


Abstract
MeCP2 is a methyl-CpG binding protein that can repress transcription of nearby genes. In humans, mutations in the MECP2 gene are the major cause of Rett syndrome. By searching expressed sequence tag (EST) databases we have found a novel MeCP2 splice isoform (MeCP2α) which encodes a distinct N-terminus. We demonstrate that the MeCP2α mRNA splice variant is more abundant than the previously annotated MeCP2 mRNA (MeCP2β) in mouse tissues and human brain. Furthermore, MeCP2β mRNA has an upstream open reading frame that inhibits its translation. As a result of these differences, >90% of MeCP2 in mouse brain is MeCP2α. Both protein isoforms are nuclear and colocalize with densely methylated heterochromatic foci in mouse cells. The presence of a previously unknown MeCP2 isoform has implications for the genetic screening of Rett syndrome patients and for studies of the functional significance of MeCP2.
INTRODUCTION
In vertebrate genomes, 5-methylcytosine (m5C) accounts for ∼1% of all DNA bases. The minor base arises by post-synthetic modification of cytosine, usually in the context of a CpG dinucleotide that is symmetrically methylated on both strands of DNA. A family of proteins that specifically bind to a methylated CpG pair share a conserved methyl-CpG binding domain (MBD) (1). MeCP2 is the founder member of the MBD protein family and is present in all tested vertebrates. Currently two conserved functional domains have been mapped in MeCP2: the MBD (2), which specifically targets MeCP2 to methylated DNA sequences in vivo (3–5), and the transcription repression domain (TRD), which is the minimal domain required to repress transcription in vitro and in vivo (5–10). MeCP2 repression is sensitive to histone deacetylase (HDAC) inhibitor TSA, indicating that deacetylation may contribute to repression (11,12). GST pulldowns and partial multiprotein complex purification from Xenopus laevis oocytes suggest that the TRD interacts with the mSin3A/HDAC co-repressor complex (11,12). As TSA partially alleviates repression, HDAC-independent mechanisms of repression are likely to exist. For example, MeCP2 has been shown to associate with histone methyltransferase activity (13). Other HDAC-independent transcriptional repression mechanisms, involving interactions of the TRD with basal transcriptional repression machinery, have also been proposed (14).
Mutations in the MECP2 gene are the primary cause of Rett syndrome (15), a neurological disorder that occurs in one in 10 000–22 000 female births. After an initial window of normal development, girls acquire a variety of symptoms including microcephaly, autism, ataxia, stereotypic hand movements, seizures and hyperventilation (16). A wide spectrum of mutations have been mapped throughout the MECP2 gene, including sites outside the MBD and TRD regions (17). MeCP2 is also essential in mice, as Mecp2-null animals have a period of normal postnatal development followed by hindlimb clasping, irregular breathing, reduced mobility and death at around 8 weeks of age (18,19). Mice with Mecp2 mutations phenotypically mimic several features of Rett syndrome (18–20).
Initially, MeCP2 protein was purified from rat brain as a single 84 kDa protein based on its binding to methylated CpG (21). The results of Edman degradation allowed the design of a degenerate probe which was then used to screen a cDNA library and isolate full-length MeCP2 cDNA (21). Later, the mouse Mecp2 gene was mapped to Xq28 and shown to be subject to X-inactivation (22). Further analysis of the Mecp2 transcript identified alternative polyadenylation sites that give rise to two main mRNA variants with different 3′UTRs (10 kb and 2 kb) (23). At this time Mecp2 was annotated as a three-exon gene, with all exons contributing to the protein (23). Sequencing of the Mecp2 genomic locus in mouse and human combined with more detailed bioinformatic analysis revealed an additional upstream non-coding exon (24). Mecp2 is therefore currently recognized as a four-exon gene. Here we report the additional complication that MeCP2 is subject to alternative splicing generating two different N-termini, one of which is significantly more abundant than the other.
