

Abstract
DxnB2 and BphD are meta-cleavage product (MCP) hydrolases that catalyze C-C bond hydrolysis of the biphenyl metabolite 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoic acid (HOPDA). BphD is a bottleneck in the bacterial degradation of polychlorinated biphenyls (PCBs) by the Bph catabolic pathway due in part to inhibition by 3-Cl HOPDAs. By contrast, DxnB2 from Sphingomonas wittichii RW1 catalyzes the hydrolysis of 3-Cl HOPDAs more efficiently. X-ray crystallographic studies of the catalytically inactive S105A variant of DxnB2 complexed with 3-Cl HOPDA revealed a binding mode in which C1 through C6 of the dienoate are coplanar. The chlorine substituent is accommodated by a hydrophobic pocket that is larger than the homologous site in BphDLB400 from Burkholderia xenovorans LB400. The planar binding mode observed in the crystalline complex was consistent with the hyper- and hypsochromically-shifted absorption spectra of 3-Cl and 3,9,11-triCl HOPDA, respectively, bound to S105A in solution. Moreover, ESred, an intermediate possessing a bathochromically-shifted spectrum observed in the turnover of HOPDA, was not detected, suggesting that substrate destabilization was rate-limiting in the turnover of these PCB metabolites. Interestingly, electron density for the first α-helix of the lid domain was poorly defined in the dimeric DxnB2 structures, unlike in the tetrameric BphDLB400. Structural comparison of MCP hydrolases identified the NC-loop, connecting the lid to the α/β-hydrolase core domain, as a determinant in oligomeric state and suggests its involvement in catalysis. Finally, an increased mobility of the DxnB2 lid may contribute to the enzyme’s ability to hydrolyze PCB metabolites, highlighting how lid architecture contributes to substrate specificity in α/β-hydrolases.
INTRODUCTION
Polychlorinated biphenyls (PCBs) are aromatic xenobiotic compounds that were widely used in the twentieth century in a variety of industrial applications. While bans on the production and use of PCBs began during the 1970s, approximately 10% of all PCBs (~2.8 × 108 kg), remain in mobile environmental reservoirs(1). PCB exposure has been linked to a range of humans diseases including cancers, developmental and neurological disorders(2). Improved environmental bioremediation strategies would minimize continued exposure to these toxic bioaccumulative compounds. To this end, aerobic, biphenyl-utilizing bacteria, first reported in 1970 (3), have been studied for their ability to cometabolize PCBs(4). Although the ability of bacteria to transform different PCB congeners is largely strain-dependent, reflecting the substrate specificities of the first four enzymes of biphenyl (Bph) catabolic pathway(5), the strain-independent accumulation of particularly recalcitrant metabolites has been observed(6). The latter include 3-chloro-2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoic acids (3-Cl HOPDAs).
In the Bph pathway, HOPDA is hydrolyzed by BphD to yield benzoic acid and 2-hydroxy-2,4-pentadienoic acid (HPD). HOPDA and related compounds result from the dioxygenase-mediated cleavage of catechols and are known as meta-cleavage products (MCPs); thus BphD is an MCP hydrolase. BphD was identified as a key determinant in the transformation of PCBs based on at least three observations: (i) yellow-colored metabolites, 3-Cl HOPDAs, accumulated during bacterial growth in the presence of 2,4′-chlorobiphenyls(7); (ii) accumulation of 3-Cl HOPDAs was strain-independent(6); and (iii) 3-Cl HOPDAs inhibit BphD(8, 9). Accordingly, the identification of DxnB2, an MCP hydrolase from Sphingomonas wittichii RW1 that possesses 8 to 80-fold greater specificity for 3-Cl HOPDA relative to bona fide BphD orthologs represents a promising lead in the expansion of the cometabolic potential of the Bph pathway(10).
