. 6C and D; maximal rate of rise, P < 0.001; maximal rate of fall, P < 0.001). These changes suggest that not only outward K+ current but also voltage-dependent Na+ current (INa) was affected by the expression of R424H mutant, because the maximal rate of rise of the action potential has been used as an index of the inward INa (Hodgkin & Katz, 1949).
To clarify the reduction of the maximal rate of rise by R424H mutant expression, INa was recorded from PCs in voltage-clamp conditions (Supplemental Fig. S5). Expression of R424H mutant subunits significantly reduced INa compared with the control group, without any changes in the voltage dependence of activation and inactivation. As the INa of cultured PCs is distributed through the cell body and axons (Fry et al. 2007), the reduction in INa would be due to the smaller cell body (Table 1, ‘Membrane capacitance’) and impaired neurite extension of R424H-expressing PCs. The expression of R424H mutant subunits did not affect the threshold current, threshold potential or action potential amplitude of PCs compared with expression of GFP (Fig. 6E–G). These results strongly suggest that expression of R424H mutant subunits inhibited the activation of the endogenous mKv3.3 channels, resulting in a reduction of the maximal rate of fall and then in the broadening of action potential duration in PCs.
We next examined the firing properties evoked by long current injection (200 ms duration). Most GFP-expressing PCs (82.9%) showed tonic firing of action potentials (tonic type) in response to depolarizing current injection (Fig. 7A and filled column of GFP in Fig. 7E), which is consistent with previous reports on the firing pattern of cultured PCs (Tabata et al. 2000; Harada et al. 2006). The other PCs (17.1%) showed a few spikes (up to three spikes) in response to current injection ranging from 0 to 200 pA in 20 pA increments and did not fire tonically during the 200 ms depolarizing pulses (onset type; open column of GFP in Fig. 7E). However, approximately half of the R424H mutant-expressing PCs exhibited onset-type firing (53.6%; Fig. 7C and E), and the remaining PCs exhibited tonic-type firing (46.4%; Fig. 7D and E). The percentages of firing types in R424H mutant-expressing PCs differed significantly from those in GFP- and WT-expressing PCs (Fig. 7E; P < 0.001 in both pairs by the χ2 test). The firing frequencies of tonic-type neurons in the three groups were plotted against the injected current (Fig. 7F). Wild-type-expressing PCs showed the highest frequencies, in the range of 40–200 pA (Fig. 7B and F), demonstrating that lentivirally expressed WT subunits contributed to the generation of narrow action potentials by accelerating the falling phase (Fig. 7A and D and Supplemental Fig. S3E). In tonic-type neurons, firing frequencies in R424H mutant-expressing PCs were significantly lower than those in GFP-expressing PCs, in the range of 180–200 pA (Fig. 7F; P < 0.05 at 180 and 200 pA depolarization). These results demonstrate that expression of R424H mutant subunits changed the ratio of tonic-firing PCs and reduced PC excitability in response to depolarization.
R424H mutant-expressing PCs show higher [Ca2+]i, and blockade of P/Q-type Ca2+ channels rescues the PC death and dendritic maldevelopment caused by the mutant subunits
The expression of R424H mutant subunits in cerebellar cultures caused PC death. We hypothesized that the cell death was caused by excessive Ca2+ influx through the following steps. As cultured PCs spontaneously generate action potentials, which increase basal [Ca2+]i (Schilling et al. 1991; Supplemental Fig. S4B and C and Table 1), the broadening of action potentials by R424H mutant expression would cause increased Ca2+ influx via excessive activation of voltage-gated Ca2+ channels. This influx would lead to a defect of Ca2+ homeostasis in PCs, resulting in the cell death as a part of a stress response (Orrenius et al. 2003). Indeed, excessive Ca2+ influx triggered by the blockade of K+ channels has been shown to induce cell death in several types of cells (Kim et al. 2000; Lajdova et al. 2004; Wang et al. 2011). To test this hypothesis, we performed calcium imaging (Fig. 8) and then rescue experiments of PC death using ω-agatoxin IVA, a specific blocker for the P/Q-type voltage-gated Ca2+ channels that mediate predominant Ca2+ currents in PCs (Mintz & Bean, 1993; Gillard et al. 1997; Fig. 9).
Figure 9. Pharmacological blockade of P/Q-type Ca2+ channels rescues the PC death and dendritic maldevelopment caused by expression of R424H mutant.