MATERIALS AND METHODS
Bioinformatics
Different isoforms encoding expressed sequence tags (ESTs) were found using the NCBI BLAST program. Alignment of genomic DNA with EST sequences was performed with GeneQuest (DNAStar). Protein alignments were done using ClustalW and T-coffee programs. Alignments were displayed with GeneDoc software.
RNA purification, analysis and RT–PCR
Total RNA from tissue culture cells was purified using TRI-Reagent (Sigma) according to the manufacturer’s recommendations. Prior to cDNA synthesis RNA was treated with RQ1 RNase-Free DNase (Promega). cDNA was synthesized by annealing 5 µg of total RNA and 5 µg random hexanucleotides (Amersham) at 70°C for 5 min. Then RT mix [final 1× reaction buffer, 40 U RNAasin ribonuclease inhibitor (Promega), 1 mM dNTP] was added and the solution was incubated for 5 min at 25°C. After addition of M-MLV reverse transcriptase (RNase H Minus, Promega) the 25 µl reaction mix was incubated for 10 min at 25°C and 1 h at 37°C. The reverse transcriptase was inactivated by incubating for 10 min at 70°C. The reaction mixture was diluted to 500 µl, and 2.5 µl was used for PCRs. Mouse exon-specific PCR was carried out using primers me11d (GGTAAAACCCGTCCGGAAAATG) and me31r (TTCAGTGGCTTGTCTCTGAG) at Ta = 61°C. Human exon-specific PCR was performed using me11d and hme31r (CTTGAGGGGTTTGTCCTTGAG) primers at Ta = 61°C. Human brain cDNA was purchased from Ambion (FirstChoice PCR-Ready). Northern blots were performed using standard procedures as described in the Hybond-N+ (Amersham) membrane users’ manual. After extraction, RNA was treated with DNase I to remove transfected plasmid. Northern blots were probed with a 1.5 kbp NcoI and NotI fragment of MeCP2 cDNA.
Embryonic stem cell differentiation
Embryonic stem (ES) cells were differentiated as described elsewhere (25). In brief, mouse ES cells were plated on non-adhesive plates without LIF. In these conditions ES cells form embryoid bodies which contain different cell lineage progenitors. Later, embryoid bodies were cultured in the presence of retinoic acid, which was shown to increase the efficiency of neuronal differentiation. In the final stage embryoid bodies were dissociated and cells were plated on serum-free N2 medium which promotes final differentiation and survival of neuronal cells.
Mutagenesis
Site-directed mutagenesis used the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturers’ recommendations. Primers used for mutagenesis were mumed (CCCGTCCGGAAAAAGGCCGCCGCTGCCGCC) and mumer (GGCGGCAGCGGCGGCCTTT TTCCGGACGGG).
Vector construction and transfections
EST clones (BI078224 and BG922233) were received from the I.M.A.G.E. consortium. Plasmids used for transfection are based on pRL-SV40 (Promega). Rluc gene was excised using NheI and XbaI sites and replaced with appropriate MeCP2 splice variant cDNA. All transfections were done with Lipofectamine reagent (Invitrogen) according to the manufacturer’s recommendations. Cells were collected 48 h after transfection.
Western blotting
Western blotting was according to standard protocols. Transfected cells were harvested by scraping, boiled in SDS loading buffer and resolved on 8% SDS–PAGE gels. Mouse brain nuclear extract was kindly provided by Rob Klose. Brain nuclei were purified from whole mouse brain and nuclear proteins were extracted with 400 mM NaCl. Anti-MeCP2 antibodies were purchased from Upstate Biotechnology (no. 07–013).
Immunohistochemistry
Transfected cells were fixed in 4% paraformaldehyde in PBS for 20 min. After two washes with PBS, cells were permeabilized with 0.2% Triton X-100 for 10 min. Blocking was performed in 3% BSA in PBS for 30 min. MeCP2 antibody (Upstate Biotechnology) was diluted 1:200 in blocking solution and incubated with slides for 1 h. After washing three times in PBS, slides were incubated with fluorescein anti-rabbit IgG (Vector Laboratories) at 1:100 dilution. After incubation for 1 h, slides were washed and mounted in Vectaschield mounting medium with DAPI (Vector Laboratories). Slides were examined using a Zeiss microscope.