The MCP hydrolases catalyze C-C bond cleavage through an unusual nucleophilic mechanism that is dependent on a substrate ketonization (Figure 1)(11, 12). As members of the α/β-hydrolase superfamily, the MCP hydrolases possess a Ser-His-Asp triad and an ‘oxyanion hole’ that catalyze the hydrolytic reaction. This catalytic machinery also spatially divides the active site into two subsites. The non-polar (NP)-subsite accommodates the variable MCP moiety: a phenyl ring in the case of HOPDAs. The polar (P)-subsite includes five conserved residues: Gly35, Asn43, Asn104, Arg180 and Trp256 (DxnB2 numbering). These interact with the substrate’s dienoate moiety and have been implicated in the ketonization reaction(13). The MCP hydrolases possess a lid domain consisting of 4 α-helices (α5-α8) inserted between β6 and α9 of the α/β-hydrolase core. Both the α/β-hydrolase core and the MCP hydrolase lid domains are required to furnish the complete suite of catalytic residues.
Figure 1.
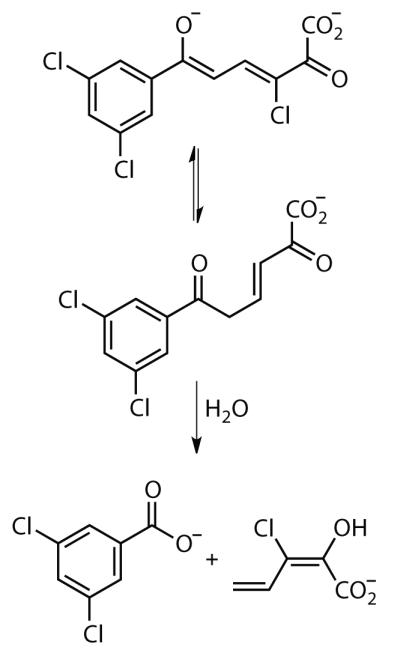
The ketonization and hydrolysis of 3,9,11-triCl HOPDA catalyzed by the MCP hydrolases.
The mechanism of inhibition by 3-Cl HOPDA has been studied extensively for BphD from Burkholderia xenovorans LB400 (BphDLB400). Steady/state kinetic studies revealed that 3-Cl HOPDA competitively inhibits BphDLB400 (Kic = 0.57 μM) and is ultimately hydrolyzed 470-times slower than HOPDA(8). A crystal structure of the BphDLB400 S112A:3-Cl HOPDA binary complex revealed a bound conformation of the HOPDA in which C1 through C6 of the dienoate were coplanar(14). This non-productive binding mode is apparently stabilized by an interaction between a hydrophobic pocket and the chlorine substituent. This contrasts to the productive, non-planar conformation observed in the BphDLB400 S112A:HOPDA binary complex(15), which has been correlated to an enzyme-destabilized species that is characterized by a bathochromically-shifted absorption spectrum observed both in solution and in the crystalline state(15). This orange-colored species (λmax ~ 470 – 500 nm) resembles a catalytically relevant intermediate named ESred that precedes substrate ketonization(11, 15, 16). Under no tested conditions has a similarly red-shifted species been observed for the 3-Cl HOPDAs. However, kinetic studies on a 3-F HOPDA revealed a small population of ESred during turnover(14). Taken together, the studies on BphDLB400 indicate that the 3-Cl substituent inhibits access to the destabilized ESred, an intermediate that is susceptible to protonation, and subsequently, bond cleavage.
The ability of DxnB2 to overcome the apparent inhibition of 3-Cl HOPDAs is poorly understood. In this work, we investigated the ability of DxnB2 to hydrolyze 3-Cl and 3,9,11-triCl HOPDA, the latter of which is a biologically relevant PCB metabolite. Crystal structures of DxnB2, DxnB2 S105A, and a DxnB2 S105A:3-Cl HOPDA binary complex were utilized for a comparative study of structural determinants for variations in catalytic efficiency. Transient-state kinetic analyses of the DxnB2-mediated turnover of 3-Cl HOPDAs were performed under single turnover conditions to identify reaction intermediates and the rate-determining step in catalysis. A structural basis for the PCB-metabolite specificity of DxnB2 is forwarded.
