

Abstract
In this study, we have showed that GCNT2, a gene-encoding glucosaminyl (N-acetyl) transferase 2, I-branching enzyme, is overexpressed in highly metastatic breast cancer cell lines of human and mouse origin and basal-like breast tumor samples. GCNT2 expression is also significantly correlated to the metastatic phenotype in breast tumor samples. Functional studies showed that ectopic expression of GCNT2 enhances cell detachment, adhesion to endothelial cells, cell migration and invasion in vitro, and lung metastasis of breast cancer cells in vivo. Knockdown of GCNT2 expression decreases cell migration and invasion in vitro and lung metastasis in vivo. We have further shown the involvement of GCNT2 in the epithelial-to-mesenchymal transition (EMT). Specifically, the expression of E-cadherin is significantly changed upon GCNT2 expression at the protein level but not at the RNA level. Moreover, we have shown that GCNT2 is a direct target of the TGF-β–smad pathway and that change in GCNT2 expression modulates EMT induced by TGF-β1 treatment. Finally, we have shown that diminution of the glycosyltransferase activity of I-branching β-1, 6-N-acetylglucosaminyl transferase 2 (GCNT2) abrogates its cell migration and invasion-promoting function and synergistic effect with TGF-β to induce EMT. Our study for the first time showed that GCNT2 is a novel gene contributing to breast cancer metastasis with preferential expression in basal-like breast cancer. Moreover, we discovered that involvement of GCNT2 in EMT and TGF-β signaling, and further glycosylation modification of E-cadherin by GCNT2, are the underlying integrative mechanisms for breast cancer metastasis, implying that blocking TGF-β/GCNT2 signaling is a promising approach for targeting metastatic breast cancer.
Introduction
Metastatic breast cancer is generally considered incurable. The primary reason for this is because gene targets underlying the metastatic process have not been clearly defined, further hindering the development of targeted therapies (1, 2). This is especially true for basal-like breast cancer, which exhibits an aggressive and early pattern of distant metastasis phenotypes (3, 4). In an effort to identify novel genes that play essential roles in breast cancer metastasis, our group conducted a cross-species integrative expression profiling assay, which combines the use of cell models of human and mouse origins and the microarray expression technique. A total of 34 genes, among which 22 genes are upregulated and 12 are downregulated, were identified with marked expression level difference between highly and poorly metastatic cell lines (5). I-branching β-1, 6-N-acetylglucosaminyl transferase 2 (GCNT2), a member of the β-1, 6-N-acetylglucosaminyltransferase family that plays a critical role in glycosylation, is a novel metastasis-related gene candidate.
Over the past decade, glycosylation alterations of proteins and lipids have been implicated in malignant transformation. For example, increased expression of MGAT5, the gene-encoding mannosyl (α-1, 6-)-glycoprotein β-1, 6-N-acetyl-glucosaminyltransferase that adds β1, 6 GlcNAc branching to N-glycans, is correlated with the progression of invasive malignancies (6, 7). Conversely, suppression of tumor growth and metastasis was observed in Mgat5-deficient mice (8). Mgat5 promotes malignant transformation and invasiveness of cancers by generating β1, 6-poly N-acetyllactosamine on N-glycans of certain signaling molecules (e.g., epidermal growth factor receptor and Integrin) such that they are retained on the cell surface longer and augment their signals for proliferation and invasion of cancer cells (9, 10). On the other hand, MGAT5 is antagonized by MGAT3, another GlcNAc transferase that adds bisecting GlcNAc to N-glycans. The presence of bisecting GlcNAc inhibits the formation of β1, 6-N-acetyllactosamine by MGAT5. Therefore, MGAT3 acts as a tumor metastasis suppressor (11-13). Moreover, a subset of malignant human breast cancer cell lines was found to have elevated MGAT5 expression. Consistently, the production of MGAT5, as detected by lectin L-PHA, was found to be increased in a subset of primary breast cancers and correlated with their invasiveness (14, 15). Furthermore, GCNT1, which encodes a core 2 β1, 6-N-acetyl-glucosaminyltransferase, is overexpressed in various human cancers, including prostate, colorectal, and pancreatic cancers (16, 17). Specifically, GCNT1 overexpression enhances the metastatic potential of the testicular germ cell tumor (18). However, as the homologous gene of GCNT1, the precise role of GCNT2 in breast tumorigenesis and metastasis remains elusive.
In this study, we have shown that the expression of GCNT2 is closely related to basal-like and metastatic phenotypes in both breast cancer cell lines of human and mouse origins and human breast tumor samples. We then intensively studied the functional role of GCNT2 in typical oncogenic properties including cell proliferation, colony formation, migration and invasion in vitro with multiple cell lines, and lung metastasis in vivo by using an experimental animal model. We then showed that GCNT2 plays a role in epithelial-to-mesenchymal transition (EMT). We also found that GCNT2 is regulated by TGF-β1 and is required for TGF-β1–induced EMT. Finally, we showed that enzymatic activity is required for cell migration, invasion, and the EMT-promoting function of GCNT2.
Materials and Methods
Cell lines and cell culture
Human breast cancer cell lines, MDA-MB 231, MDA-MB 435, MDA-MB 361, MCF7, and BT20 were obtained from American Type Culture Collection (ATCC) and were grown by ATCC recommendations. SUM1315, SUM159, and SUM149 were obtained from Dr. Stephen P. Ethier’s laboratory and cultured as previously described (19). Three mouse breast cancer cell lines, 67NR, 168FARN, and 4T1 were cultured as previously reported (20). Human and mouse mammary epithelial cell lines HMLE and EpRas were generously provided by Robert A. Weinberg of MIT, and cultured as previously mentioned (21). NMuMG cell was provided by Lindsey D. Mayo at Indiana University, and cultured with Dulbecco’s modified Eagle’s medium medium with 4.5 g/L glucose, 10 mg/mL insulin, and 10% FBS. All cell lines were authenticated upon receipt by comparing them to the original morphologic and growth characteristics.
Tissue microarray and immunohistochemistry assay
A breast cancer tissue array, 150 cores including 75 cases of normal, reactive, premalignant, and malignant tissues of the breast in duplicates were purchased from Pantomics. Standard procedures were used for the immunohistochemistry (IHC) assay. The GCNT2 antibody was purchased from Sigma (Sigma-Aldrich) and the working dilution is 1:200.
Establishment of stable cell lines
One set of mouse short-hairpin RNAs (shRNA) and the human full-length cDNA for GCNT2 was purchased from Open Biosystems. Two mutant forms of GCNT2 were created by using the Stratagene’s QuikChange kit based on wild-type GCNT2. The Trans-Lentiviral Packaging System and the ViraPower Lentiviral Expression System (Invitrogen) were used to produce shRNA and overexpressing lentiviruses, respectively. shRNA lentiviruses were used to silence GCNT2 expression in 4T1 and NMuMG cells and overexpressing lentiviruses were introduced into HMLE, EpRas, MCF7, Madin Darby canine kidney (MDCK), and NMuMG cells. Stable cells were generated after 2 weeks of antibiotic selection (22, 23).