RESULTS
Identification of an MeCP2 splice variant in EST databases
We searched mouse EST databases for cDNA sequences encoding MeCP2. Alignments revealed that ESTs can be grouped into two categories depending on the presence (BY107013, BI409371) or absence (CA980031, BY244111) of exon 2. Human MECP2 cDNAs with (BC11612, BM923600) and without (BG706068, BI458175) exon 2 also exist in human EST databases. As exon 2 contains the ATG for the initiation of translation, we considered the possibility that the transcript lacking exon 2 is a non-coding RNA. However exon 1 contains an ATG that in the absence of exon 2 initiates a potential open reading frame (ORF) of 501 amino acids (aa) in mice and 498 aa in humans (Table 1). We designate the new exon 2-minus isoform as MeCP2α and the previously described isoform containing exon 2 as MeCP2β. Alignment of α and β predicted protein sequences demonstrates identity except at the extreme N-terminus (Fig. 1a and b). The MeCP2α N-terminus contains polyalanine and polyglycine repeat tracts encoded by GCC and GGA trinucleotides. Comparison of these isoforms with other vertebrate MeCP2 sequences showed that MeCP2α shares a polyalanine tract, serine-glycine residues and EERL motifs with Xenopus and zebrafish MeCP2 sequences, whereas MeCP2β lacks these features. The alignment suggested that MeCP2α more closely resembles the ancestral form of MeCP2 than does MeCP2β (Fig. 1c). We have searched the non-mammalian EST databases and failed to find any β-isoform-like EST. Furthermore, examination of zebrafish and Fugu rubripes genomic sequences surrounding the MeCP2 gene revealed exon 1 (α), but no potential exon 2 (β). We conclude that the β isoform is either absent or highly diverged compared with the mammalian β sequence.
Table 1. Physical characteristics of human and mouse MeCP2 isoforms.
| MW (kDa) | Length (aa) | pI | Charge at pH 7 | |
|---|---|---|---|---|
| mMECP2α | 53.6 | 501 | 9.89 | 34.9 |
| hMECP2α | 53.3 | 498 | 9.88 | 34.8 |
| mMECP2β | 52.3 | 484 | 9.96 | 37.8 |
| hMECP2β | 52.4 | 486 | 9.95 | 37.8 |
Figure 1.

Alternative splicing of MeCP2 mRNA. (a) Previously described MeCP2β is encoded when all known exons are sequentially spliced. (b) The novel MeCP2α isoform arises when exon 1 is spliced onto exon 3, skipping exon 2. Shaded boxes are protein coding and open boxes are non-coding. ESTs suggesting the occurrence of each isoform in vivo are mapped as lines above the genomic DNA. (c) Alignment of mouse and human MeCP2β and MeCP2α N-termini with zebrafish and frog MeCP2. MeCP2α is more similar to zebrafish and frog orthologs than is MeCP2β.
MeCP2α is the major mRNA splice variant in vivo
To confirm in silico evidence and assess the relative abundance of the different MeCP2 isoforms in vivo, we performed semiquantitative RT–PCR analysis of cDNA derived from different mouse tissues. The two primers were designed to anneal to exon 1 and exon 3, respectively (Fig. 2a). Control amplification of mixed MeCP2α and MeCP2β encoding plasmids showed no significant amplification bias towards the shorter product (Fig. 2a). RT–PCR analysis demonstrated that MeCP2α is more abundant than MeCP2β in most tissues. Relative abundance of MeCP2α was highest in the brain, thymus and lung, whereas an approximately 1:1 isoform ratio was seen in testis and liver. The RT–PCRs (32 cycles) were not saturating and therefore were responsive to template concentration. This was verified for each tissue sample by showing that fewer (30) or more (34) cycles generated less or more product, respectively (data not shown). Similar semiquantitative RT–PCRs provided evidence that both splice variants also exist in human brain; again, the predominant form of mRNA encodes MECP2α (Fig. 2b).