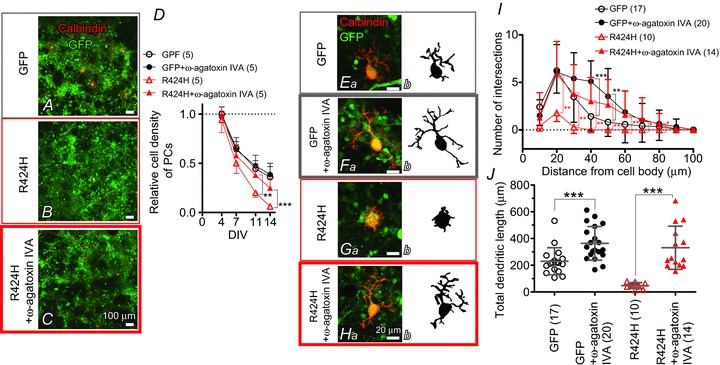
A–C, cerebellar cultures expressing GFP alone (A) or R424H mutant with GFP (B and C). The cultures were immunostained for calbindin at DIV 14. In C, ω-agatoxin IVA was added to the culture medium every other day from DIV 2. D, relative cell density of PCs plotted as a function of DIV. The density was normalized to the value of PCs expressing GFP alone at DIV 4. E–H, calbindin-immunolabelled PCs expressing GFP alone (Ea and Fa) or R424H mutant subunits with GFP (Ga and Ha) at DIV 14. Morphologies of PCs are depicted in the right-hand panels for clarity. In F and H, ω-agatoxin IVA was added. I, and J, summary of dendrite complexity measured by Sholl analysis (I) and of total dendritic length (J). *P < 0.05, **P < 0.01, and ***P < 0.001.
Calcium imaging was performed using cerebellar cultures loaded with fura-2 AM (see Methods). After a baseline recording (measurement of basal [Ca2+]i) for 8 min, cerebellar cultures were depolarized for 5 min by perfusion of high-K+ ACSF (Fig. 8B). In basal conditions, [Ca2+]i in R424H mutant-expressing PCs was approximately four times higher than in the control group (Fig. 8Ca; P < 0.001 in R424H versus GFP and in R424H versus WT). There were no significant differences in high-K+-induced [Ca2+]i elevation (∼200 nm) between PCs expressing the R424H mutant and those expressing GFP alone or expressing WT subunits (Fig. 8Cb). The basal [Ca2+]i of granule cells infected with the lentiviruses was also measured, and there were no significant differences between R424H mutant-expressing and control cultures (GFP, 71.2 ± 41.2 nm, n= 86; WT, 69.7 ± 38.0 nm, n= 118; R424H, 63.6 ± 32.9 nm, n= 72; P= 0.365 between GFP and R424H; P= 0.423 between WT and R424H).
To examine whether the elevated basal [Ca2+]i induced PC death in R424H mutant-expressing cultures, we performed a similar experiment to that in Fig. 2. In this experiment, ω-agatoxin IVA (0.2 μm) was added to the culture medium at DIV 2 (see Methods), and WT-expressing cultures were omitted to simplify the experimental design. Treatment of GFP-expressing cultures with ω-agatoxin IVA did not affect relative PC density (Fig. 9D, filled circles) but increased the branch number and the total length of dendrites at DIV 14 (Fig. 9E and F; filled circles in Fig. 9I and J), in good agreement with a previous report (Schilling et al. 1991; see Discussion). Treatment of R424H mutant-expressing cultures with ω-agatoxin IVA significantly increased relative PC density at DIV 11 and 14 (Fig. 9D, red triangles) and significantly rescued dendritic development in PCs (Fig. 9G and H; red triangles in Fig. 9I and J). These results clearly indicate that P/Q-type Ca2+ channels play a critical role in the PC death and impairment of dendrite development caused by R424H mutant expression, and support our hypothesis.
Discussion
In this study, we found that the expression of R424H mutant subunits in cerebellar cultures significantly impaired dendritic development and survival in PCs (Figs 2 and 3). Prior to cell death, R424H mutant-expressing PCs showed broadened action potential waveforms, altered firing properties and elevated basal [Ca2+]i (Figs 678). Moreover, chronic inhibition of P/Q-type Ca2+ channels by ω-agatoxin IVA rescued the PC death and dendritic maldevelopment caused by expression of R424H mutant subunits (Fig. 9). This is the first report to show that a missense mutation found in SCA13 patients induces maldevelopment of PC dendrites and eventually PC death, most probably due to elevated basal [Ca2+]i in PCs.