Cell proliferation, colony formation, cell invasion, and migration assays
All these assays were done as previously described (23).
Western blotting
A Western blot was done according to regular protocol (5, 22).
Tumorigenesis and metastasis assay
For EpRas cells, 2.5 × 105 cells were injected subcutaneously into the mammary glands of 5-week-old, female NCR Nu/Nu mice (Taconics). For 4T1 cells, 2.5 × 104 cells were injected into the mammary gland of female BALB/C mice (The Jackson Laboratory). Eight to 10 animals per group were used in every experiment. Four weeks later after tumor cell injections, mice were sacrificed and tumors were removed and weighed individually. Lungs were collected and embedded into paraffin blocks. Standard hematoxylin and eosin (H&E) staining of paraffin-embedded tissue was done for histologic examination of metastases. The lung metastasis fields in individual mice were counted as a percentile of the whole lung section area under the parallel dissection scope. All experimental animal protocols were approved by the Institutional Animal Care and Use Committee.
Statistical analysis
Data are presented as mean ± SD. Statistical analysis was done by using a student’s t test for significance. P value less than 0.05 was considered significant. The Cochran–Armitage test was used to analyze the statistically significant linear trend between the expression of GCNT2 and metastasis phenotypes.
Results
GCNT2 is overexpressed in highly metastatic breast cancer and its expression correlates with adverse pathologic phenotypes
To confirm GCNT2 upregulation in metastatic breast cancer cells, we first determined the expression of 3 isoforms of GCNT2 (24) in a set of human breast cancer cell lines with different metastatic characteristics. All 3 isoforms of GCNT2 have a very similar expression pattern. High GCNT2 expression was shown in most highly metastatic cancer cell lines (all of them are basal-like cells) and low expression of GCNT2 was seen in most nonmetastatic or low-metastatic cell lines (MDA361 and MCF7 are luminal type and BT20 is basal like; Fig. 1A). However, isoform B and C have significantly low expression levels compared with isoform A (Supplementary Fig. S1). Isoform A of GCNT2 is then used for the detailed functional study. Moreover, correlation of high expression of GCNT2 and metastatic phenotype was also confirmed in mouse model cell lines (Fig. 1B). Overall, these results suggest that overexpression of GCNT2 correlates to metastatic characteristics in breast cancer cell lines.
Figure 1.
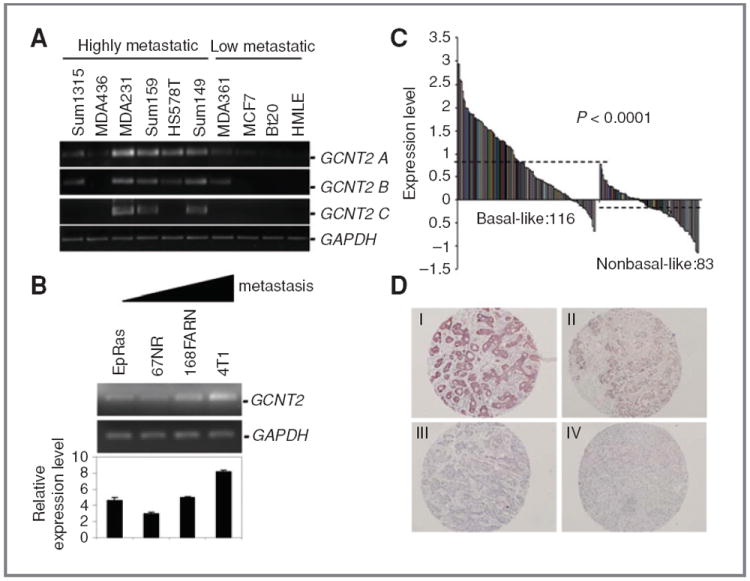
Expression pattern of GCNT2 in breast cancer. A, the expression pattern of 3 isoforms of GCNT2 in human breast cancer cell lines. The highly (+) and low (−) metastatic breast cancer cell lines are marked as indicated. B, GCNT2 RNA expression in a set of mouse cell lines with different metastatic capabilities. The triangular bar indicates the increase in metastatic capability. C, the expression level of GCNT2 in breast tumor samples (basal-like sample, 116 cases and nonbasal-like sample, 83 cases). Dashed lines indicate the average expression level. D, expression of GCNT2 protein in breast tumors. The panels show representative figures of the IHC assay. I is an example of strong staining. II is intermediate staining. III is weak staining. IV is no staining.
To investigate the expression pattern of GCNT2 in clinical breast tumor samples, we first analyzed a breast cancer gene expression dataset obtained from the Netherlands Cancer Institute, which contains 116 basal-like breast tumors and 83 nonbasal-like breast tumors (25). We found that the levels of GCNT2 expression were significantly higher in basal-like breast tumors than in nonbasal-like breast tumors (P < 0.0001; Fig. 1C). We then conducted an IHC to investigate the GCNT2 expression in breast tumors by using a tissue microarray and a pan-isoform GCNT2 antibody. The Cochran–Armitage trend test reveals that there is a statistically significant linear correlation between the expression of GCNT2 and metastasis phenotypes (P = 0.006) in breast cancer (Fig. 1D, Supplementary Fig. S2). Furthermore, numerous independent microarray data sets showed that a high level of GCNT2 expression is strongly correlated with basal-like tumors (26), BRCA1 mutation tumors (27), and estrogen receptor or progesterone receptor-negative tumors (28, 29). Moreover, expression of GCNT2 increased concomitantly with breast cancer grade (Ref. 30; Supplementary Fig. S3). Overall, our laboratory findings are consistent with the results from data mining, suggesting that upregulation of GCNT2 expression contributes to adverse pathologic phenotypes and progression of breast cancer.
The effect of GCNT2 overexpression on oncogenic properties
To determine whether GCNT2 expression affects regular oncogenic properties, we established GCNT2 A isoform overexpression model cell lines based on HMLE, EpRas, and MCF7 cells using a lentivirus expression system (Fig. 2A, Supplementary Figs. S4 and S5). We compared cell proliferation and colony formation of the HMLE, EpRas, and MCF7 cells carrying either GCNT2 or control vector. Our results showed that both gene and control vector overexpressing cells originated from all 3 cell lines grew at similar rates and had similar colony number in vitro (Fig. 2A and B, Supplementary Figs. S4 and S5). Similar results were also observed in MDCK cells (Supplementary Fig. S6). These results clearly show that the ectopic expression of GCNT2 has minimal effects on the proliferation and colony formation of mammary epithelial cells.