Figure 2.

Relative abundance of splice variant mRNAs in mouse tissue, human brain and differentiating ES cells. (a) A mouse tissue cDNA panel was analysed by semiquantitative PCR with primers that anneal to exons 1 and 3 (short arrows on map). The α isoform was more abundant in lung, thymus, brain and heart. Amplification of an equimolar mixture of α- and β-encoding plasmids indicated no preference for amplification of either PCR band. (b) Human brain cDNA showed dominance of the α isoform mRNA. (c) ES cells were differentiated to give embryoid bodies and neuronal cells in culture. EB4 and EB8 refer to embryoid bodies on day 4 and day 8 of differentiation. 4DN2 refers to day 4 after embryoid bodies were split and plated on serum-free neurobasal medium.
To analyse the abundance of the two isoforms during cellular differentiation, we differentiated mouse ES cells into neurons. Samples from stages of differentiation were assayed for the different isoforms. Semiquantitative RT–PCR showed that MeCP2α mRNA is more abundant than MeCP2β mRNA in ES cells and the proportion of α mRNA appeared to increase during neuronal differentiation (Fig. 2c).
The MeCP2α splice variant is more efficiently translated in vivo
Having established that the MeCP2α mRNA splice isoform is abundant in mouse tissues and human brain, we next asked whether MeCP2α mRNA is translated in vivo. To address this, we transfected tail fibroblasts from Mecp2-null mice (18) with plasmid constructs containing α- or β-isoform cDNA (Fig. 3a). Western blotting with a C-terminal MeCP2 antibody showed that MeCP2α is successfully translated, but we consistently observed only very low amounts of MeCP2β in independent transfections (Fig. 3b). As a potential explanation, we noted a short ORF (39 aa) within the 5′UTR region of MeCP2β that could potentially interfere with its translation. The short ORF starts from the AUG that initiates translation of the MeCP2α isoform, but in MeCP2β mRNA this ORF terminates due to alternative splicing 55 nt (in mouse) upstream of the bona fide MeCP2β translation start site. To test for interference of the upstream ORF, we introduced a point mutation that changed the upstream ATG to AAG (Fig. 3a) and expressed the wild-type and mutant versions of MeCP2β in fibroblasts. Northern blots probed with MeCP2 cDNA showed that similar amounts of mRNA were produced from transfected wild-type MeCP2α, MeCP2β and mutant MeCP2β plasmids (Fig. 3c, lanes 1–3). As before, MeCP2α was efficiently synthesized, but a negligible amount of MeCP2β protein was translated from the wild-type cDNA construct. Mutation of the upstream ATG, however, led to a dramatic increase in the amount of translated MeCP2β (Fig. 3c, lanes 1–3).
Figure 3.

MeCP2β is inefficiently translated to give a protein that can be separated from MeCP2α by PAGE. (a) Diagrams of MeCP2α, MeCP2β and mutated MeCP2β constructs used for transfection of Mecp2-null tail fibroblasts. An SV-40 promoter and artificial intron is followed by the MeCP2 5′UTR, coding sequence, 3′UTR, MeCP2 polyadenylation signal and SV40 polyadenylation signal. MeCP2 exons are numbered below each map. The start codon of the wild-type upstream ORF in MeCP2β is labeled as ATG (middle diagram) and is mutated to AAG in the MeCP2β ATG-AAG construct (bottom diagram). (b) Weak expression of the MeCP2β construct in transfected cells. Two independent plasmid preparations were used for each type of transfection (MeCP2α, lanes 1 and 2; MeCP2β, lanes 3 and 4). The product of the MeCP2β plasmid was barely visible in lane 4 only. (c) Western blot analysis (upper panel) of transfected Mecp2-null mouse fibroblasts and native MeCP2 from mouse brain nuclear extracts. MeCP2 protein isoforms (asterisks) migrated at different sizes on this 8% PAGE gel (compare lanes 1, 3 and 4). The upstream ORF inhibited the translation of MeCP2β (lanes 2 and 3). MeCP2α is the predominant isoform in mouse brain nuclear extract (lanes 8, 9 and 10). The northern blot (lower panel) showed that different levels of translated MeCP2 protein are not due to differential transcription of the constructs, as similar amounts of MeCP2 mRNA were seen in lanes 1–3. In the absence of transfection, no endogenous MeCP2 RNA is detectable in these Mecp2-null cells.