Biophysical properties of R424H mutant channels
The biophysical properties of hKv3.3 channels with the R423H mutation, which corresponds to the R424H mutation in mKv3.3 channels, have been previously reported (Figueroa et al. 2010; Minassian et al. 2012). Our results in Supplemental Fig. S2 agreed well with the previous reports and suggest that the properties of R424H mutant mKv3.3 were essentially identical to those of R423H-mutant hKv3.3. Moreover, we found that coexpression of R424H mutant and WT subunits accelerated the inactivation kinetics and slowed recovery from inactivation compared with expression of WT subunits alone (Fig. 1). Therefore, we predict that the properties we found in R424H mutant mKv3.3 are shared with R423H mutant hKv3.3.
We confirmed that homomeric R424H mutant channels showed negligible currents and that R424H mutant subunits exerted a dominant-negative influence on WT mKv3.3 channels in Xenopus oocytes (Supplemental Fig. S2A and B; Figueroa et al. 2010, 2011). Very recently, Zhao et al. ()2013 reported that in heterologous expression systems using Chinese hamster ovary cells, the surface protein level of R423H mutant hKv3.3 channels is 30% of that of WT hKv3.3 and that the conductance density of the mutant is 16% of that of the WT. Therefore, we cannot exclude the possibility that the reduced surface expression of mKv3.3 channels by the mutation would also contribute to the broadening of action potentials (Fig. 6) and lower firing frequency (Fig. 7) in transduced PCs. However, the reduction of the conductance density cannot be explained fully by the reduced surface protein expression.
To explain the negligible activity and dominant-negative property of R424H mutant channels, we propose two hypothetical mechanisms. First, the positively charged arginine at position 424 in mKv3.3 may be a critical residue in the S4 segment, serving as a part of the voltage sensor domain (Seoh et al. 1996). The partial disruption of the sensor domain by R424H mutation would make the subunits less sensitive to membrane voltage changes, resulting in the loss of channel function. Second, an arginine residue at position 174 in the S4 segment of KAT1, which is a voltage-gated K+ channel in Arabidopsis, plays an essential role in the appropriate integration of the S3 and S4 segment into the endoplasmic reticulum membrane (Sato et al. 2003). Given that the R174 is homologous to R424 in mKv3.3, defective membrane insertion of R424H mutant subunits could occur in Xenopus oocytes, leading to a defect in channel activity.
Purkinje cell death by R424H mutant expression and the inhibition by blockade of P/Q-type voltage-gated Ca2+ channels
In this study, we revealed that expression of R424H mutant subunits caused cell death and impaired dendritic growth in PCs (Figs 2 and 3) and that these effects were reversed by the blockade of P/Q-type Ca2+ channels (Fig. 9). Addition of ω-agatoxin IVA also enhanced dendritic elongation in PCs expressing GFP alone (Fig. 9E and F; filled circles in Fig. 9I and J). Together with a previous report showing that chronic application of TTX in cerebellar cultures caused dendritic elongation in PCs (Schilling et al. 1991), activation of P/Q-type Ca2+ channels by neuronal activity may adversely influence dendritic elongation in PCs. Addition of ω-agatoxin IVA in R424H mutant-expressing cultures did not completely restore PC survival rates (Fig. 9D). This may be because some Ca2+ currents in cultured PCs are mediated by Ca2+ channels other than the P/Q-type (Gillard et al. 1997), and activation of these channels may contribute to PC death. We therefore performed the same rescue experiments using CdCl2 (0.2 mm; a non-selective Ca2+ channel blocker) or a combination of ω-agatoxin IVA and verapamil hydrochloride (0.02 mm; an L-type Ca2+ channel blocker), but these chemicals markedly deteriorated the viability and development of cerebellar cultures within 3 DIV (data not shown).
In contrast to PCs, there were no significant decreases in the numbers of granule cells upon R424H mutant expression (Fig. 2E). This may be because granule cells do not express endogenous mKv3.3 channels with which R424H mutant subunits form oligomeric channels (Supplemental Fig. S3C), resulting in the absence of the dominant-negative influence on the endogenous channels by the expression of mutant channel subunits.