MATERIALS AND METHODS
Chemicals
3-Chloro (3-Cl) and 3,9,11-trichloro (3,9,11-triCl) HOPDAs were generated enzymatically from 4-chloro-2,3-dihydroxybiphenyl and 4,3′,5′-trichloro-2,3-dihydroxybiphenyl, respectively, using 2,3-dihydroxybiphenyl dioxygenase, as previously described(8). All other chemicals were of analytical grade.
Mutagenesis, protein production and purification
Site-directed mutagenesis was performed using a 5′-phosphorylated primer, 5′-PO4-CCCTGATCGGCAACGCCATGGG-3′ to amplify a single-stranded plasmid carrying the desired codon substitution followed by the use of the QuikChange Multi Site-Directed Mutatgenesis Kit (Stratgene, La Jolla, CA). DxnB2 was produced in Escherichia coli as previously described(13). Harvested cells overproducing the recombinant enzymes were resuspended in 20 mM HEPES, pH 7.5 (Buffer A), and lysed at 4 °C using an Emulsi Flex-C5 homogenizer (Avestin Inc, Ottawa, ON). Debris was removed by ultracentrifugation at 50000g for 45 min at 4 °C. The supernatant was passed through a 0.45 μm filter before being loaded onto a 28 mL Source 15Q column, which was equilibrated in Buffer A containing 35 mM NaCl and operated at 15 ml/min. Proteins were eluted using a linear gradient from 35 to 200 mM NaCl in 15 column volumes. Fractions containing DxnB2, which centered near 130 mM NaCl, were pooled and concentrated using an Amicon stirred cell concentrator fitted with an YM30 membrane (Millipore, Billerica, MA). The concentrated fractions were then brought to 1 M (NH4)2SO4 at pH 7.5, and stirred for 12 hours at 4 °C. The resulting precipitate was washed with three 25 mL volumes of Buffer A containing 1 M (NH4)2SO4 before dissolution in 20 mM TAPS, pH 8.5. The purified DxnB2 preparations (> 99% homogeneity based on SDS-PAGE) were then exchanged into potassium phosphate buffer (I = 0.1 M), pH 7.5, concentrated to > 20 mg/ml, and flash frozen in liquid nitrogen. Approximately 80 mg of purified DxnB2 was obtained per litre of cell culture, and all enzyme preparations were stored at −80 °C. The enzyme concentration used in each assay was determined using the molar absorptivity of DxnB2 (ε280 nm = 36.2 mM−1cm−1), calculated from the A280 nm of a sample whose concentration was determined by amino acid analysis at the Advanced Protein Technology Centre (Hospital for Sick Children, Toronto, ON). BphD(17) and HsaD(18) were produced and purified as previously described.
Enzyme-substrate complex formation
ES complexes were formed by mixing 40 μM DxnB2 S105A with 10 μM 3-Cl or 3,9,11-triCl HOPDA in potassium phosphate (I = 0.1 M), pH 7.5. Electronic absorption spectra of the substrates alone and in complex were recorded using a Cary5000 spectrophotometer equipped with a thermostatted cuvette holder maintained at 25 °C.
Stopped-flow spectrophotometry
Experiments were conducted using an SX.18MV stopped-flow reaction analyzer (Applied Photophysics Ltd., Leatherhead, UK) outfitted with a circulating water bath maintained at 25 ± 0.1 °C. Kinetic measurements were performed using potassium phosphate (I = 0.1 M) ± 30% sucrose (w/v), pH 7.5. A photodiode array was initially used to determine the λmax of detectable intermediates formed during each reaction. The reported rate data are derived from single wavelength measurements using a 4.96 nm/mm bandpass monochromator with the slit width set at 0.5 mm. Single turnover experiments were performed using a 4:1 ratio of DxnB2 to substrate: 16 μM DxnB2 was reacted with 4 μM 3-Cl HOPDA or 3,9,11-triCl HOPDA. All reactions were performed using freshly prepared reagents for each replicate. Each replicate was an average of at least five shots from the stopped-flow syringes. The Applied Photophysics Pro-Data Software was employed for non-linear curve fitting and the kinetic parameters are reported as the average of and standard deviation between three replicates.