Figure 2.
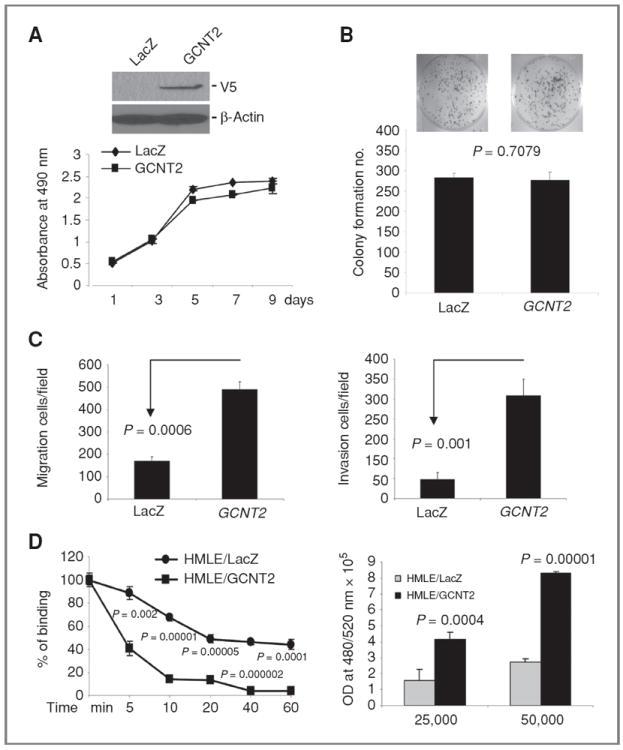
The effect of GCNT2 on cell proliferation, colony formation, cell migration, invasion, cell detachment, and attachment. A, ectopic expression of GCNT2 in HMLE cells detected by Western blotting (top). The effect of ectopic expression of GCNT2 on cell proliferation (low). B, the effect of ectopic expression of GCNT2 on colony formation in HMLE cells. C, the effect of ectopic expression of GCNT2 on cell migration (left) and invasion (right) in HMLE cells. D, the effect of GCNT2 on cell detachment and attachment. The left panel shows that ectopic expression of GCNT2 led to an increase in detachment of HMLE cells from plates when treated with EDTA (0.02%) at different times. The right panel shows that ectopic expression of GCNT2 led to an increase of cell adhesion to HUVEC cells. 25,000 and 50,000 HMLE-derived cells were plated on top of the monolayer of HUVEC cells.
We then checked the effect of GCNT2 on cell migration and invasion by using a BD Biocoat migration chamber and matrigel invasion chamber. We found that ectopic expression of GCNT2 in HMLE, EpRas, and MCF7 cells led to a significant increase in cell migration and invasion (Fig. 2C, Supplementary Figs. S4, S5, and S7). MDCK cells with ectopic expression of GCNT2 also showed significant increase in cell migration (Supplementary Fig. S6). Furthermore, we found that overexpression of GCNT2 significantly increased the detachment of HMLE cells from the plate (P < 0.002; Fig. 2D, left). In addition, the adhesion of HMLE cells to human umbilical vein endothelial cell (HUVEC) was also found significantly increased in cells with GCNT2 expression (Fig. 2D, right). This result is very similar to what is reported for Snail and MGAT5 genes (31, 32).
The effect of GCNT2 expression knockdown on oncogenic properties
On the basis of highly metastatic 4T1 cells, which also have high GCNT2 expression, we established the GCNT2 A isoform knockdown model cell line. Two clones (4T1/shRNA1 and 4T1/shRNA5) with significant inhibition of GCNT2 expression, as shown by reverse transcriptase PCR (RT-PCR; there is no specific antibody for A isoform; Fig. 3A), were used to check the effect of GCNT2 knockdown on cell proliferation and colony formation. We found that 2 clones with GCNT2 expression knockdown showed a marked decrease in cell proliferation and colony formation compared with nontarget control cells (Fig. 3B and C). We further found that 4T1 cells with GCNT2 knockdown showed a significant decrease in migration (80%, P = 0.0001) and invasion (80%, P = 0.002; Fig. 3D and Supplementary Fig. S8).
Figure 3.
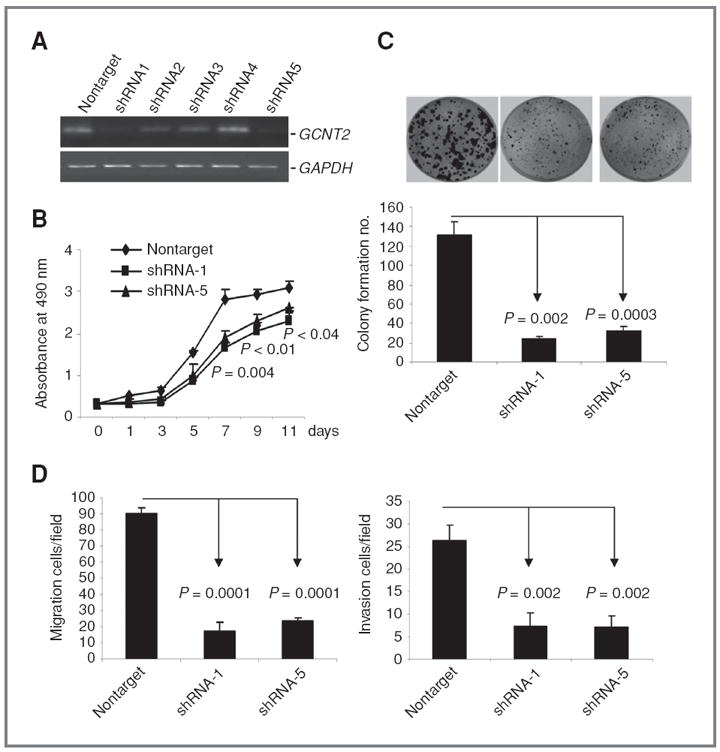
The effect of knockdown of GCNT2 on cell proliferation, colony formation, cell proliferation, and invasion. A, knockdown of GCNT2 in 4T1 cells with shRNA. Two clones (shRNA1 and shRNA5) show significant reduction of GCNT2 expression. B, two clones with significant GCNT2 expression knockdown show a marked decrease in cell proliferation rates compared with nontarget control by MTT assay. C, two clones with significant GCNT2 expression knockdown show a marked decrease in colony formation compared with nontarget control. D, two clones with significant GCNT2 expression knockdown show a marked decrease in migration (left) and invasion (right) compared with the nontarget control.