Comparison with preceding papers on Kv3.3 knockout mice and zebrafish expressing mutant Kv3.3
In contrast to the impaired dendritic development in R424H mutant-expressing PCs (Fig. 3Cb′′ and D), the cerebellum of Kv3.3 knockout mice shows neither dendritic shrinkage of PCs nor cerebellar atrophy (Zagha et al. 2010). Furthermore, the knockout mice display only moderate motor dysfunction and no ataxic phenotype, although SCA13 patients show severe ataxia (Joho et al. 2006; Hurlock et al. 2008; Waters & Pulst, 2008; Figueroa et al. 2010). This difference may be attributable to the following factors. In PCs, mKv3.3 is thought to form heteromultimeric channels by assembling with Kv3.1 and/or Kv3.4 (Goldman-Wohl et al. 1994; Weiser et al. 1994), and Kv3 channels contribute to repolarization of both somatic Na+ spikes and dendritic Ca2+ spikes (McKay & Turner, 2004). Genetic elimination of Kv3.3 subunits may be insufficient to exhibit the dendritic shrinkage and severe ataxia phenotypes because of functional compensation by Kv3.1 and Kv3.4 in PCs (Goldman-Wohl et al. 1994; Weiser et al. 1994; Martina et al. 2003). We detected the expression of Kv3.4 subunits in cultured PCs (Supplemental Fig. S3D). It is therefore reasonable to hypothesize that R424H mutant subunits form heteromultimeric channels not only with endogenous mKv3.3 but also with other members of Kv3.3, including Kv3.4, resulting in total inhibition of the K+ channel activity in PCs. This may account for the differences in the morphological phenotypes of PCs in our results versus the knockout mice.
Zebrafish expressing infant-onset mutant zebrafish Kv3.3 (homologous to the F448L mutant in SCA13 patients) in spinal motoneurons show defective axonal pathfinding (Issa et al., 2012). Indeed, the zebrafish is an interesting model for understanding the effects of mutant Kv3.3 expression in spinal motoneurons. However, as they used a motoneuron-specific enhancer of Mnx1 (Hb9) gene, the exogenous proteins were not expressed in the cerebellar neurons. To examine the effects of mutant Kv3.3 in the cerebellum, it would be necessary to express the mutant protein directly in the cerebellar neurons using a different method.
Comparison of our culture results with SCA13 patients harbouring the R423H mutation
Spinocerebellar ataxia type 13 patients harbouring the R423H mutation generally show early-onset, slow-progressive ataxia and cerebellar atrophy (Figueroa et al. 2010, 2011). Our immunohistochemical analyses demonstrated that expression of R424H mutant subunits impaired dendritic development and induced cell death in cultured PCs (Figs 2 and 3). Those defects may be responsible for the cerebellar atrophy and ataxia observed in SCA13 patients, although it is necessary to verify that similar impairments are also observed in post-mortem cerebellum of the patients.
In functional aspects, we found that expression of R424H mutant subunits significantly decreased outward current mediated by voltage-gated K+ channels, reduced sEPSCs, broadened action potentials and altered firing properties (Figs 4567 and Table 2), suggesting the existence of similar functional changes in SCA13 patients. As PCs are the sole output neurons from the cerebellar cortex and make inhibitory synaptic contacts directly onto neurons in the deep cerebellar nuclei and the vestibular nuclei in the brainstem, PCs play crucial roles in motor co-ordination (Zheng & Raman, 2010). Accordingly, it is easily assumed that the reduction of spontaneous excitatory inputs and the changed firing properties in PCs disrupt synaptic transmission to neurons in the deep cerebellar nuclei and vestibular nuclei, resulting in impaired motor co-ordination. To examine the effects of the R424H mutation on electrophysiological properties of PCs and animal behaviour, we tried expressing R424H mutant subunits in PCs in vivo by directly injecting the virus solution into mouse cerebellar cortex as described in our previous papers (Torashima et al. 2006, 2008; Shuvaev et al. 2011). However, despite the presence of the infection, sufficient overexpression of mKv3.3 channels and apparent ataxia were not observed (data not shown). This may be because endogenous mKv3.3 proteins are abundantly expressed in PCs and the overexpression failed to reach the endogenous protein level. Efficient reduction of K+ currents in PCs in vivo as observed in cultured PCs may be attained by using a different type of viral vector, such as adeno-associated virus vectors (Nathanson et al. 2009). Alternatively, K+ currents in PCs in vivo may be effectively decreased using viral vector-mediated expressions of R424H mutant subunits in Kv3.3−/− or Kv3.3+/− mice, which express no mKv3.3 proteins or only half the normal amount.
Currently, three different missense mutations in hKv3.3 channels have been reported from distinct pedigrees, and the disease onset and clinical phenotypes also differ among them (Waters et al. 2006; Figueroa et al. 2010, 2011). In the present study, we focused on only one mutation (R423H in hKv3.3) because of the drastic changes it induced in channel properties in the Xenopus oocyte expression system and its early-onset phenotype in SCA13 patients. Further studies of the effects of other mutants (R420H and F448L in hKv3.3) on cultured PCs may provide explanations for the differences in the disease phenotypes.