Our results indicate that the MeCP2α mRNA is more abundant than MeCP2β mRNA, but also that MeCP2β mRNA is inefficiently translated. Together, these findings suggest that MeCP2α protein will be much more abundant than MeCP2β in vivo. To test this prediction, we took advantage of the different sizes of the α and β protein isoforms (Table 1). A high-resolution SDS–PAGE gel of in vivo translated α and β forms confirmed that they migrate differently (Fig. 3c). Cotransfection of equal amounts of α- and β-isoform expression constructs showed that MeCP2α is greatly over- represented among the translation products (Fig. 3c, lanes 4 and 5). To investigate whether native MeCP2 also contains predominantly MeCP2α, we loaded different amounts of mouse brain nuclear extract on the same gel (Fig. 3c, lanes 8–10). MeCP2β protein could be detected only with higher amounts of nuclear extract loaded. We estimate conservatively that the MeCP2α protein is at least 10-fold more abundant than MeCP2β in mouse brain extracts. An alternative hypothetical explanation for the difference might be that MeCP2β is poorly extracted from brain nuclei compared with MeCP2α. Given our evidence that MeCP2β mRNA is not only less abundant in brain, but also much less well translated than MeCP2α mRNA, we consider it likely that the data accurately reflect the rarity of the β isoform in vivo.
Localization of different MeCP2 isoforms in mouse cells
The majority of methylated DNA in mouse cell nuclei is in repeated major satellite sequences, which exhibit punctate staining with DAPI. In mouse cells MeCP2β has previously been shown to colocalize with DAPI bright spots in a DNA methylation-dependent manner (4). To compare localization of the different MeCP2 isoforms in mouse cells, we transfected Mecp2-null tail fibroblasts with plasmids expressing the α or β isoforms and stained with an MeCP2 antibody that recognizes the invariant C-terminal domain. Both isoforms were nuclear and colocalized with DAPI bright spots (Fig. 4). The construct encoding the β isoform included the upstream ORF that inhibits translation. Accordingly the number of cells that expressed detectable protein was very low. By comparison, parallel transfections with the α construct gave a much higher percentage of nuclei with easily detectable punctate expression of MeCP2. A few α transfected cells expressed very high protein levels that resulted in saturating fluorescence throughout the nucleus. We suspect that these cells and the few β cells expressing detectable βMeCP2 (Fig. 4, top panel) received by chance a large dose of the transfected expression construct. These experiments reveal no functional difference between α and β MeCP2 isoforms at the level of cellular localization.
Figure 4.
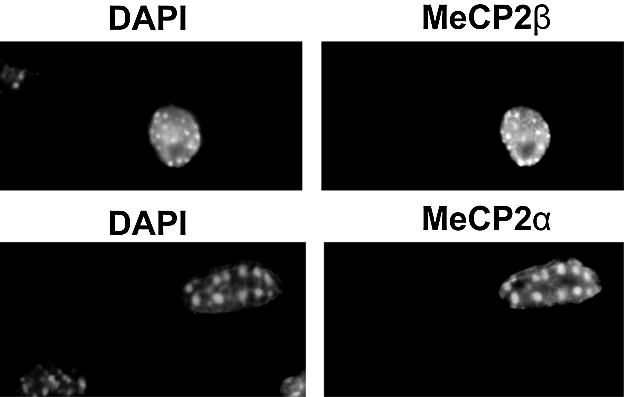
Both MeCP2 isoforms colocalized with methyl-CpG-rich DAPI bright spots when Mecp2-null fibroblasts were transfected with wild-type MeCP2α and MeCP2β expression constructs.
