

Abstract
Neurotoxic microglial-neuronal interactions have been implicated in the pathogenesis of various neurodegenerative diseases such as Alzheimer’s disease, and vitamin E has been shown to have direct neuroprotective effects. To determine whether vitamin E also has indirect neuroprotective effects through suppression of microglial activation, we used a microglial-neuronal coculture. Lipopolysaccharide (LPS) treatment of a microglial cell line (N9) induced a time-dependent activation of both p38 mitogen-activated protein kinase (p38 MAPK) and nuclear factor-κB (NFκB), with consequent increases in interleukin-1α (IL-1α), tumor necrosis factor-α (TNF-α), and nitric oxide (NO) production. Differentiated neuronal cells (PC12 cells treated with nerve growth factor) exhibited marked loss of processes and decreased survival when cocultured with LPS-activated microglia. Preincubation of microglia with vitamin E diminished this neurotoxic effect, independently of direct effects of the antioxidant on the neuronal cells. Microglial NO production and the induction of IL-1α and TNFα expression also were attenuated by vitamin E. Such antiinflammatory effects of vitamin E were correlated with suppression of p38 MAPK and NFκB activation and were mimicked by an inhibition of either p38 MAPK (by SB203580) or NFκB (by decoy oligonucleotides). These results suggest that, in addition to the beneficial effects of providing direct antioxidant protection to neurons reported by others, vitamin E may provide neuroprotection in vivo through suppression of signaling events necessary for microglial activation.
Keywords: Alzheimer’s disease, interleukin-1, NFκB, nitric oxide, p38 mitogen-activated protein kinase, tumor necrosis factor, vitamin E
Brain responses to injury include activation of glia (microglia and astrocytes) and consequent release of proinflammatory cytokines, such as interleukin-1 (IL-1) and tumor necrosis factor-α (TNF-α), and other glia-derived factors, such as nitric oxide (NO). A synergistic neurotoxic effect of IL-1 and TNF-α has been shown in mixed glial—neuronal cultures (Jeohn et al., 1998), and these potentially neurotoxic effects may, in turn, aggravate further neuronal injury (Dawson et al., 1991; Chao et al., 1992). Evidence is accumulating that activated microglia contribute to the neuropathological changes in Alzheimer’s disease (AD) through overexpression of IL-1 (Griffin et al., 1989, 1998; Mrak and Griffin, 2000). A direct effect of microglia-derived IL-1 has been demonstrated in the acetylcholine system, where IL-1 directly induces acetylcholine esterase expression and activity (Li et al., 2000b). Microglia activation has also been suggested to have effects on other neurotransmitter systems, such as glutamate (Barger and Basile, 2001), γ-aminobutyric acid (Scali et al., 1999), and serotonin (Tsai and McNulty, 1997). These studies suggest that understanding glial-neuronal interactions and mechanisms involved in regulation of microglial activation are important steps toward identification of therapeutic targets and potential development of treatment strategies for neurodegenerative conditions.
Several lines of evidence suggest that vitamin E protects neurons against oxidative stress by scavenging the free radicals (Halks-Miller et al., 1986; Goodman and Mattson, 1994; Behl, 1999; Butterfield et al., 1999). In animal models, vitamin E reduces the degeneration of hippocampal cells after cerebral ischemia (Hara et al., 1990) and delays neurological deterioration in transgenic mice expressing human variants of β-amyloid precursor protein (Grundman, 2000). Moreover, one clinical trial suggested that vitamin E might help prevent the development of AD or slow further deterioration (Sano et al., 1997) These neuroprotective properties of vitamin E have been attributed largely to prevention of free radical-induced neurotoxicity. However, vitamin E has additional actions that might be pertinent. These include increasing longevity and reducing age-related diseases through influences on the immune system, mediated through reduction of prostaglandin E2 (PGE2) synthesis (Meydani et al., 1995), inhibition of secretion of macrophage migration-inhibitory factor from macrophages (Sakamoto et al., 1998), and inhibition of monocytic cell adhesion (Erl et al., 1997). These findings suggest that a component of the neuroprotective effects of vitamin E may involve the macrophage equivalent in the brain: microglia.
When activated by a neurodegenerative stimulant, microglia increase production of excitotoxic amino acids, NO, and proinflammatory cytokines such as IL-1 and TNFα. Two signal transduction molecules, p38 mitogen-activated protein kinase (p38 MAPK) and nuclear factor κB (NFκB), are critical for the increase in cytokine gene expression and NO production (Nick et al., 1999; Yamakawa et al., 1999). The p38 MAPK is reported to phosphorylate (activate) various transcription factors, including NFκB (Raingeaud et al., 1995; Carter et al., 1999; Nick et al., 1999). NFκB-binding sites have been found in the promoter regions of iNOS (Xie et al., 1993), IL-1 (Hiscott et al., 1993), and TNF-α (Goldfeld et al., 1990), and the expression of these genes has been shown to be regulated by activation of NFκB (Akama et al., 1998; Nick et al., 1999). Interestingly, vitamin E-induced inhibition of monocytic cell adhesion (Erl et al., 1997) and of in vitro TNF-α transcription (Nakamura et al., 1998) is associated with decreased NFκB activation, suggesting that these effects are mediated through vitamin E modulation of cell signal transduction events.