GCNT2 contributes to distal metastasis in vivo
We then injected EpRas cells expressing either control vector or GCNT2 into the mammary glands of nude mice and examined their lungs for metastasis 4 weeks after injection. Detailed quantification was conducted with H&E staining on the lung sections. In lung sections from 10 mice injected with the EpRas cell expressing control vector, only 3 cases showed a more than 2% metastatic tumor in whole sections. In contrast, the metastatic area made up at an average of 9% of the sections in all 10 lung sections from mice injected with EpRas cells overexpressing GCNT2 (Fig. 4A, P = 0.002). Moreover, the tumor burden in the control vector group and the GCNT2 overexpression group had no significant difference during the 4 weeks (Fig. 4B). This data, along with the result of in vitro analysis, confirms a role of GCNT2 as a metastasispromoting gene.
Figure 4.
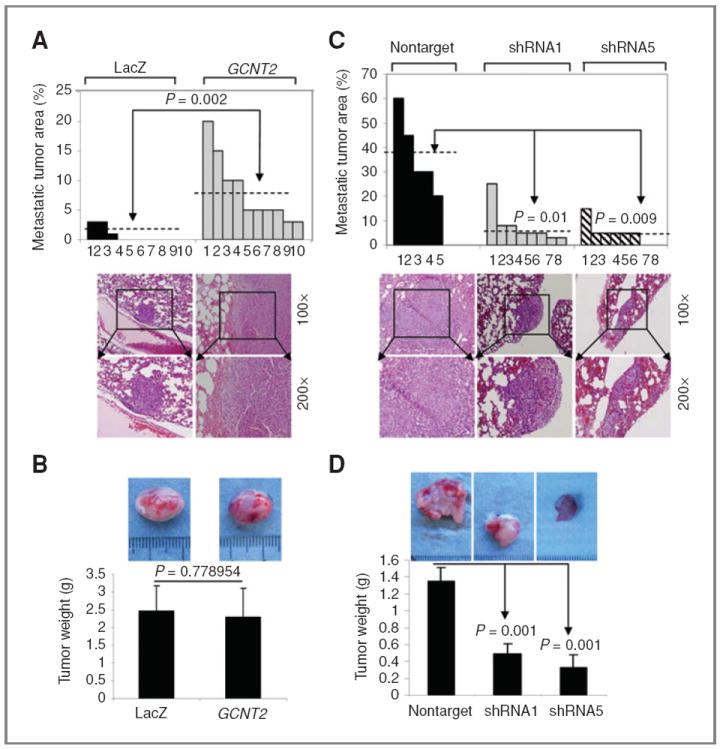
The effect of GCNT2 on distal metastasis in vivo. A and C, quantification of metastasis in vivo. A, 2 populations of EpRas cells with the vector (LacZ) or the GCNT2 gene were used in 10 mice, respectively. C, 4T1 cells with nontarget control and GCNT2 shRNA1 and shRNA5 were used in 5 and 8 mice, respectively. Representative H&E staining pictures of lung sections from mice injected with genetically modified EpRas or 4T1-derived cells are shown as low panels (magnification, 100× and 200×). B, representative figures of the tumor burdens for EpRas cells with vector control and GCNT2 expression. No significant difference was observed (P = 0.778954). D, representative figures of the tumor burdens for 4T1 cells with nontarget control, shRNA clone1, and shRNA clone5. Significant decrease in tumor weight was observed in GCNT2 knockdown clones.
To further validate the role of GCNT2 in spontaneous metastasis, 2 clones of 4T1 derivative cells with GCNT2 knockdown and a nontarget control were injected into the fat pads of BALB/C mice. We observed that 4T1 cells with nontarget control have a similar metastatic propensity as the parental 4T1 cells (30%–40% lung involvement), which showed an average of 37% metastasis tumor in the lung sections. In contrast, the 4T1 cells with GCNT2 knockdown showed an average of 7.75% and 5% metastatic tumor in the lung section for the shRNA1 clone (P = 0.01) and the shRNA5 clone (P = 0.009), respectively (Fig. 4C). Moreover, tumor burdens in 4T1 cells with GCNT2 knockdown also had significant shrinkage compared with nontarget control (Fig. 4D).
GCNT2 is involved in EMT
We next investigated whether GCNT2 is involved in the EMT process using established model cell lines with GCNT2 overexpression and knockdown. We first observed that downregulation of GCNT2 caused increased levels of epithelial marker E-cadherin and α, β-catenin and decreased levels of mesenchymal markers fibronectin and vimentin in 4T1 cells (Fig. 5A, left and Supplementary Fig. S9). Moreover, downregulation of GCNT2 led to a significant cellular morphologic shift from mesenchymal to epithelial in 4T1 cells, which is showed by a more spindle like, scattered distribution in 4T1/vector cells, and a more cobblestone-like appearance in 4T1/shRNA1 cells (Fig. 5B). Conversely, we observed that GCNT2 overexpression led to reduced levels of epithelial markers E-cadherin and α-catenin, and an increased level of the mesenchymal marker fibronectin in HMLE cells (Fig. 5A, right and Supplementary Fig. S9). No marked morphologic change was shown in this condition. Similarly, we observed significant inhibition of E-cadherin expression, but no other markers, in the MDCK cell without marked morphologic change when GCNT2 was expressed (Supplementary Fig. S10). These data strongly suggest the involvement of GCNT2 in the EMT process. However, ectopic expression of GCNT2 alone is not sufficient to trigger a complete EMT.
Figure 5.
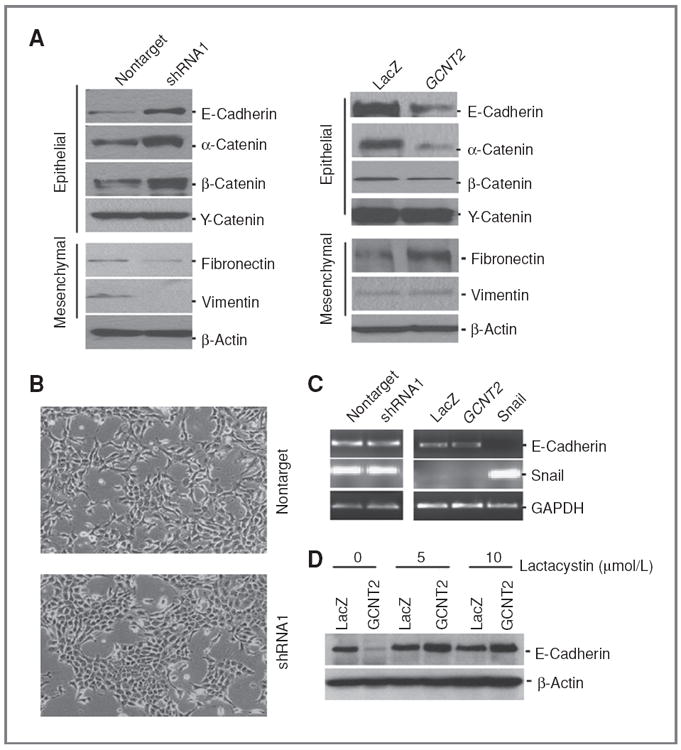
GCNT2 is actively involved in EMT. A, the effect of GCNT2 on the EMT program. Left panels show the effect of knockdown of GCNT2 expression on the EMT program in 4T1 cells. Right panels show the effect of ectopic expression of GCNT2 on the EMT program in HMLE cells. B, representative pictures show morphologic change after knockdown of GCNT2 in 4T1 cells. C, the expression of E-cadherin at the RNA level by knockdown of GCNT2 in 4T1 cells (left) and ectopic expression of GCNT2 in HMLE cells (right). The Snail gene is used as a control. D, the effect of lactacystin on E-cadherin protein level in cells with and without GCNT2 expression.