Size Exclusion Chromatography Multi-angle Light Scattering (SEC-MALS)
SEC-MALS experiments were performed using an AKTA Purifier (GE Healthcare) fitted with Wyatt Technology Corporation SEC column and guard column (WTC-030S5 and WTC-030S5G). The protein concentrations used were between 1.5 and 2.5 mg/ml, prepared in 20 mM HEPES, pH 7.5. Multi-angle light scattering and refractive index was measured using the miniDAWN TREOS and Optilab T-rEX detectors (Wyatt Technology Corporation, Santa Barbara, CA). Calibration was performed using bovine serum albumin.
Crystallization and manipulation of crystals
Crystals were grown using the sitting drop vapor diffusion method at 20°C. Protein samples, 8 to 9 mg/ml in 20 mM HEPES, pH 7.5, were mixed 1:1 with the reservoir, and equilibrated against 500 μL of the reservoir solution. Wild-type (WT) DxnB2 crystals were obtained from a grid of ammonium sulfate (1.0 – 2.5 M) containing 0.1 M Tris, pH 8.2 and 6 – 10% ethanol. Crystals of the S105A variant were obtained from a grid of sodium malonate (1.5 – 2.4 M), pH 6.5/7.0. Crystalline ES complexes were prepared by incubating crystals of S105A in a 30 μL droplet of reservoir solution containing 3-Cl HOPDA at a concentration of ~15 mM for 15 to 20 minutes at 20°C. Prior to flash freezing, crystals were transferred into 30 μL droplets of reservoir solution augmented with a suitable cryoprotectant. The cryoprotectants employed for WT and S105A were 20% glycerol and 2.1 M sodium malonate, respectively. Cryoprotectant solutions for ES complexes contained ~ 5 mM 3-Cl HOPDA.
Structure Determination and Refinement
Diffraction experiments were performed under cryogenic conditions (100 K) at the Advanced Photon Source, Argonne National Laboratory, Argonne, IL, USA: beamlines 14-BM-C and 23-ID-D were used for the WT and S105A crystals, respectively. Typical datasets included 300 frames, collected with a 0.5° rotation per frame at an X-ray wavelength of 1.0 Å. Exposure times were 6 s per degree. Diffraction images were recorded using MarMosaic 300 CCD detectors (Mar USA, Inc., Evanston, IL). Programs from the HKL2000 suite were utilized to process the images and scale and merge intensities(19).
The structure of WT DxnB2 was determined using programs from the CCP4 program package(20). Initial phases were obtained by the molecular replacement method. An initial search model was derived from the structure of a homologue, CarC (PDB ID: 1J1I)(21). The sequence of DxnB2 was incorporated using the CCP4 program CHAINSAW, which pruned non-conserved residues to the last common atom. Molecular replacement calculations performed with the program MOLREP(22) using reflections to 3.5 Å resolution placed one monomer in the asymmetric unit (initial R = 50.9%). After preliminary rigid body and restrained atomic parameter refinement R and Rfree were 36% and 42%, respectively. The electron density was discontinuous for residues 140 – 158 of the lid domain. A revised search model was prepared for a second round of molecular replacement by deleting the entire lid domain, incorporating distal atoms from non-conserved residues, and rebuilding other residues. The structure of WT served as the search model for phasing of the S105A variant by molecular replacement.
Initial solutions obtained from molecular replacement underwent rigid body refinement and multiple cycles of restrained atomic parameter refinement using REFMAC(23). Residues and substrates were manually built into the electron density utilizing molecular graphics programs O(24) and COOT(25). Coordinates for 3-Cl HOPDA and sulfate were generated using PRODRG(26). Refinement restraint libraries were generated using LIBCHECK from the CCP4 package.