In this study, we used a neuronal—microglial coculture system to define vitamin E effects on the cellular and molecular mechanisms responsible for microglial activation and cytokine expression and to assess the potential benefit of vitamin E in ameliorating detrimental interaction between activated microglia and neurons.
MATERIALS AND METHODS
Cell Cultures
A cultured pheochromocytoma cell line (PC12 cells; ATCC, Rockville, MD) was maintained in 5% fetal bovine serum (FBS; Gibco, Grand Island, NY) and 10% horse serum in RPMI 1640 medium (Gibco). Neuronal differentiated PC12 cells were induced using mouse 2.5S nerve growth factor (NGF; Sigma, St. Louis, MO; 50 ng/ml) in 1% horse serum in RPMI 1640 medium for 7–10 days. The N9 mouse microglial cell line (Corradin et al., 1993) was maintained in 10% FBS in Dulbecco’s modified Eagle’s medium (DMEM). Cell survival was assessed using a 3-[4–5-dimethythiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) assay (Li et al., 1998). Vitamin E (α-tocopherol; Sigma) was dissolved in 100% ethanol as a stock solution, with final experimental ethanol concentrations ≤0.1%. Pure ethanol was used as a vehicle for experimental controls. N9 cell cultures were transferred to serum-free medium at least 24 hr before treatment with vitamin E or lipopolysaccharide (LPS; Sigma). In experiments using the p38 MAPK inhibitor SB203580 (Calbiochem, La Jolla, CA), this was added to final concentrations of 0.5—10 μM 1 hr before stimulation with LPS.
Microglial-Neuronal Co culture
Coculture of neuronal PC12 and N9 microglia was performed as previously described (Li et al., 2000b). N9 cells were grown in culture inserts (pore size 0.4 (μm) and were treated with either LPS (30 ng/ml for 2 hr) with or without preincubation with vitamin E (50 μM for 60 min). The N9 cell culture inserts were washed twice with fresh culture medium and then cocultured with NGF-differentiated PC12 cell culture dishes for 24 hr in serum-free RPMI medium. In this coculture system, the N9 microglial cells do not directly contact PC12 cells but rather may communicate with them through the semipermeable membrane.
RNA Isolation
Total RNA was extracted from cultured cells using TriReagent RNA (Molecular Research Center Inc., Cincinnati, OH) according to the manufacturer’s instructions. Integrity of isolated RNA was verified on agarose gels with ethidium bromide staining, and quantification of isolated RNA was performed using spectrophotometric absorbance at 260 nm. RNA samples employed in this study had 260/280 ratios of 1.7—2.0.
Reverse Transcription Reaction and Polymerase Chain Reaction Amplification
RT-PCR was performed as previously described (Li et al., 2000b). Briefly, for comparisons of mRNA levels among different RNA samples, RT were performed simultaneously using reagents from a single master mix. PCR was performed using reagents from Clontech (Palo Alto, CA). The sequences of primers for mouse IL-1α, TNF-α, iNOS, and G3PDH used in this study are listed in Table 1. PCR amplification was performed through 26 (for IL-1α), 28 (for TNF-α), or 28 (for iNOS) cycles at 94°C for 45 sec, 60°C for 45 sec, and 72°C for 45 sec. For G3PDH, amplification was performed through 26 cycles at 94°C for 45 sec, 55°C for 45 sec, and 72°C for 45 sec. PCR was stopped by final extension for 10 min at 72°C. The PCR cycles for each target gene were determined by sampling 5 μl aliquots every third cycle from cycle 21 onward. The indicated cycles were verified to be within the linear range of product accumulation for the specific PCR. Equal volumes of reaction mixture from each sample were loaded onto 1.5% agarose gels, and fluorescent images were digitally captured for analysis of intensity with NIH Image software. Levels of IL-1α, TNF-α, and iNOS mRNA were normalized relative to that from G3PDH in the same sample.
TABLE I.