We then conducted RT-PCR to investigate E-cadherin expression upon overexpression and siRNA knockdown of GCNT2. We found that neither overexpression of GCNT2 in HMLE cells nor knockdown of GCNT2 with shRNA in 4T1 cells changed the expression of E-cadherin at the RNA level (Fig. 5C). In contrast, ectopic expression of the Snail gene significantly inhibited expression of E-cadherin at the RNA level. In addition, HMLE cells with and without GCNT2 overexpression were treated with different doses of proteasome inhibitor lactacystin. We found that the decrease of E-cadherin protein expression caused by GCNT2 expression was inhibited by the treatment of lactacystin (Fig. 5D). These data, along with the data obtained above, suggest that GCNT2 modulates the expression of E-cadherin through a posttranslational mechanism, which is completely different from the mechanistic action of transcription factors (33, 34).
GCNT2 is involved in TGF-β signaling and multiple cell signaling pathways
To investigate whether GCNT2 is involved in TGF-β signaling, we treated the NMuMG cell with TGF-β1 and detected an increase of GCNT2 gene expression (Fig. 6A). Furthermore, we found that treatment of TGF-β led to the activation of pSmad2 and SB431542 (TβR-II inhibitor) treatment totally erased the activation of pSmad2 in TGF-β–treated and untreated cells (Supplementary Fig. S11A). More importantly, GCNT2 expression was found concomitantly changed with the activation of pSmad2 (Supplementary Fig. S11B), suggesting GCNT2 expression is TGF-β/Smad dependent. We next knocked down GCNT2 expression in NMuMG cells with shRNA technique and produced 3 clones with a significant decrease of GCNT2 expression (Fig. 6B). Nontarget control cells and shRNA1 clone cells (with most significant GCNT2 knockdown) were then treated with TGF-β1. We found that TGF-β1–induced significant EMT in nontarget control cells within 5 days, but not in shRNA1 clone cells (Fig. 6C). Furthermore, E-cadherin expression was inhibited in nontarget cells with TGF-β1 treatment. However, E-cadherin expression was unchanged in shRNA1 clone cells with GCNT2 knockdown (Fig. 6D). Conversely, ectopic expression of wild-type GCNT2, along with TGF-β1 treatment, induces significant morphologic change of EMT and corresponding molecular changes, such as decreased Ecadherin expression in NMuMG cells compared with vector control cells (Supplementary Fig. S12). All these results indicate that GCNT2 is involved in and required for TGF-β1–induced EMT.
Figure 6.
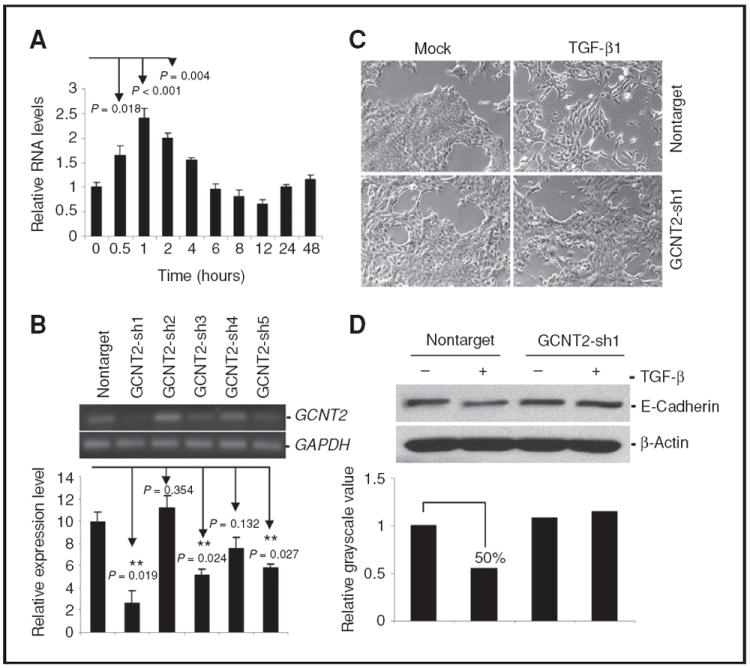
The involvement of GCNT2 in TGF-β signaling. A, induction of GCNT2 by TGF-β1. NMuMG cells were treated with 5 ng/mL TGF-β1 for 2 days. GCNT2 expression is confirmed by quantitative real-time RT-PCR. B, knockdown of GCNT2 in NMuMG cells with shRNA was confirmed by RT-PCR (top) and real-time RTPCR (bottom). C, representative pictures show the morphologic change after treatment of TGF-β1 in NMuMG cells with nontarget vector and shRNA1. D, Western blotting results show the effect of GCNT2 on E-cadherin expression after TGF-β1 treatment in NMuMG cells with nontarget control and shRNA1. The lower panel shows the quantification of the signal intensity.
In addition, we checked the effect of GCNT2 on common cell signaling pathways. Our data showed that overexpression of GCNT2 led to the activation of the ERK and AKT signaling pathway, which is represented by upregulation of pAKT, pSrc, pS6K, and pERK in HMLE and EpRas cells (Supplementary Fig. S13). Conversely, knockdown of GCNT2 in 4T1 cells led to the inhibition of AKT and ERK signaling.