Comparison of MCP hydrolases
Superposition of MCP hydrolase structures was performed using the STAMP algorithm implemented in Multiseq, a plugin available in VMD(27-29) or tools available in COOT. The common names, biological sources and PDB codes of the aligned enzymes are: BphDLB400 from B. xenovorans LB400 (2OG1, chain A)(16), BphDRHA1 from Rhodococcus jostii RHA1 (1C4X)(30), CarC from Janthinobacterium sp. strain J3 (1J1I)(21), CumD from Pseudomonas fluorescens IP01 (1IUP)(31), DxnB2 from Sphingomonas witichii RW1 (this study), HsaD from Mycobacterium tuberculosis H37Rv (2VF2, chain A)(32) and MhpC from E. coli W3110 (1U2E, chain A)(33).
RESULTS
Stable enzyme-substrate complexes
Substitution of the catalytic serine to alanine in the MCP hydrolases results in a catalytically impaired enzyme. However, these variants bind substrates in a manner that closely reproduces spectroscopic signatures of a reaction intermediate that precedes chemistry in the wild type enzyme. For example, the binding of HOPDA to catalytic serine-to-alanine variants of BphDLB400 and DxnB2 resulted in bathochromic shifts of the enolate spectra by 72 and 66 nm, respectively, which mirrored the 58 and 53 nm perturbations that characterized ESred under single turnover conditions(11, 15, 16). Accordingly, DxnB2 S105A was used to investigate the ability of this enzyme to destabilize 3-Cl HOPDAs. The electronic absorption spectra of 10 μM 3-Cl or 3,9,11-triCl HOPDA was measured in the presence or absence of 40 μM DxnB2 S105A in potassium phosphate (I = 0.1 M), pH 7.5 at 25 °C. Binding of 3-Cl HOPDA to DxnB2 S105A resulted in a hyperchromically-shifted spectrum (Figure 2A) whereas the DxnB2 S105A:3,9,11-triCl HOPDA complex resulted in a 21 nm hypsochromic shift from 438 to 417 nm (Figure 2B). Neither complex revealed a detectable population of a red-shifted species associated with substrate destabilization(16, 18).
Figure 2.

Electronic absorption spectra of 3-Cl HOPDAs in solution or in complex with 40 μM DxnB2 S105A in potassium phosphate (I = 0.1 M), pH 7.5 at 25 °C. (A) 10 μM 3-Cl HOPDA and (B) 10 μM 3,9,11-triCl HOPDA.
Single turnover of 3-Cl HOPDAs by DxnB2
In order to gain kinetic insight into the ability of DxnB2 to overcome the apparent inhibitory or stabilizing effect attributed to the 3-chloro substituent, hydrolysis of 3-Cl and 3,9,11-triCl HOPDA was monitored under single turnover conditions. Specifically, 16 μM DxnB2 was reacted with 4 μM substrate in potassium phosphate (I = 0.1 M), pH 7.5 at 25 °C. A photodiode array was utilized to identify intermediates (Figure 3 inset). For more accurate rate determination, reactions were monitored at single wavelengths. While decay of the HOPDA enolates (λmax 432 or 438 nm) was readily monitored, the formation of 3-Cl-HPD (λmax ~ 244 nm) could not be distinguished from other underlying processes, and the resulting multiphasic kinetic traces could not be reliably interpreted (Figure S1). To distinguish intra- and intermolecular events, additional reactions were performed in the presence of a viscogen, 30% sucrose (w/v).
Figure 3.

Results from representative stopped-flow experiments demonstrating the turnover of 4 μM 3-Cl HOPDAs by 16 μM DxnB2 in potassium phosphate (I = 0.1 M), pH 7.5 at 25 °C. (A) Hydrolysis of 3-Cl HOPDA monitored at 432 nm. (B) Hydrolysis of 3,9,11-triCl HOPDA monitored at 417 and 438 nm. For each substrate, the accumulation of intermediates as revealed by monitoring time-dependent changes to the visible spectrum from 300 to 600 nm with a PDA is inset.