Primers Sequence for Detection and Quantification of Mouse cDNA
| Name | Sequence (5′ → 3′) | Size of fragment (bp) |
|---|---|---|
| IL-1α 5′ | AAGATGTCCAACTTCACCTTCAAGGAGAGCCG | 491 |
| IL-1α 3′ | AGGTCGGTCTCACTACCTGTGATGAGTTTTGG | |
| TNF-α 5′ | TTCTGTCTACTGAACTTCGGGGTGATCGGTCC | 354 |
| TNF-α 3′ | GTATGAGATAGCAAATCGGCTGACGGTGTGGG | |
| iNOS 5′ | CCCTTCCGAAGTTTCTGGCAGCAGC | 496 |
| iNOS 3′ | GGCTGTCAGAGCCTCGTGGCTTTGG | |
| G3PDH 5′ | ACCACAGTCCATGCCATCAC | 452 |
| G3PDH 3′ | TCCACCACCCTGTTGCTGTA |
Western Immunoblot Assay
Proteins were extracted using lysis buffer, containing 50 mM Tris HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate (SDS) and were quantified using a micro-BCA assay reagent kit (Pierce, Rockford, IL) as previously described (Li et al., 1998). Aliquots (40 μg each) were loaded onto a 10% SDS-polyacrylamide gel and subjected to electrophoresis at 90 V for 1.5 hr and transferred to Immobilon-P membranes. Membranes were incubated with polyclonal antiphosphospecific p38 MAPK or p38 MAPK antibody (New England Biolabs, Beverly, MA) overnight at 4°C and visualized using the Western-Light Chemiluminescent Detection System (Tropix, Inc., Bedford, MA). Films were digitized and analyzed using NIH Image software, version 1.60.
Electrophoretic Mobility Shift Assay
Nuclear extracts for the EMSA and gel shift assay were performed as previously described (Li et al., 2000a). A double-stranded DNA oligonucleotide sequence fitting the consensus for NFκB binding (5′-AGT TGA GGG GAC TTT CCC AGG C-3′) was used for gel shift assays. The oligonucleotide was end labeled with γ[32P]ATP (300 Ci/mmol; Amersham, Arlington Heights, IL) and T4 polynucleotide kinase (Promega/Serva, Heidelberg, Germany) and purified on a Sephadex G-50 column. Two micrograms of nuclear protein from each sample were incubated with 32P-labeled double-stranded oligonucleotide. The complexes were resolved by nondenaturing gel electrophoresis (6% acrylamide, 0.25 X TBE) and visualized by autoradiography. Antibodies against p50 (Santa Cruz, Santa Cruz, CA) and p65 (Upstate Biologicals, Lake Placid, NY) components of NFκB were used to confirm identity of the κB-binding proteins.
NFκB “Decoy” Oligonucleotide Treatments
Decoy treatment was performed as previously described (Li et al, 2000a). In brief, the phosphothioated oligonucleotides containing an NFκB binding sequence (5′-CCA AGG GGA CTT TCC ATG G-3′) and a control oligonucleotide of scrambled sequence (5′-GAC CAT GTC GTG CAG TAG C-3′; Oligos Etc., Wilsonville, OR) were hybridized with their complements to create double-stranded oligonucleotides. Oligonucleotides were incubated at 1 μM with 47 μg/ml lipofectamine (Gibco) in culture medium for 30 min. These mixtures were added directly to the existing medium of the appropriate cultures for 3 hr. Then, the culture medium was completely replaced with fresh medium containing LPS or vehicle control. After a 24 hr treatment, the cultures were harvested for RNA extraction and RT-PCR assay.
Nitrite Assay
Nitrite assay was performed as previously described (Barger and Harmon, 1997). Culture medium from N9 cells following different treatments was removed and mixed with Griess reagent A and Griess reagent B. The color reaction was measured by absorbance at 540 nm and the value represented as percentage of the released nitrite in untreated cultures.
RESULTS
Vitamin E Inhibits Induction of NO, IL-1α, and TNF-α in LPS-Activated Microglia
LPS-induced activation of N9 cells resulted in significant time-dependent increases in production and release of nitrite, a stable derivative of NO, into the culture medium (Fig. 1). NO production was increased after 12 hr, but not after 6 hr, of exposure of N9 cells to LPS. Further increases were seen after 24 hr of LPS treatment. LPS treatment significantly increased N9 cell expression of IL-lα, TNF-α, and iNOS mRNA, as determined by RT-PCR (Fig. 2A). IL-1α expression increased threefold, TNF-α ninefold, and iNOS eightfold after 24-hr treatment of N9 cells with LPS (30 ng/ml; Fig. 2B).
Fig. 1.

Vitamin E inhibits microglial NO release. Microglia were preincubated with vitamin E (50 μM) for 1 hr and then treated with LPS (30 ng/ml) for the indicated time periods. The amount of NO was determined by measuring the amount of nitrite converted from NO in the media as described in Materials and Methods. Values are expressed as mean ± SEM of four to six samples. **P < 0.01.
Fig. 2.
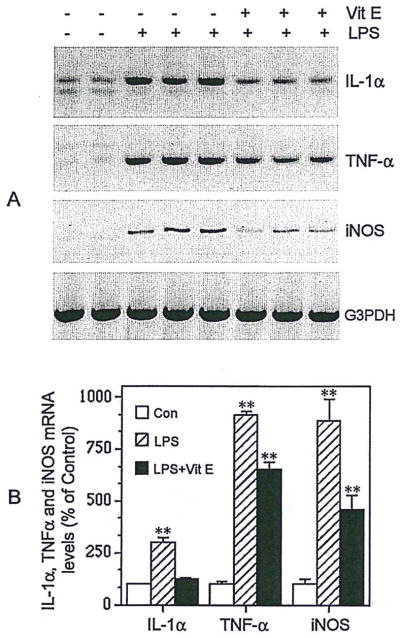
Vitamin E inhibits microglial expression of IL-1α, TNF-α, and iNOS. Illustration (A) and quantification (B) of transcriptional levels of IL-1α, TNF-α, and iNOS in LPS-stimulated microglia, with or without vitamin E pretreatment. RNAs were extracted from these cultures, and RT-PCR was performed to detect the mRNA levels of IL-1α, TNF-α, and iNOS. Values are expressed as mean ± SEM of four to six samples. **P < 0.01.