Glycosyltransferase activity is required for GCNT2 in promoting cell migration, invasion, and EMT
According to published genetic studies, substitutions of G1049A (Gly350Glu) and G1154A (Arg385His) in exon 3 of GCNT2 are known to result in an i phenotype (i blood group antigen) and congenital cataracts in patients, which is a result of enzymatic activity elimination of GCNT2 (35). To test whether enzymatic activity is required for the metastasispromoting function of GCNT2, we introduced G1049A and G1154A mutations into the wild-type GCNT2 and generated corresponding lentiviruses. Stable cell models were then established with this virus to infect the EpRas and NMuMG cells (Fig. 7A). Further studies showed that ectopic expression of mutant GCNT2 into EpRas cells does not alter cell migration and invasion compared with vector control. These results are markedly different from results obtained with wild-type GCNT2 expression (Fig. 7B and Supplementary Fig. S14A). Furthermore, ectopic expression of mutant GCNT2 in cells with wild-type GCNT2 could not inhibit the promoting effect of wild-type GCNT2 on cell migration and invasion, suggesting that mutant GCNT2 does not have a dominant negative effect (Supplementary Fig. S14B). This result is consistent with the genetic evidence showing that patients with I phenotype must have 2 mutant GCNT2 allele (35). In addition, we found that only the expression of wild-type GCNT2 but not the mutant GCNT2 in the GCNT2 knockdown cells can sensitize the NMuMG cells to TGF-β1 treatment, which is shown by changing of the E-cadherin expression along with the morphologic alterations (Fig. 7C and D). Taken together, these results suggest that the metastasis-promoting effect of GCNT2 is dependent on its glycosyltransferase activity.
Figure 7.
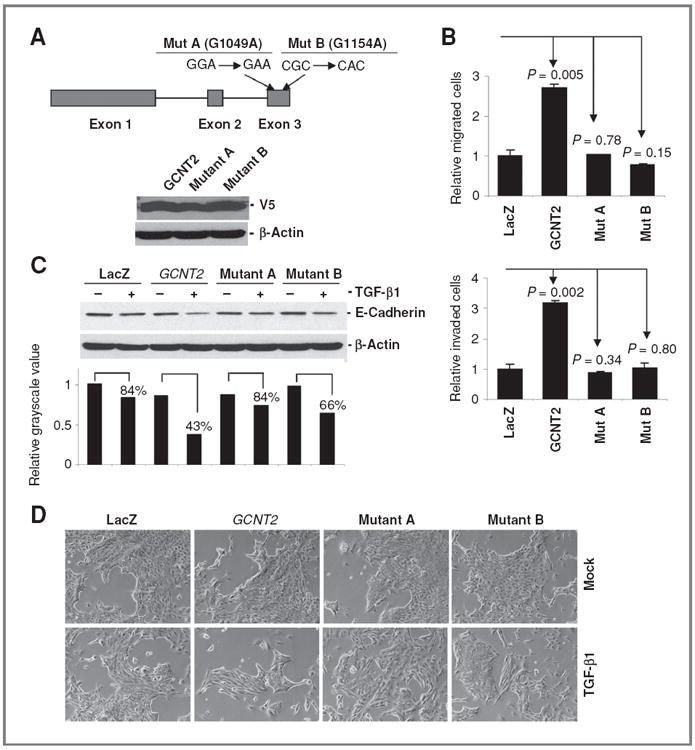
Glycosyltransferase activity is required for cell migration, invasion, and the EMTpromoting function of GCNT2. A, the schematic structure of GCNT2 and the position of the 2 mutations in the molecule (top). The lower panel shows the expression of wild type and 2 mutant forms of GCNT2 in EpRas cells detected by Western blot with V5 antibody. B, quantitative analysis of the effect of wild type and 2 mutant forms of GCNT2 on migration and invasion in EpRas cells. C, Western blot shows the expression of E-cadherin in NMuMG cells with GCNT2 knockdown after expression of LacZ, GCNT2 wild type and mutant forms. A total of 5 ng/mL TGF-β1 was used for 3 days. The quantitative analysis was done by comparison of the band intensity of TGF-β–treated cells versus the nontreated cells. D, morphologic analysis of the NMuMG GCNT2 knockdown cells with and without TGF-β1 treatment. Cells with wild type, 2 mutant forms of GCNT2 and LacZ control were compared.
Discussion
Glycosylation is a common posttranslational modification and is involved in a variety of physiologic and pathologic events (36). Glycosyltransferase is one of the enzymes that catalyze the glycosylation reactions by adding sugar chains to various complex carbohydrates, such as glycoproteins, glycolipids, and proteoglycans. About 170 glycosyltransferases have been cloned (37). However, only a few of them, including MGAT5, MGAT3, and GCNT1, have been associated with cancer metastasis. Our study is the first to identify GCNT2, which encodes the enzyme responsible for formation of the blood group I antigen, has a critical biological function in modulating EMT and promoting breast cancer metastasis. Therefore, our study of GCNT2 in breast cancer metastasis will substantially complement previous studies on other glycosyltransferases and will establish a new paradigm in the metastasis research field.
The biological function of GCNT2 is to branch poly-N-lactosamine chains by adding an N-acetylglucosaminyl residue via β-1-6 linkage to a galactosyl residue (38). Linear poly-N-acetyllactosamines bear the i blood group antigen, whereas the branched molecules bear the I blood group antigen. These i and I antigens are not only present in red blood cells but also on the surface of most human cells (39). Interestingly, I antigen was also found to be present in the soluble glycoproteins in various body fluids including milk, saliva, plasma, urine, and ovarian cyst fluid (39, 40). Specifically, potential roles for GCNT2 in human breast cancers are also suggested by the observation made more than 20 years ago that I antigens in the sera of breast cancer patients were increased and the I antigen levels correlated with the breast carcinoma stages (41-43). The results of our study thus provide direct biological explanation for the long-standing observation and suggest that the overexpression of GCNT2, along with breast cancer progression, lead to the abundant expression of protein with I antigen. These proteins with I antigen may be further released into various bodily fluids, including serum. In this regard, direct measurement of the I antigen in body fluids of breast cancer patients may be a promising noninvasive approach to detect metastasis.
The paradox of the TGF-β role in cancer progression has been well documented (44, 45). Recently, TGF-β signaling induced EMT is recognized as an emerging mechanism underlying its metastasis-promoting function (46). In this context, targeting TGF-β signaling has been a promising therapeutic strategy for aggressive cancer including metastatic basal-like breast cancer (47-50). Our current study showed that GCNT2 is regulated by EMT signaling, such as TGF-β, and enhances EMT and breast cancer metastasis. These results suggest that targeting GCNT2 will provide an alternative option in targeting TGF-β signaling. However, the regulatory mechanism of TGF-β on GCNT2 is worthy of further investigation. More importantly, it can be expected that the targets for posttranslational modification by GCNT2 are not limited to E-cadherin. Many other molecules involved in EMT, cell morphology and cell-to-cell interaction could also be the targets of GCNT2. The changes of these molecules could function together with E-cadherin to contribute to EMT and further cell migration, invasion, and metastasis. Therefore, our study provides an integrative molecular mechanism that combines the TGF-β signaling and downstream glycosylation modification for breast cancer metastasis. Further in-depth analysis of this mechanism will provide new insight into breast cancer metastasis and will result in novel therapeutic strategies in targeting this disease for which effective therapy is currently unavailable.