The hydrolysis of both substrates by DxnB2 was biphasic (Table 1; Figure 3). The first observable kinetic phase, 1/τ1, was attributed to ES complex formation, which occurred at rates of 220 and 26 s−1 for the mono- and trichlorinated substrate, respectively. In both cases, the presence of sucrose slowed this intermolecular event by 1.3 to 2-fold. For 3-Cl HOPDA, ES complex formation was accompanied by a small hypochromic perturbation whereas the spectrum of 3,9,11-triCl HOPDA was hypsochromically-shifted by 21 nm as in the stable S105A complex. Following accumulation of ES complexes, the respective signals decayed at rates of 0.152 and 0.056 s−1. The observed insensitivity of 1/τ2 to sucrose and the relatively large loss in amplitude (~ 90%), corresponding to the dissipation of the HOPDA enolate chromophore, support the assignment of this kinetic step to on-enzyme chemistry. A more precise assignment of 1/τ2 to acylation limited by the rate of substrate ketonization was recently described for DxnB2(11). However, the absence of ESred from the kinetic traces suggests that formation of this intermediate is rate-limiting to the hydrolysis of 3-Cl HOPDAs.
Table 1.
Kinetic values for the DxnB2-mediated hydrolysis of 3-Cl and 3,9,11-triCl HOPDAa
| Substrate | λ (nm) | k1 (s−1) [Amp (%)] | k2 (s−1) [Amp (%)] | k cat b | ||
|---|---|---|---|---|---|---|
| 3-Cl | 432 | 220 ± 20 | [11.5 ± 0.3] | 0.152 ± 0.001 | [88 ± 2] | 0.25 ± 0.1 |
| + 30% sucrose | 432 | 100 ± 10 | [7.4 ± 0.3] | 0.152 ± 0.001 | [92.6 ± 0.7] | |
|
| ||||||
| 3,9,11 triCl- | 417 | 26 ± 1 | [12.2 ± 0.6] | 0.056 ± 0.001 | [88 ± 2] | |
| 438 | 22 ± 2 | [32 ± 1] | 0.056 ± 0.001 | [68 ± 1] | 0.13 ± 0.1 | |
| + 30% sucrose | 417 | 18 ± 2 | [12 ± 1] | 0.054 ± 0.001 | [88 ± 2] | |
| 438 | 16.9 ± 0.2 | [32.9 ± 0.8] | 0.054 ± 0.001 | [67 ± 2] | ||
|
| ||||||
| HOPDAC | 434 | > 500 | 69.4 ± 0.1 | |||
Overall structure and oligomeric state of DxnB2
The stable DxnB2 S105A ES complexes and kinetic behavior of the wild type enzyme paralleled studies on BphDLB400, and therefore provided limited insight into the apparent increased specificity for 3-Cl HOPDAs. These results, together with the classification of DxnB2 and BphDLB400 into phylogenetically distinct MCP hydrolase subfamilies or classes(10), inspired the investigation of the tertiary and quaternary structure of DxnB2 using X-ray crystallography (Table 2) and SEC-MALS analysis.
Table 2.