DISCUSSION
We report a new splice variant of the MeCP2 gene which we designate MeCP2α. MeCP2α is more similar to frog and zebrafish MeCP2 sequences than the currently known isoform MeCP2β. These findings suggest that MeCP2α is more closely related to the ancestral form of MeCP2 and that the appearance of exon 2 may be a relatively recent event in the evolution of the mammalian gene. The differing size and charge of α and β isoforms permits their separation by gel electrophoresis. This allowed us to demonstrate that both isoforms exist in mouse brain, but that MeCP2α is by far the dominant form. The predominance of MeCP2α can be partly accounted for by the greater abundance of its transcript. In addition, we demonstrate translational interference by an upstream ORF in mRNA of the β isoform. Translational interference by upstream ORFs is well established and has been shown to depend on the distance between the upstream ORF and the AUG of the downstream ORF and also on the structure of 5′UTR RNA (26–29). It is not known whether translational interference of this kind can be modulated in vertebrates as a means of regulating protein synthesis.
The existence of the new isoform has implications for the study of Rett syndrome. Exon 1 was previously thought to be non-coding and has therefore been excluded from many mutational screening programmes. Our finding emphasizes the need for routine inclusion of exon 1 in these screens. Because MeCP2α is the predominant isoform, introduction of a nonsense or frameshift mutation would remove >90% of total MeCP2. This may result in classical Rett syndrome, a milder variant form of Rett syndrome or a related condition such as autism or X-linked mental retardation. We note that the MeCP2α N-terminus contains polyalanine and polyglycine sequences that are encoded by repeated GCC and GGA codons respectively. Expansion of a GCC trinucleotide sequence in the FMR2 gene is reported to cause FRAXE mental retardation (30) and an equivalent expansion may in theory contribute to Rett syndrome. It is noteworthy that no Rett syndrome mutations in exon 2 have been described. It is possible that exon 2 mutations, which would only affect MeCP2β, are compensated by the more abundant MeCP2α isoform and would therefore have a much less severe phenotypic consequence.
Most previous research on MeCP2 function has utilized the MeCP2β isoform, which we now report to be a minor form in vivo. We found that MeCP2 localization in mouse cells is the same for both isoforms, at least in cultured cells. Also, the alternative N-terminus is located outside the previously described functional domains MBD and TRD. It is therefore unlikely that either MBD or TRD function is affected by the N-terminus. Indeed, human MECP2β alone was able to successfully rescue MeCP2 deficiency in frog embryos, whose endogenous protein more closely resembles MeCP2α (31). Consequently, we expect that the functions of α and β isoforms may overlap significantly. On the other hand, it cannot be ruled out that the two isoforms exert somewhat distinct functions in vivo. For example, the MeCP2α N-terminus contains a conserved serine residue that is absent in MeCP2β and which could be a target of phosphorylation. Recently, MeCP2 phosphorylation has been shown to accompany induction of bdnf transcription in cultured mouse neurons (32,33). It may be of future interest to determine the functional significance of differing MeCP2 N-termini by the creation of isoform-specific gene disruptions in mice.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Jim Selfridge for the mouse tissue panel RNA, Rob Klose for mouse brain nuclear extract and comments on the manuscript, Meng Li (ISCR, Edinburgh) for practical advice on ES cell differentiation, Cathy Abbott for helpful discussions and Helle Jorgensen for a critical reading of the manuscript. S.K. is the Darwin Trust scholar. The research was funded by the Wellcome Trust.