Possible significance of this study
We developed an in vitro SCA13 model using mouse cerebellar cultures and lentivirus vector-mediated gene expression. This model has advantages over in vivo models, such as transgenic mice, in the ease of controlling culture conditions by applying chemical compounds. Therefore, this model would be useful in screening drugs for SCA13 and in detailed investigations of the signalling cascades that promote the observed cell death. Given that blockade of P/Q-type Ca2+ channels rescued the phenotypes found in this research, the channel blockers may be potential therapeutic drugs for SCA13. Furthermore, this culture method, in combination with virus-mediated gene expression, may be applicable to the study of other types of hereditary spinocerebellar ataxia.
Acknowledgments
The lentiviral vector and MSCV promoter were provided by St Jude Children’s Research Hospital and the American National Red Cross, respectively. We thank Dr L. K. Kaczmarek for mouse Kv3.3 cDNA and Dr K. Nakajo for technical advice and valuable comments on electrophysiological recording from Xenopus oocytes.
Glossary
- ACSF
artificial cerebrospinal fluid
- AF
AlexaFluor
- a.u.
arbitrary unit
- calbindin
calbindin D-28k
- DIV
days in vitro
- DNQX
6,7-dinitroquinoxaline-2,3-dione
- G
conductance
- GFP
green fluorescent protein
- hKv3.3
human Kv3.3
- INa
Na+ current
- k
a slope factor
- mKv3.3
mouse Kv3.3
- MSCV
murine embryonic stem cell virus
- PBS-XCG
PBS containing 0.3% Triton X-100, 0.12%λ-carrageenan, 1% goat serum and 0.02% sodium azide
- PCs
Purkinje cells
- P2A
2A peptide sequence from porcine teschovirus-1
- Rmax
maximal fluorescence ratio
- Rmin
minimal fluorescence ratio
- RT
room temperature
- SCA13
spinocerebellar ataxia type 13
- sEPSCs
spontaneous excitatory postsynaptic currents
- τacti
activation time constant
- τinacti
inactivation time constant
- τrecove
recovery time constant
- VSV-G
vesicular stomatitis virus G protein
- WT
wild-type
Additional Information
Competing interests
None declared.
Author contributions
T.I. and H.H. conceived and designed experiments. T.I. and Y.M. performed experiments. T.I. collected and analysed data. T.I., Y.S. and H.H. wrote the paper.
Funding
This work was supported by Health Labour Sciences Research Grant (T.I.), JSPS KAKENHI grant numbers 24790230 (T.I.) and 19670003 (H.H.), and JSPS Funding Program for Next Generation World-Leading Researchers (LS021 to H.H.).
References
- Armstrong CM, Bezanilla F. Charge movement associated with the opening and closing of the activation gates of the Na channels. J Gen Physiol. 1974;63:533–552. doi: 10.1085/jgp.63.5.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM. From molecule to malady. Nature. 2006;440:440–447. doi: 10.1038/nature04707. [DOI] [PubMed] [Google Scholar]
- Chang SY, Zagha E, Kwon ES, Ozaita A, Bobik M, Martone ME, Ellisman MH, Heintz N, Rudy B. Distribution of Kv3.3 potassium channel subunits in distinct neuronal populations of mouse brain. J Comp Neurol. 2007;502:953–972. doi: 10.1002/cne.21353. [DOI] [PubMed] [Google Scholar]
- Costa PF, Emilio MG, Fernandes PL, Ferreira HG, Ferreira KG. Determination of ionic permeability coefficients of the plasma membrane of Xenopus laevis oocytes under voltage clamp. J Physiol. 1989;413:199–211. doi: 10.1113/jphysiol.1989.sp017649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai R, Kronengold J, Mei J, Forman SA, Kaczmarek LK. Protein kinase C modulates inactivation of Kv3.3 channels. J Biol Chem. 2008;283:22283–22294. doi: 10.1074/jbc.M801663200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erisir A, Lau D, Rudy B, Leonard CS. Function of specific K+ channels in sustained high-frequency firing of fast-spiking neocortical interneurons. J Neurophysiol. 1999;82:2476–2489. doi: 10.1152/jn.1999.82.5.2476. [DOI] [PubMed] [Google Scholar]