Supplementary Material
Acknowledgments
We thank Dr. Robert A. Weinberg, professor of Biology at MIT; Dr. Stephen P. Ethier, professor of Pathology at Wayne State University; and Dr. Lindsey D. Mayo at Indiana University for providing technical advice and agents. We also thank Ms. Elizabeth A. Katz of Marketing & Communications Department of Barbara Ann Karmanos Cancer Institute for editing our manuscript.
Grant Support
This work was supported in part by the Susan G. Komen grant KG080465 (G. Wu), the NOMIC grant from the Karmanos Cancer Institute (G. Wu), and awards for young university teachers (2006jql141zd) and Natural Science Key Foundation (KJ2010A299) from Department of Education, Anhui Province, People’s Republic of China (H. Zhang). The Biostatistics Core and the Biorepository Core of the Karmanos Cancer Institute are supported by grant number P30-CA022453-29.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369:1742–57. doi: 10.1016/S0140-6736(07)60781-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weigelt B, Wessels LF, Bosma AJ, Glas AM, Nuyten DS, He YD, et al. No common denominator for breast cancer lymph node metastasis. Br J Cancer. 2005;93:924–32. doi: 10.1038/sj.bjc.6602794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haupt B, Ro JY, Schwartz MR. Basal-like breast carcinoma: a phenotypically distinct entity. Arch Pathol Lab Med. 2010;134:130–3. doi: 10.5858/134.1.130. [DOI] [PubMed] [Google Scholar]
- 4.Korsching E, Jeffrey SS, Meinerz W, Decker T, Boecker W, Buerger H. Basal carcinoma of the breast revisited: an old entity with new interpretations. J Clin Pathol. 2008;61:553–60. doi: 10.1136/jcp.2008.055475. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, Meng F, Liu G, Zhang B, Zhu J, Wu F, et al. Forkhead transcription factor foxq1 promotes epithelial-mesenchymal transition and breast cancer metastasis. Cancer Res. 2011;71:1292–301. doi: 10.1158/0008-5472.CAN-10-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dennis JW, Granovsky M, Warren CE. Glycoprotein glycosylation and cancer progression. Biochim Biophys Acta. 1999;1473:21–34. doi: 10.1016/s0304-4165(99)00167-1. [DOI] [PubMed] [Google Scholar]
- 7.Murata K, Miyoshi E, Kameyama M, Ishikawa O, Kabuto T, Sasaki Y, et al. Expression of N-acetylglucosaminyltransferase V in colorectal cancer correlates with metastasis and poor prognosis. Clin Cancer Res. 2000;6:1772–7. [PubMed] [Google Scholar]
- 8.Granovsky M, Fata J, Pawling J, Muller WJ, Khokha R, Dennis JW. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat Med. 2000;6:306–12. doi: 10.1038/73163. [DOI] [PubMed] [Google Scholar]
- 9.Guo HB, Lee I, Kamar M, Pierce M. N-acetylglucosaminyltransferase V expression levels regulate cadherin-associated homotypic cell-cell adhesion and intracellular signaling pathways. J Biol Chem. 2003;278:52412–24. doi: 10.1074/jbc.M308837200. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Y, Li J, Xing Y, Wang J, Lu C, Xin X, et al. N-acetylglucosaminyltransferase V mediates cell migration and invasion of mouse mammary tumor cells 4TO7 via RhoA and Rac1 signaling pathway. Mol Cell Biochem. 2008;309:199–208. doi: 10.1007/s11010-007-9656-6. [DOI] [PubMed] [Google Scholar]
- 11.Iijima J, Zhao Y, Isaji T, Kameyama A, Nakaya S, Wang X, et al. Cell-cell interaction-dependent regulation of N-acetylglucosaminyltransferase III and the bisected N-glycans in GE11 epithelial cells. Involvement of E-cadherin-mediated cell adhesion. J Biol Chem. 2006;281:13038–46. doi: 10.1074/jbc.M601961200. [DOI] [PubMed] [Google Scholar]
- 12.Taniguchi N, Miyoshi E, Ko JH, Ikeda Y, Ihara Y. Implication of N-acetylglucosaminyltransferases III and V in cancer: gene regulation and signaling mechanism. Biochim Biophys Acta. 1999;1455:287–300. doi: 10.1016/s0925-4439(99)00066-6. [DOI] [PubMed] [Google Scholar]
- 13.Yoshimura M, Nishikawa A, Ihara Y, Taniguchi S, Taniguchi N. Suppression of lung metastasis of B16 mouse melanoma by N-acetylglucosaminyltransferase III gene transfection. Proc Natl Acad Sci U S A. 1995;92:8754–8. doi: 10.1073/pnas.92.19.8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandes B, Sagman U, Auger M, Demetrio M, Dennis JW. Beta 1–6 branched oligosaccharides as a marker of tumor progression in human breast and colon neoplasia. Cancer Res. 1991;51:718–23. [PubMed] [Google Scholar]
- 15.Taeda Y, Nose M, Hiraizumi S, Ohuchi N. Expression of L-PHA-binding proteins in breast cancer: reconstitution and molecular characterization of beta 1–6 branched oligosaccharides in three-dimensional cell culture. Breast Cancer Res Treat. 1996;38:313–24. doi: 10.1007/BF01806151. [DOI] [PubMed] [Google Scholar]
- 16.Beum PV, Singh J, Burdick M, Hollingsworth MA, Cheng PW. Expression of core 2 beta-1,6-N-acetylglucosaminyltransferase in a human pancreatic cancer cell line results in altered expression of MUC1 tumor-associated epitopes. J Biol Chem. 1999;274:24641–8. doi: 10.1074/jbc.274.35.24641. [DOI] [PubMed] [Google Scholar]
- 17.Shimodaira K, Nakayama J, Nakamura N, Hasebe O, Katsuyama T, Fukuda M. Carcinoma-associated expression of core 2 beta-1,6-N-acetylglucosaminyltransferase gene in human colorectal cancer: role of O-glycans in tumor progression. Cancer Res. 1997;57:5201–6. [PubMed] [Google Scholar]
- 18.Hatakeyama S, Kyan A, Yamamoto H, Okamoto A, Sugiyama N, Suzuki Y, et al. Core 2 N-acetylglucosaminyltransferase-1 expression induces aggressive potential of testicular germ cell tumor. Int J Cancer. 2010;127:1052–9. doi: 10.1002/ijc.25117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuperwasser C, Dessain S, Bierbaum BE, Garnet D, Sperandio K, Gauvin GP, et al. A mouse model of human breast cancer metastasis to human bone. Cancer Res. 2005;65:6130–8. doi: 10.1158/0008-5472.CAN-04-1408. [DOI] [PubMed] [Google Scholar]
- 20.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52:1399–405. [PubMed] [Google Scholar]