Incubation of N9 cells with 50 μM vitamin E for 24 hr significantly inhibited LPS-induced NO production (68%; Fig. 1) and also reduced expression of IL-1α (89%), TNF-α (32%), and iNOS (55%; Fig. 2). These inhibitory effects of vitamin E pretreatment also were demonstrable at vitamin E concentrations ranging from 10 to 500 μM (data not shown).
Vitamin E Inhibits Microglial Activation by Down-Regulating the Phosphorylation of p38 MAP Kinase and Suppressing Activation of NFκB
In view of evidence that p38 MAP kinase is critical for LPS-induced cytokine gene expression in human monocytes (Carter et al., 1999) and neutrophils (Nick et al., 1999), we examined the possibility that the effects of vitamin E were exerted upstream of these signal transduction components. The levels of phosphorylated p38 MAPK in N9 cells increased following a 30 min LPS treatment (10 ng/ml). This effect persisted for 60 min (Fig. 3A), gradually decreasing over a 12 hr period (Fig. 3A). Total (phosphorylated plus nonphosphorylated) p38 MAPK levels were unchanged by these treatments (Fig. 3A). Pretreatment of N9 cells with vitamin E (50 μM) significantly inhibited LPS-induced increases in p38 MAPK phosphorylation (81% inhibition; P < 0.05) but did not affect total p38 MAPK protein levels (Fig. 3B).
Fig. 3.

Vitamin E decreases phosphorylation of p38 MAPK. Illustrations of time course of LPS-induced microglial p38 MAPK activation (Phos-p38) and the level of total p38 MAPK (Total-p38) (A) and vitamin E inhibition of p38 MAPK phosphorylation in microglia stimulated by LPS for 1 hr (B) (typical of three separate experiments).
LPS treatment (10 ng/ml) of N9 cells induced significant NFκB DNA-binding activity (Fig. 4A) within 2 hr; both p50 and p65 subunits were shown to contribute to the observed NFκB DNA-binding activity (data not shown). Pretreatment with vitamin E significantly reduced LPS-induced increases in NFκB activity (Fig. 4B).
Fig. 4.
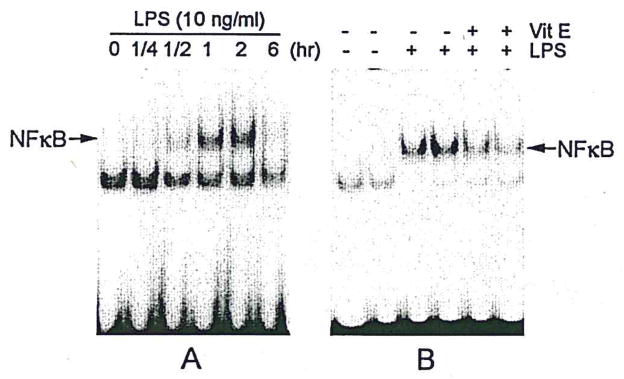
Vitamin E inhibits NFκB activation. Illustrations of time course of LPS-induced microglial NFκB activation (A) and inhibition of NFκB binding activity by vitamin E pretreatment of microglia stimulated with LPS for 2 hr (B) (typical of three separate experiments).
To determine whether activation of both p38 MAPK and NFκB contributes to induction of proinflammatory cytokine expression in N9 microglia, we used SB203580, an inhibitor of p38 MAPK, or a “decoy” approach of NFκB to inhibit the activation of p38 MAPK or NFκB, respectively, before LPS treatment of microglial cultures. Pretreatment of N9 cells with the p38 MAPK inhibitor SB203580 reduced LPS-induced increases in the expression of IL-1α, TNF-α, and iNOS (Fig. 5) and also reduced LPS-induced increases in NO production (data not shown). Pretreatment of N9 cells with NFκB decoy oligonucleotide produced similar results (Fig. 6). Because p38 MAPK directly regulates NFκB activation in neutrophils (Nick et al., 1999), but not monocytes (Carter et al., 1999), we asked whether the p38 MAPK could be a direct upstream regulator for activation of NFκB in N9 microglia. Preincubation of N9 cells with SB203580 (10 μM) had no discernible effect on LPS-induced NFκB binding activity (Fig. 7), suggesting that p38 MAPK does not directly induce activation of NFκB in microglia. This is consistent with the differential effects of NFκB on neutrophils (Nick et al., 1999) and monocytes (Carter et al., 1999). Thus, although both p38 MAPK and NFκB apparently play crucial roles in the process of microglial activation, they may do so through separate, but parallel, signal transduction pathways governing cytokine expression in microglia.