- Figueroa KP, Minassian NA, Stevanin G, Waters M, Garibyan V, Forlani S, Strzelczyk A, Bürk K, Brice A, Dürr A, Papazian DM, Pulst SM. KCNC3: phenotype, mutations, channel biophysics—a study of 260 familial ataxia patients. Hum Mutat. 2010;31:191–196. doi: 10.1002/humu.21165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa KP, Waters MF, Garibyan V, Bird TD, Gomez CM, Ranum LP, Minassian NA, Papazian DM, Pulst SM. Frequency of KCNC3 DNA variants as causes of spinocerebellar ataxia 13 (SCA13) PLoS One. 2011;6:e17811. doi: 10.1371/journal.pone.0017811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry M, Boegle AK, Maue RA. Differentiated pattern of sodium channel expression in dissociated Purkinje neurons maintained in long-term culture. J Neurochem. 2007;101:737–748. doi: 10.1111/j.1471-4159.2007.04470.x. [DOI] [PubMed] [Google Scholar]
- Gillard SE, Volsen SG, Smith W, Beattie RE, Bleakman D, Lodge D. Identification of pore-forming subunit of P-type calcium channels: an antisense study on rat cerebellar Purkinje cells in culture. Neuropharmacology. 1997;36:405–409. doi: 10.1016/s0028-3908(97)00046-4. [DOI] [PubMed] [Google Scholar]
- Gimenez-Cassina A, Lim F, Diaz-Nido J. Gene transfer into Purkinje cells using herpesviral amplicon vectors in cerebellar cultures. Neurochem Int. 2007;50:181–188. doi: 10.1016/j.neuint.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Goldman-Wohl DS, Chan E, Baird D, Heintz N. Kv3.3b: a novel Shaw type potassium channel expressed in terminally differentiated cerebellar Purkinje cells and deep cerebellar nuclei. J Neurosci. 1994;14:511–522. doi: 10.1523/JNEUROSCI.14-02-00511.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hanawa H, Hematti P, Keyvanfar K, Metzger ME, Krouse A, Donahue RE, Kepes S, Gray J, Dunbar CE, Persons DA, Nienhuis AW. Efficient gene transfer into rhesus repopulating hematopoietic stem cells using a simian immunodeficiency virus-based lentiviral vector system. Blood. 2004;103:4062–4069. doi: 10.1182/blood-2004-01-0045. [DOI] [PubMed] [Google Scholar]
- Harada KH, Ishii TM, Takatsuka K, Koizumi A, Ohmori H. Effects of perfluorooctane sulfonate on action potentials and currents in cultured rat cerebellar Purkinje cells. Biochem Biophys Res Commun. 2006;351:240–245. doi: 10.1016/j.bbrc.2006.10.038. [DOI] [PubMed] [Google Scholar]
- Hawley RG, Lieu FH, Fong AZ, Hawley TS. Versatile retroviral vectors for potential use in gene therapy. Gene Ther. 1994;1:136–138. [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. Sunderland, MA: Sinauer; 2001. [Google Scholar]
- Hirai H, Launey T. The regulatory connection between the activity of granule cell NMDA receptors and dendritic differentiation of cerebellar Purkinje cells. J Neurosci. 2000;20:5217–5224. doi: 10.1523/JNEUROSCI.20-14-05217.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T, Kasono K. Spatial distribution of excitatory and inhibitory synapses on a Purkinje cell in a rat cerebellar culture. J Neurophysiol. 1993;70:1316–1325. doi: 10.1152/jn.1993.70.4.1316. [DOI] [PubMed] [Google Scholar]
- Hirano T, Kubo Y, Wu MM. Cerebellar granule cells in culture: monosynaptic connections with Purkinje cells and ionic currents. Proc Natl Acad Sci U S A. 1986;83:4957–4961. doi: 10.1073/pnas.83.13.4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Katz B. The effect of sodium ions on the electrical activity of giant axon of the squid. J Physiol. 1949;108:37–77. doi: 10.1113/jphysiol.1949.sp004310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlock EC, McMahon A, Joho RH. Purkinje-cell-restricted restoration of Kv3.3 function restores complex spikes and rescues motor coordination in Kcnc3 mutants. J Neurosci. 2008;28:4640–4648. doi: 10.1523/JNEUROSCI.5486-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa FA, Mock AF, Sagasti A, Papazian DM. Spinocerebellar ataxia type 13 mutation that is associated with disease onset in infancy disrupts axonal pathfinding during neuronal development. Dis Model Mech. 2012;5:921–929. doi: 10.1242/dmm.010157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joho RH, Street C, Matsushita S, Knöpfel T. Behavioral motor dysfunction in Kv3-type potassium channel-deficient mice. Genes Brain Behav. 2006;5:472–482. doi: 10.1111/j.1601-183X.2005.00184.x. [DOI] [PubMed] [Google Scholar]