REFERENCES
- 1.Hendrich B. and Bird,A. (1998) Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol., 18, 6538–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nan X., Meehan,R.R. and Bird,A. (1993) Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res., 21, 4886–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El Osta A. and Wolffe,A.P. (2001) Analysis of chromatin-immunopurified MeCP2-associated fragments. Biochem. Biophys. Res. Commun., 289, 733–737. [DOI] [PubMed] [Google Scholar]
- 4.Nan X., Tate,P., Li,E. and Bird,A. (1996) DNA methylation specifies chromosomal localization of MeCP2. Mol. Cell Biol., 16, 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lorincz M.C., Schubeler,D. and Groudine,M. (2001) Methylation-mediated proviral silencing is associated with MeCP2 recruitment and localized histone H3 deacetylation. Mol. Cell. Biol., 21, 7913–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nan X., Campoy,F.J. and Bird,A. (1997) MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell, 88, 471–481. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen C.T., Gonzales,F.A. and Jones,P.A. (2001) Altered chromatin structure associated with methylation-induced gene silencing in cancer cells: correlation of accessibility, methylation, MeCP2 binding and acetylation. Nucleic Acids Res., 29, 4598–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.El Osta A., Baker,E.K. and Wolffe,A.P. (2001) Profiling methyl-CpG specific determinants on transcriptionally silent chromatin. Mol. Biol. Rep., 28, 209–215. [DOI] [PubMed] [Google Scholar]
- 9.Drewell R.A., Goddard,C.J., Thomas,J.O. and Surani,M.A. (2002) Methylation-dependent silencing at the H19 imprinting control region by MeCP2. Nucleic Acids Res., 30, 1139–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rietveld L.E., Caldenhoven,E. and Stunnenberg,H.G. (2002) In vivo repression of an erythroid-specific gene by distinct corepressor complexes. EMBO J., 21, 1389–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nan X., Ng,H.H., Johnson,C.A., Laherty,C.D., Turner,B.M., Eisenman,R.N. and Bird,A. (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature, 393, 386–389. [DOI] [PubMed] [Google Scholar]
- 12.Jones P.L., Veenstra,G.J., Wade,P.A., Vermaak,D., Kass,S.U., Landsberger,N., Strouboulis,J. and Wolffe,A.P. (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nature Genet., 19, 187–191. [DOI] [PubMed] [Google Scholar]
- 13.Fuks F., Hurd,P.J., Wolf,D., Nan,X., Bird,A.P. and Kouzarides,T. (2003) The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem., 278, 4035–4040. [DOI] [PubMed] [Google Scholar]
- 14.Kaludov N.K. and Wolffe,A.P. (2000) MeCP2 driven transcriptional repression in vitro: selectivity for methylated DNA, action at a distance and contacts with the basal transcription machinery. Nucleic Acids Res., 28, 1921–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amir R.E., Van den Veyver, I, Wan,M., Tran,C.Q., Francke,U. and Zoghbi,H.Y. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genet., 23, 185–188. [DOI] [PubMed] [Google Scholar]
- 16.Hagberg B. (2002) Clinical manifestations and stages of Rett syndrome. Ment. Retard. Dev. Disabil. Res. Rev., 8, 61–65. [DOI] [PubMed] [Google Scholar]
- 17.Kriaucionis S. and Bird,A. (2003) DNA methylation and Rett syndrome. Hum. Mol. Genet., 12 (Spec. No. 2), R221–R227. [DOI] [PubMed] [Google Scholar]
- 18.Guy J., Hendrich,B., Holmes,M., Martin,J.E. and Bird,A. (2001) A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nature Genet., 27, 322–326. [DOI] [PubMed] [Google Scholar]