Fig. 5.

Inhibition of p38 MAPK reduces IL-1α, TNF-α, and iNOS expression. Microglia were incubated for 1 hr with a specific inhibitor of p38 MAPK (SB 203580, 10 μM) and then treated with LPS (10 ng/ml) for 24 hr (typical of three separate experiments).
Fig. 6.

NFκB decoy DNA suppresses IL-1α, TNF-α, and iNOS expression. Microglia were treated for 3 hr with either NFκB DNA decoy (Dec DNA) or scrambled control DNA (Con DNA) and then stimulated with LPS (30 ng/ml) for 24 hr (typical of three separate experiments).
Fig. 7.
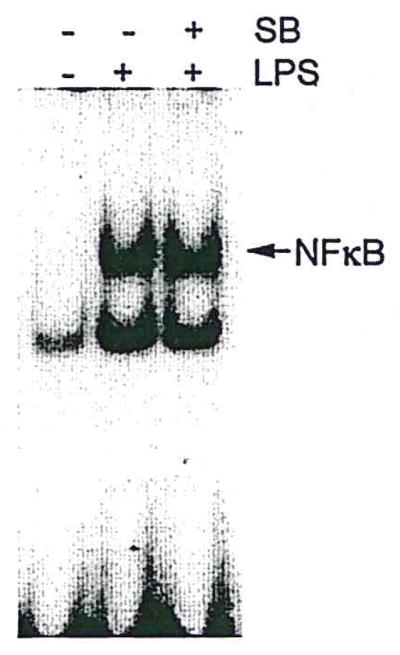
p38 MAPK inhibitor does not alter NFκB binding activity. LPS activation of microglia (2 hr) was unchanged by treatment with SB203580 (1 hr). Nuclear protein was isolated from these cultures, and EMSA was performed as described in Materials and Methods (typical of four separate experiments).
Vitamin E Directly Suppresses the Neurotoxic Effects of LPS-Activated Microglia
To show more directly the ways in which vitamin E may influence neuroprotection through modulation of microglial responses, we utilized a culture arrangement that allowed microglia to be pretreated with vitamin E alone, or LPS alone, or pretreated with vitamin E and then stimulated with LPS. This independent treatment of microglia allowed us separate microglial responses to vitamin E and LPS from effects of these agents on neurons. For this, N9 microglia were plated in basket-type culture-well inserts, and then pretreated with vitamin E alone or vitamin E, followed by a brief exposure to LPS. These culture-well inserts, containing the pretreated N9 cells, were washed thoroughly with fresh medium before placement in coculture with PC12 cells. In these cocultures, pretreatment of N9 cells with LPS alone resulted in neurodegeneration, including marked loss of PC12 cell processes (Fig. 8C,D) and significantly decreased PC12 cell survival (Fig. 9). In contrast, vitamin E pretreatment of LPS stimulated N9 cells ameliorated not only the loss of processes (Fig. 8D,F) but also the detrimental effect of LPS-treated N9 cells on PC12 cell survival (Fig. 9).
Fig. 8.

Vitamin E reduces toxic effects of activated microglia on neuronal cells. Phase-contrast photomicrographs of differentiated PC12 cells before (A,C,E) or after 24 hr of (B,D,F) coculture with N9 microglia. Before coculture, N9 cells were pretreated for 2 hr with LPS (30 ng/ml) alone (LPS) or LPS plus vitamin E (50 μM) pretreatment (LPS + Vit E) or without treatment (CON), and then the N9 culture inserts were completely washed with fresh medium and cocultured with differentiated PC12 cells. Scale bar = 50 μm.
Fig. 9.
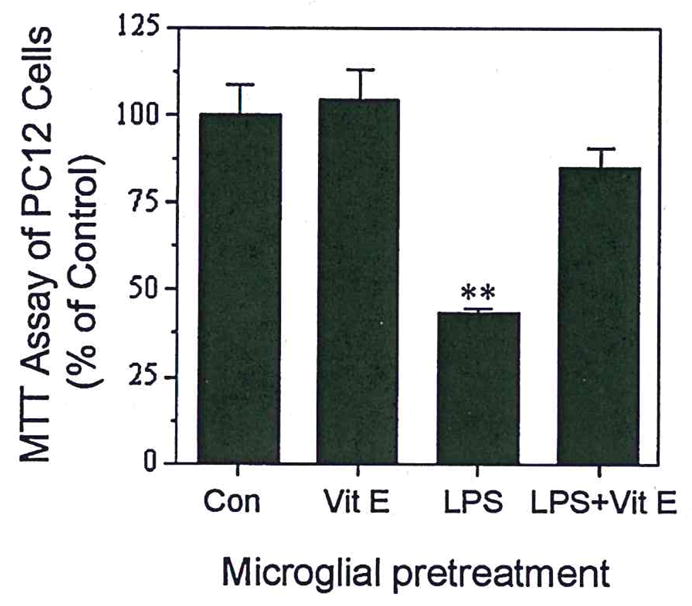
Vitamin E promotes survival of PC 12 cells cocultured with N9 microglia. N9 microglia were preincubated with LPS (30 ng/ml), vitamin E (50 μM), or LPS plus vitamin E pretreatment for 2 hr, and then the N9 culture inserts were completely washed with fresh medium and cocultured with differentiated PC12 cells for 24 hr. MTT was added to the PC12 cell culture 2 hr before harvesting. Values are expressed as mean ± SEM of 6–12 samples. **P < 0.001.