- Kim JA, Kang YS, Jung MW, Kang GH, Lee SH, Lee YS. Ca2+ influx mediates apoptosis induced by 4-aminopyridine, a K+ channel blocker, in HepG2 human hepatoblastoma cells. Pharmacology. 2000;60:74–81. doi: 10.1159/000028350. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Murata Y. Control of rectification and permeation by two distinct sites after the second transmembrane region in Kir2.1 K+ channel. J Physiol. 2001;531:645–660. doi: 10.1111/j.1469-7793.2001.0645h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajdova I, Chorvat D, Jr, Spustova V, Chorvatova A. 4-Aminopyridine activates calcium influx through modulation of the pore-forming purinergic receptor in human peripheral blood mononuclear cells. Can J Physiol Pharmacol. 2004;82:50–56. doi: 10.1139/y03-128. [DOI] [PubMed] [Google Scholar]
- McKay BE, Turner RW. Kv3 K+ channels enable burst output in rat cerebellar Purkinje cells. Eur J Neurosci. 2004;20:729–739. doi: 10.1111/j.1460-9568.2004.03539.x. [DOI] [PubMed] [Google Scholar]
- MacKinnon R. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature. 1991;350:232–235. doi: 10.1038/350232a0. [DOI] [PubMed] [Google Scholar]
- Martina M, Yao GL, Bean BP. Properties and functional role of voltage-dependent potassium channels in dendrites of rat cerebellar Purkinje neurons. J Neurosci. 2003;23:5698–5707. doi: 10.1523/JNEUROSCI.23-13-05698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor JR, Merlo D, Jones A, Wisden W, Randall AD. Mouse cerebellar granule cell differentiation: electrical activity regulates the GABAA receptor α6 subunit gene. J Neurosci. 1998;18:2822–2833. doi: 10.1523/JNEUROSCI.18-08-02822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikuni T, Uesaka N, Okuno H, Hirai H, Deisseroth K, Bito H, Kano M. Arc/Arg3.1 is a postsynaptic mediator of activity-dependent synapse elimination in the developing cerebellum. Neuron. 2013;78:1024–1035. doi: 10.1016/j.neuron.2013.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minassian NA, Lin MC, Papazian DM. Altered Kv3.3 channel gating in early-onset spinocerebellar ataxia type 13. J Physiol. 2012;590:1599–1614. doi: 10.1113/jphysiol.2012.228205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz IM, Bean BP. Block of calcium channels in rat neurons by synthetic omega-Aga-IVA. Neuropharmacology. 1993;32:1161–1169. doi: 10.1016/0028-3908(93)90010-z. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Nathanson JL, Yanagawa Y, Obata K, Callaway EM. Preferential labelling of inhibitory and excitatory cortical neurons by endogenous tropism of adeno-associated virus and lentivirus vectors. Neuroscience. 2009;161:441–450. doi: 10.1016/j.neuroscience.2009.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium–apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Rae JL, Shepard AR. Kv3.3 potassium channels in lens epithelium and corneal endothelium. Exp Eye Res. 2000;70:339–348. doi: 10.1006/exer.1999.0796. [DOI] [PubMed] [Google Scholar]
- Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 2001;24:517–526. doi: 10.1016/s0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- Sato M, Suzuki K, Yamazaki H, Nakanishi S. A pivotal role of calcineurin signalling in development and maturation of postnatal cerebellar granule cells. Proc Natl Acad Sci U S A. 2005;102:5874–5879. doi: 10.1073/pnas.0501972102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Sakaguchi M, Goshima S, Nakamura T, Uozumi N. Molecular dissection of the contribution of negatively and positively charged residues in S2, S3, and S4 to the final membrane topology of the voltage sensor in the K+ channel, KAT1. J Biol Chem. 2003;278:13227–13234. doi: 10.1074/jbc.M300431200. [DOI] [PubMed] [Google Scholar]