- 19.Chen R.Z., Akbarian,S., Tudor,M. and Jaenisch,R. (2001) Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nature Genet., 27, 327–331. [DOI] [PubMed] [Google Scholar]
- 20.Shahbazian M., Young,J., Yuva-Paylor,L., Spencer,C., Antalffy,B., Noebels,J., Armstrong,D., Paylor,R. and Zoghbi,H. (2002) Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron, 35, 243–254. [DOI] [PubMed] [Google Scholar]
- 21.Lewis J.D., Meehan,R.R., Henzel,W.J., Maurer-Fogy,I., Jeppesen,P., Klein,F. and Bird,A. (1992) Purification, sequence and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell, 69, 905–914. [DOI] [PubMed] [Google Scholar]
- 22.Quaderi N.A., Meehan,R.R., Tate,P.H., Cross,S.H., Bird,A.P., Chatterjee,A., Herman,G.E. and Brown,S.D. (1994) Genetic and physical mapping of a gene encoding a methyl CpG binding protein, Mecp2, to the mouse X chromosome. Genomics, 22, 648–651. [DOI] [PubMed] [Google Scholar]
- 23.Coy J.F., Sedlacek,Z., Bachner,D., Delius,H. and Poustka,A. (1999) A complex pattern of evolutionary conservation and alternative polyadenylation within the long 3”-untranslated region of the methyl-CpG-binding protein 2 gene (MeCP2) suggests a regulatory role in gene expression. Hum. Mol. Genet., 8, 1253–1262. [DOI] [PubMed] [Google Scholar]
- 24.Reichwald K., Thiesen,J., Wiehe,T., Weitzel,J., Poustka,W.A., Rosenthal,A., Platzer,M., Stratling,W.H. and Kioschis,P. (2000) Comparative sequence analysis of the MECP2-locus in human and mouse reveals new transcribed regions. Mamm. Genome, 11, 182–190. [DOI] [PubMed] [Google Scholar]
- 25.Li M., Pevny,L., Lovell-Badge,R. and Smith,A. (1998) Generation of purified neural precursors from embryonic stem cells by lineage selection. Curr. Biol., 8, 971–974. [DOI] [PubMed] [Google Scholar]
- 26.Kozak M. (2001) Constraints on reinitiation of translation in mammals. Nucleic Acids Res., 29, 5226–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Child S.J., Miller,M.K. and Geballe,A.P. (1999) Translational control by an upstream open reading frame in the HER-2/neu transcript. J. Biol. Chem., 274, 24335–24341. [DOI] [PubMed] [Google Scholar]
- 28.Xu G., Rabadan-Diehl,C., Nikodemova,M., Wynn,P., Spiess,J. and Aguilera,G. (2001) Inhibition of corticotropin releasing hormone type-1 receptor translation by an upstream AUG triplet in the 5′ untranslated region. Mol. Pharmacol., 59, 485–492. [DOI] [PubMed] [Google Scholar]
- 29.Arrick B.A., Lee,A.L., Grendell,R.L. and Derynck,R. (1991) Inhibition of translation of transforming growth factor-beta 3 mRNA by its 5′ untranslated region. Mol. Cell. Biol., 11, 4306–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knight S.J., Flannery,A.V., Hirst,M.C., Campbell,L., Christodoulou,Z., Phelps,S.R., Pointon,J., Middleton-Price,H.R., Barnicoat,A., Pembrey,M.E. et al. (1993) Trinucleotide repeat amplification and hypermethylation of a CpG island in FRAXE mental retardation. Cell, 74, 127–134. [DOI] [PubMed] [Google Scholar]
- 31.Stancheva I., Collins,A.L., Van den Veyver, I, Zoghbi,H. and Meehan,R.R. (2003) A mutant form of MeCP2 protein associated with human Rett syndrome cannot be displaced from methylated DNA by notch in Xenopus embryos. Mol. Cell, 12, 425–435. [DOI] [PubMed] [Google Scholar]
- 32.Martinowich K., Hattori,D., Wu,H., Fouse,S., He,F., Hu,Y., Fan,G. and Sun,Y.E. (2003) DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science, 302, 890–893. [DOI] [PubMed] [Google Scholar]
- 33.Chen W.G., Chang,Q., Lin,Y., Meissner,A., West,A.E., Griffith,E.C., Jaenisch,R. and Greenberg,M.E. (2003) Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science, 302, 885–889. [DOI] [PubMed] [Google Scholar]