DISCUSSION
In the present work, we found that vitamin E significantly suppressed LPS-induced microglial activation, associated NO production, and induction of IL-1α, TNF-α, and iNOS expression. Furthermore, this vitamin E-mediated suppression of microglial activation significantly reduced the toxic effects of activated microglia on neurons. Our findings that vitamin E inhibits p38 MAPK phosphorylation and NFκB activity in LPS-treated microglia suggest that suppression of these signal steps associated with microglial activation are important in microglial toxicity and in vitamin E neuroprotection. This suggestion is supported by our findings that conventional pharmacological approaches, which also inhibit p38 MAPK or NFκB, result in significant reductions of LPS-induced microglial cytokine and NO production. These data showing vitamin E-mediated decreases in proinflammatory events in microglia provide an additional explanation for the neuroprotective actions of vitamin E in in vivo situations in which there is a potential for glial-neuronal interactions.
The neuroprotective roles of vitamin E have been well documented both in vivo and in vitro (Halks-Miller et al., 1986; Goodman and Mattson, 1994; Behl, 1999; Grundman, 2000). To date, most speculation about possible roles of vitamin E in neuroprotection has focused on direct protection of neurons against toxic free radicals, specifically, protection of neuronal cell membranes, trapping of free radicals, and interruption of deleterious oxidative redox chain reactions. Beyond its recognized antioxidant functions in neurons, vitamin E also regulates immune functions in monocytes and macrophages (Erl et al., 1997; Nakamura et al., 1998; Sakamoto et al., 1998). This suggests that the direct antioxidant neuroprotective effects of vitamin E might be attributable in part to vitamin E effects on microglia, the macrophage equivalent in the brain. Our results show an ameliorating effect of vitamin E on the neurotoxicity of microglia in a coculture system that allowed for vitamin E exposure to microglia only, without vitamin E exposure to neurons. Thus, the neuroprotective effects of vitamin E shown here may be directly attributable to vitamin E effects on microglia.
We provide evidence that the effects of vitamin E involve suppression of basic signal transduction events involved in microglial activation and cytokine expression. The vitamin E-induced inhibition of microglial responses to LPS was correlated with suppression of the activation of p38 MAPK and NFκB, both of which regulate cytokine and iNOS expression (Lee et al., 1994). In the present study, indices of activation associated with gene regulation were suppressed by SB203580, a pharmacological inhibitor of p38 MAPK. Likewise, suppression of NFκB activity with decoy oligonucleotides was sufficient to inhibit LPS-induced microglial responses. In some cell types, p38 MAPK directly regulates NFκB activation and consequent cytokine expression; in other cell types, it does not. For instance, p38 MAPK directly regulates NFκB in neutrophils (Nick et al., 1999) but not in monocytes (Carter et al., 1999). Our observation that the p38 MAPK inhibitor SB203580 did not significantly decrease NFκB binding activity in LPS-activated microglia suggests the existence of cell-specific signal transduction pathways and that in microglia, as in monocytes (Carter et al., 1999), p38 MAPK is not a direct upstream regulator of NFκB.
The toxic effects of microglia on neuronal processes and cell survival and the ameliorating effect of vitamin E demonstrated here may occur in vivo by synergistic effects of cytokines, NO production, free radicals, or other previously described microglia-derived toxic factors (Brenneman et al., 1993; Hensley et al., 1994; Meda et al., 1995; Giulian et al., 1996; Jeohn et al., 1998; Barger and Basile, 2001). Therefore, the ability of vitamin E to attenuate the neurotoxicity of microglia may include mechanisms that are both dependent on and independent of gene expression.
The LPS-induced microglial increases in cytokine expression observed here were significant and prolonged, persisting even after a transient exposure to LPS. Thorough washing of LPS-activated microglia did not abolish their expression of neurotoxicity, of cytokines, or of activation of signaling pathways. These results imply that activation elicits stable changes in microglial phenotype, including persistent gene expression of neurotoxic factors related to activation of p38 MAPK and increased binding activity of NFκB. These activation-related events in microglia and the neurotoxic consequences suggest that further enhancement of microglial activation would occur as a response to the neurotoxicity. This idea is consistent with the concept (Griffin et al., 1989) that persistently activated microglia in neurodegenerative conditions such as Alzheimer’s disease participate in a self-propagating cytokine cycle of cellular and molecular events that are neurodegenerative in nature (Griffin et al., 1998; for review see Griffin et al., 2000). Because vitamin E suppresses microglial intracellular signaling steps involved in activation, further propagation of this cycle would be limited. In such a scenario, vitamin E might be effective in ameliorating microglia-derived aspects of neurodegeneration even after the initiation of a condition such as Alzheimer’s disease.