- Sawada Y, Kajiwara G, Iizuka A, Takayama K, Shuvaev AN, Koyama C, Hirai H. High transgene expression by lentiviral vectors causes maldevelopment of Purkinje cells in vivo. Cerebellum. 2010;9:291–302. doi: 10.1007/s12311-010-0161-1. [DOI] [PubMed] [Google Scholar]
- Schilling K, Dickinson MH, Connor JA, Morgan JI. Electrical activity in cerebellar cultures determines Purkinje cell dendritic growth patterns. Neuron. 1991;7:891–902. doi: 10.1016/0896-6273(91)90335-w. [DOI] [PubMed] [Google Scholar]
- Seoh SA, Sigg D, Papazian DM, Bezanilla F. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Shuvaev AN, Horiuchi H, Seki T, Goenawan H, Irie T, Iizuka A, Sakai N, Hirai H. Mutant PKCγ in spinocerebellar ataxia type 14 disrupts synapse elimination and long-term depression in Purkinje cells in vivo. J Neurosci. 2011;31:14324–14334. doi: 10.1523/JNEUROSCI.5530-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- Tabata T, Sawada S, Araki K, Bono Y, Furuya S, Kano M. A reliable method for culture of dissociated mouse cerebellar cells enriched for Purkinje neurons. J Neurosci Methods. 2000;104:45–53. doi: 10.1016/s0165-0270(00)00323-x. [DOI] [PubMed] [Google Scholar]
- Takayama K, Torashima T, Horiuchi H, Hirai H. Purkinje-cell-preferential transduction by lentiviral vectors with the murine stem cell virus promoter. Neurosci Lett. 2008;443:7–11. doi: 10.1016/j.neulet.2008.07.058. [DOI] [PubMed] [Google Scholar]
- Torashima T, Iizuka A, Horiuchi H, Mitsumura K, Yamasaki M, Koyama C, Takayama K, Iino M, Watanabe M, Hirai H. Rescue of abnormal phenotypes in δ2 glutamate receptor-deficient mice by the extracellular N-terminal and intracellular C-terminal domains of the δ2 glutamate receptor. Eur J Neurosci. 2009;30:355–365. doi: 10.1111/j.1460-9568.2009.06841.x. [DOI] [PubMed] [Google Scholar]
- Torashima T, Koyama C, Iizuka A, Mitsumura K, Takayama K, Yanagi S, Oue M, Yamaguchi H, Hirai H. Lentivector-mediated rescue from cerebellar ataxia in a mouse model of spinocerebellar ataxia. EMBO Rep. 2008;9:393–399. doi: 10.1038/embor.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torashima T, Okoyama S, Nishizaki T, Hirai H. In vivo transduction of murine cerebellar Purkinje cells by HIV-derived lentiviral vectors. Brain Res. 2006;1082:11–22. doi: 10.1016/j.brainres.2006.01.104. [DOI] [PubMed] [Google Scholar]
- Wang W, Xiao J, Adachi M, Liu Z, Zhou J. 4-Aminopyridine induces apoptosis of human acute myeloid leukemia cells via increasing [Ca2+]i through P2X7 receptor pathway. Cell Physiol Biochem. 2011;28:199–208. doi: 10.1159/000331731. [DOI] [PubMed] [Google Scholar]
- Waters MF, Minassian NA, Stevanin G, Figueroa KP, Bannister JP, Nolte D, Mock AF, Evidente VG, Fee DB, Müller U, Dürr A, Brice A, Papazian DM, Pulst SM. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat Genet. 2006;38:447–451. doi: 10.1038/ng1758. [DOI] [PubMed] [Google Scholar]
- Waters MF, Pulst SM. SCA13. Cerebellum. 2008;7:165–169. doi: 10.1007/s12311-008-0039-7. [DOI] [PubMed] [Google Scholar]
- Weiser M, Vega-Saenz de Miera E, Kentros C, Moreno H, Franzen L, Hillman D, Baker H, Rudy B. Differential expression of Shaw-related K+ channels in the rat central nervous system. J Neurosci. 1994;14:949–972. doi: 10.1523/JNEUROSCI.14-03-00949.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagha E, Manita S, Ross WN, Rudy B. Dendritic Kv3.3 potassium channels in cerebellar Purkinje cells regulate generation and spatial dynamics of dendritic Ca2+ spikes. J Neurophysiol. 2010;103:3516–3525. doi: 10.1152/jn.00982.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhu J, Thornhill WB. Spinocerebellar ataxia-13 Kv3.3 potassium channels: arginine-to-histidine mutations affect both functional and protein expression on the cell surface. Biochem J. 2013;454:259–265. doi: 10.1042/BJ20130034. [DOI] [PubMed] [Google Scholar]
- Zheng N, Raman IM. Synaptic inhibition, excitation, and plasticity in neurons of the cerebellar nuclei. Cerebellum. 2010;9:56–66. doi: 10.1007/s12311-009-0140-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.