- 21.Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, Miura N, et al. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci U S A. 2007;104:10069–74. doi: 10.1073/pnas.0703900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H, Chen D, Ringler J, Chen W, Cui QC, Ethier SP, et al. Disulfiram treatment facilitates phosphoinositide 3-kinase inhibition in human breast cancer cells in vitro and in vivo. Cancer Res. 2010;70:3996–4004. doi: 10.1158/0008-5472.CAN-09-3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H, Liu G, Dziubinski M, Yang Z, Ethier SP, Wu G. Comprehensive analysis of oncogenic effects of PIK3CA mutations in human mammary epithelial cells. Breast Cancer Res Treat. 2008;112:217–27. doi: 10.1007/s10549-007-9847-6. [DOI] [PubMed] [Google Scholar]
- 24.Inaba N, Hiruma T, Togayachi A, Iwasaki H, Wang XH, Furukawa Y, et al. A novel I-branching beta-1,6-N-acetylglucosaminyltransferase involved in human blood group I antigen expression. Blood. 2003;101:2870–6. doi: 10.1182/blood-2002-09-2838. [DOI] [PubMed] [Google Scholar]
- 25.Kreike B, van Kouwenhove M, Horlings H, Weigelt B, Peterse H, Bartelink H, et al. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res. 2007;9:R65. doi: 10.1186/bcr1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richardson AL, Wang ZC, De Nicolo A, Lu X, Brown M, Miron A, et al. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9:121–32. doi: 10.1016/j.ccr.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 27.van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 28.Desmedt C, Piette F, Loi S, Wang Y, Lallemand F, Haibe-Kains B, et al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res. 2007;13:3207–14. doi: 10.1158/1078-0432.CCR-06-2765. [DOI] [PubMed] [Google Scholar]
- 29.Ginestier C, Cervera N, Finetti P, Esteyries S, Esterni B, Adelaide J, et al. Prognosis and gene expression profiling of 20q13-amplified breast cancers. Clin Cancer Res. 2006;12:4533–44. doi: 10.1158/1078-0432.CCR-05-2339. [DOI] [PubMed] [Google Scholar]
- 30.Lu X, Wang ZC, Iglehart JD, Zhang X, Richardson AL. Predicting features of breast cancer with gene expression patterns. Breast Cancer Res Treat. 2008;108:191–201. doi: 10.1007/s10549-007-9596-6. [DOI] [PubMed] [Google Scholar]
- 31.Haraguchi M, Okubo T, Miyashita Y, Miyamoto Y, Hayashi M, Crotti TN, et al. Snail regulates cell-matrix adhesion by regulation of the expression of integrins and basement membrane proteins. J Biol Chem. 2008;283:23514–23. doi: 10.1074/jbc.M801125200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Zhao JH, Zhang XY, Guo HB, Liu F, Chen HL. Relations of the type and branch of surface N-glycans to cell adhesion, migration and integrin expressions. Mol Cell Biochem. 2004;260:137–46. doi: 10.1023/b:mcbi.0000026065.84798.62. [DOI] [PubMed] [Google Scholar]
- 33.Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. 2003;116:499–511. doi: 10.1242/jcs.00224. [DOI] [PubMed] [Google Scholar]
- 34.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 35.Yu LC, Twu YC, Chou ML, Reid ME, Gray AR, Moulds JM, et al. The molecular genetics of the human I locus and molecular background explain the partial association of the adult i phenotype with congenital cataracts. Blood. 2003;101:2081–8. doi: 10.1182/blood-2002-09-2693. [DOI] [PubMed] [Google Scholar]
- 36.Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 37.Narimatsu H. Human glycogene cloning: focus on beta 3-glycosyltransferase and beta 4-glycosyltransferase families. Curr Opin Struct Biol. 2006;16:567–75. doi: 10.1016/j.sbi.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 38.Bierhuizen MF, Mattei MG, Fukuda M. Expression of the developmental I antigen by a cloned human cDNA encoding a member of a beta-1,6-N-acetylglucosaminyltransferase gene family. Genes Dev. 1993;7:468–78. doi: 10.1101/gad.7.3.468. [DOI] [PubMed] [Google Scholar]
- 39.Magnet AD, Fukuda M. Expression of the large I antigen forming beta-1,6-N-acetylglucosaminyltransferase in various tissues of adult mice. Glycobiology. 1997;7:285–95. doi: 10.1093/glycob/7.2.285. [DOI] [PubMed] [Google Scholar]
- 40.Marsh WL, Nichols ME, Allen FH., Jr Inhibition of anti-I sera by human milk. Vox Sang. 1970;18:149–54. doi: 10.1111/j.1423-0410.1970.tb01440.x. [DOI] [PubMed] [Google Scholar]
- 41.Burchell J, Wang D, Taylor-Papadimitriou J. Detection of the tumour-associated antigens recognized by the monoclonal antibodies HMFG-1 and 2 in serum from patients with breast cancer. Int J Cancer. 1984;34:763–8. doi: 10.1002/ijc.2910340605. [DOI] [PubMed] [Google Scholar]
- 42.Dube VE, Haid M, Chmiel JS, Anderson B. Serum cold agglutinin and IgM levels in breast carcinoma. Breast Cancer Res Treat. 1984;4:105–8. doi: 10.1007/BF01806392. [DOI] [PubMed] [Google Scholar]
- 43.Dube VE, Kallio P, Chmiel JS, Haid M, Hakim A. An immunosorbent assay for blood group I antigens in breast carcinoma. Clin Immunol Immunopathol. 1987;45:196–207. doi: 10.1016/0090-1229(87)90034-1. [DOI] [PubMed] [Google Scholar]
- 44.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–29. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 45.Tian M, Neil JR, Schiemann WP. Transforming growth factor-beta and the hallmarks of cancer. Cell Signal. 2010;23:951–62. doi: 10.1016/j.cellsig.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010;15:169–90. doi: 10.1007/s10911-010-9181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ganapathy V, Ge R, Grazioli A, Xie W, Banach-Petrosky W, Kang Y, et al. Targeting the transforming growth factor-beta pathway inhibits human basal-like breast cancer metastasis. Mol Cancer. 2010;9:122. doi: 10.1186/1476-4598-9-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Korpal M, Kang Y. Targeting the transforming growth factor-beta signalling pathway in metastatic cancer. Eur J Cancer. 2010;46:1232–40. doi: 10.1016/j.ejca.2010.02.040. [DOI] [PubMed] [Google Scholar]
- 49.Muraoka RS, Dumont N, Ritter CA, Dugger TC, Brantley DM, Chen J, et al. Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. J Clin Invest. 2002;109:1551–9. doi: 10.1172/JCI15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov. 2004;3:1011–22. doi: 10.1038/nrd1580. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.