Acknowledgments
Contract grant sponsor: NIA; Contract grant number: AG12411; Contract grant sponsor: NINDS; Contract grant number: NS35872; Contract sponsor: Donald W. Reynolds Foundation.
The authors thank Mr. Xianrong Mao for technical support and Ms. Pam Free for secretarial support.
References
- Akama KT, Albanese C, Pestell RG, Van Eldik LJ. Amyloid β-peptide stimulates nitric oxide production in astrocytes through an NFκB-dependent mechanism. Proc Nad Acad Sci USA. 1998;95:5795–5800. doi: 10.1073/pnas.95.10.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- Barger SW, Harmon AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature. 1997;388:878–881. doi: 10.1038/42257. [DOI] [PubMed] [Google Scholar]
- Behl C. Vitamin E and other antioxidants in neuroprotection. Int J Vitam Nutr Res. 1999;69:213–219. doi: 10.1024/0300-9831.69.3.213. [DOI] [PubMed] [Google Scholar]
- Brenneman DE, Page SW, Schultzberg M, Thomas FS, Zelazowski P, Burnet P, Avidor R, Sternberg EM. A decomposition product of a contaminant implicated in L-tryptophan eosinophilia myalgia syndrome affects spinal cord neuronal cell death and survival through stereospecific, maturation and partly interleukin-1-dependent mechanisms. J Pharmacol Exp Ther. 1993;266:1029–1035. [PubMed] [Google Scholar]
- Butterfield DA, Koppal T, Subramaniam R, Yatin S. Vitamin E as an antioxidant/free radical scavenger against amyloid β-peptide-induced oxidative stress in neocortical synaptosomal membranes and hippocampal neurons in culture: insights into Alzheimer’s disease. Rev Neurosci. 1999;10:141–149. doi: 10.1515/revneuro.1999.10.2.141. [DOI] [PubMed] [Google Scholar]
- Carter AB, Knudtson KL, Monick MM, Hunninghake GW. The p38 mitogen-activated protein kinase is required for NF-κB-dependent gene expression. The role of TATA-binding protein (TBP) J Biol Chem. 1999;274:30858–30863. doi: 10.1074/jbc.274.43.30858. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- Corradin SB, Mauel J, Donini SD, Quattrocchi E, Ricciardi-Castagnoli P. Inducible nitric oxide synthase activity of cloned murine microglial cells. Glia. 1993;7:255–262. doi: 10.1002/glia.440070309. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erl W, Weber C, Wardemann C, Weber PC. α-Tocopheryl succinate inhibits monocytic cell adhesion to endothelial cells by suppressing NF-κ B mobilization. Am J Physiol. 1997;273:H634–640. doi: 10.1152/ajpheart.1997.273.2.H634. [DOI] [PubMed] [Google Scholar]
- Giulian D, Haverkamp LJ, Yu JH, Karshin W, Tom D, Li J, Kirkpatrick J, Kuo LM, Roher AE. Specific domains of β-amyloid from Alzheimer plaque elicit neuron killing in human microglia. J Neurosci. 1996;16:6021–6037. doi: 10.1523/JNEUROSCI.16-19-06021.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfeld AE, Doyle C, Maniatis T. Human tumor necrosis factor α. gene regulation by virus and lipopolysaccharide. Proc Natl Acad Sci USA. 1990;87:9769–9773. doi: 10.1073/pnas.87.24.9769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman Y, Mattson MP. Secreted forms of β-amyloid precursor protein protect hippocampal neurons against amyloid β-peptide-induced oxidative injury. Exp Neurol. 1994;128:1–12. doi: 10.1006/exnr.1994.1107. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, Araoz C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA. 1989;86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer’s disease: the potential role of a “cytokine cycle” in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WS, Nicoll JA, Grimaldi LM, Sheng JG, Mrak RE. The pervasiveness of interleukin-1 in alzheimer pathogenesis: a role for specific polymorphisms in disease risk. Exp Gerontol. 2000;35:481–487. doi: 10.1016/s0531-5565(00)00110-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundman M. Vitamin E and Alzheimer disease: the basis for additional clinical trials. Am J Clin Nutr. 2000;71:630S–636S. doi: 10.1093/ajcn/71.2.630s. [DOI] [PubMed] [Google Scholar]
- Halks-Miller M, Henderson M, Eng LF. α-Tocopherol decreases lipid peroxidation, neuronal necrosis, and reactive gliosis in reaggregate cultures of fetal rat brain. J Neuropathol Exp Neurol. 1986;45:471–484. doi: 10.1097/00005072-198607000-00008. [DOI] [PubMed] [Google Scholar]
- Hara H, Kato H, Kogure K. Protective effect of α-tocopherol on ischemic neuronal damage in the gerbil hippocampus. Brain Res. 1990;510:335–338. doi: 10.1016/0006-8993(90)91386-u. [DOI] [PubMed] [Google Scholar]
- Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscott J, Marois J, Garoufalis J, D’Addario M, Roulston A, Kwan I, Pepin N, Lacoste J, Nguyen H, Bensi G, Fenton M. Characterization of a functional NF-κB site in the human interleukin 1 β promoter: evidence for a positive autoregulatory loop. Mol Cell Biol. 1993;13:6231–6240. doi: 10.1128/mcb.13.10.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeohn GH, Kong LY, Wilson B, Hudson P, Hong JS. Synergistic neurotoxic effects of combined treatments with cytokines in murine primary mixed neuron/glia cultures. J Neuroimmunol. 1998;85:1–10. doi: 10.1016/s0165-5728(97)00204-x. [DOI] [PubMed] [Google Scholar]
- Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens I, Fisher SH, Livi GP, White JR, Adams JL, Young PR. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang J, Sheng JG, Liu L, Barger SW, Jones RA, Van Eldik LJ, Mrak RE, Griffin WS. S100B increases levels of β-amyloid precursor protein and its encoding mRNA in rat neuronal cultures. J Neurochem. 1998;71:1421–1428. doi: 10.1046/j.1471-4159.1998.71041421.x. [DOI] [PubMed] [Google Scholar]
- Li Y, Barger SW, Liu L, Mrak RE, Griffin WS. S100B induction of the proinflammatory cytokine interleukin-6 in neurons. J Neurochem. 2000a;74:143–150. doi: 10.1046/j.1471-4159.2000.0740143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu L, Kang J, Sheng JG, Barger SW, Mrak RE, Griffin WS. Neuronal-glial interactions mediated by interleukin-1 enhance neuronal acetylcholinesterase activity and mRNA expression. J Neurosci. 2000b;20:149–155. doi: 10.1523/JNEUROSCI.20-01-00149.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by β-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Meydani SN, Wu D, Santos MS, Hayek MG. Antioxidants and immune response in aged persons: overview of present evidence. Am J Clin Nutr. 1995;62:1462S–1476S. doi: 10.1093/ajcn/62.6.1462S. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS. Interleukin-1 and the immunogenetics of Alzheimer disease. J Neuropathol Exp Neurol. 2000;59:471–476. doi: 10.1093/jnen/59.6.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Goto M, Matsumoto A, Tanaka I. Inhibition of NF-κB transcriptional activity by α-tocopheryl succinate. Biofactors. 1998;7:21–30. doi: 10.1002/biof.5520070104. [DOI] [PubMed] [Google Scholar]
- Nick JA, Avdi NJ, Young SK, Lehman LA, McDonald PP, Frasch SC, Billstrom MA, Henson PM, Johnson GL, Worthen GS. Selective activation and functional significance of p38α mitogen-activated protein kinase in lipopolysaccharide-stimulated neutrophils. J Clin Invest. 1999;103:851–858. doi: 10.1172/JCI5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- Sakamoto W, Nishihira J, Fujie K, Handa H, Ozaki M, Yukawa S. Inhibition of macrophage migration inhibitory factor secretion from macrophages by vitamin E. Biochim Biophys Acta. 1998;1404:427–434. doi: 10.1016/s0167-4889(98)00070-6. [DOI] [PubMed] [Google Scholar]
- Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E, Schneider LS, Thal LJ. A controlled trial of selegiline, α-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study [see comments] N Engl J Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- Scali C, Prosperi C, Giovannelli L, Bianchi L, Pepeu G, Casamenti F. β(1–40) Amyloid peptide injection into the nucleus basalis of rats induces microglia reaction and enhances cortical gamma-aminobutyric acid release in vivo. Brain Res. 1999;831:319–321. doi: 10.1016/s0006-8993(99)01492-4. [DOI] [PubMed] [Google Scholar]
- Tsai SY, McNulty JA. Microglia in the pineal gland of the neonatal rat: characterization and effects on pinealocyte neurite length and serotonin content. Glia. 1997;20:243–253. doi: 10.1002/(sici)1098-1136(199707)20:3<243::aid-glia8>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Xie QW, Whisnant R, Nathan C. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J Exp Med. 1993;177:1779–1784. doi: 10.1084/jem.177.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa T, Eguchi S, Matsumoto T, Yamakawa Y, Numaguchi K, Miyata I, Reynolds CM, Motley ED, Inagami T. Intracellular signaling in rat cultured vascular smooth muscle cells: roles of nuclear factor-κB and p38 mitogen-activated protein kinase on tumor necrosis factor-α production. Endocrinology. 1999;140:3562–3572. doi: 10.1210/endo.140.8.6914. [DOI] [PubMed] [Google Scholar]