Properties of the crystals, diffraction data, and refinement statistics
| Crystal properties and diffraction data | |||
| Structure | DxnB2 | DxnB2 S105A | S105A:3 Cl-HOPDA |
| PDB ID | 4LXG | 4LYD | 4LXH |
| Resolution rangea (Å) | 36.5 – 2.2 | 57.4 – 2.2 | 57.6 – 2.0 |
| Space Group | P6522 | P6522 | P6522 |
| Cell Dimensions (Å) | a = b = 67.0, c = 327.7 |
a = b = 66.3, c = 331.6 |
a = b = 66.6, c = 338.2 |
| Unique reflections | 21 400 | 21 860 | 28 184 |
| Multiplicitya | 9.9 (9.6) | 11.5 (6.9) | 7.2 (6.5) |
| Completenessa (%) | 99.5 (98.3) | 93.5 (54.3) | 98.6 (90.8) |
| Rsymma(%) | 9.8 (32.2) | 7.8 (52.7) | 9.0 (50.0) |
| Meana I/σ2 | 21.0 (6.6) | 29.1 (2.0) | 10.8 (1.9) |
|
| |||
| Refinement | |||
|
| |||
| Rfactor/Rfree | 0.20 / 0.23 | 0.21 / 0.26 | 0.21 / 0.26 |
| Model content (atoms) | |||
| Non-hydrogen atoms | 2185 | 2115 | 2220 |
| Protein | 2095 | 2053 | 2114 |
| Precipitant/HOPDAb | 10 / 0 | 0 / 0 | 1 / 17 |
| Water oxygens | 80 | 62 | 88 |
| Average Bfactors (Å2) | |||
| all atoms | 52.9 | 48.0 | 49.5 |
| protein | 52.9 | 48 | 49.1 |
| ligands/HOPDAb | 59.5 / NA | NA / NA | 57.9 / 81.5 (0.8) |
| waters | 50.9 | 46.5 | 52.9 |
| rmsdc bond lengths (Å) | 0.006 | 0.010 | 0.023 |
| rmsdc bond angles (degrees) | 1.0 | 1.5 | 1.8 |
Values for highest resolution bin in parentheses
Each ligand was modeled at full occupancy unless otherwise stated in parentheses; one molecule of sulfate = 5 atoms, HOPDA = 16 atoms + substituent atoms
rmsd is the root-mean-square deviation from restraint targets.
SEC-MALS analysis indicated that DxnB2 exists as a dimer with a molecular weight of 58 ± 2 kDa in solution (Figure S2). The DxnB2 structures reported herein contain one monomer per asymmetric unit in space group P6522, but the crystal structure includes a two-fold symmetric dimer stabilized by an extensive interface centered on an anti-parallel β interaction between the monomers’ β8 strands, as observed in other MCP hydrolases(16).
The DxnB2 monomer, as other MCP hydrolases, is composed of the α/β-hydrolase core (residues 1-129 and 204-277) and an α-helical lid domain (residues 138-203) inserted between β6 and α9 of the core. For some α/β hydrolase families(34, 35), the term NC-loop has been used to refer to a protein segment of variable length and structure connecting β6 to the first structurally conserved helix in the lid domain. Herein NC-loop refers to residues 130-145 of DxnB2 and residues 137-152 of BphDLB400. For the three reported structures, electron density maps clearly defined the backbone for all residues of the core domain except the C-terminal residue (Asp277). With the exception of a few solvent exposed residues, the electron density for core domain side chains was also clear in all structures. In contrast, the electron density was much less distinctive for several residues in the NC-loop and the beginning of the lid domain. Thus the maps for all structures clearly place residues 1-139 and residues 155-276, but the quality of the density and the content of the model varied among the crystal structures for residues 140-154. It is unclear whether this disorder reflects a functionally significant aspect of the α-helical lid in DxnB2.
Comparison of the Tertiary and Quaternary Structures of MCP hydrolases
SEC-MALS analysis indicates DxnB2 is a dimer whereas BphDLB400 and HsaD are tetramers (Figure 4A). CumD, CarC, and MhpC have also been described as dimers. However, the crystal structure of MhpC displays a tetrameric assembly of the same form as BphDLB400 and HsaD. BphDRHA1 purified as an octamer. To investigate the basis of the differences in oligomeric state, the seven structurally characterized MCP hydrolases were superposed and a structure-based sequence alignment was established (Figure S3). As noted previously(16), all of the crystal structures present a two-fold symmetric dimer mediated in part by anti-parallel β-interactions between edge strands (β8) of the monomer’s core domains; we define the associated protein surfaces as elements of the dimeric interface, the A-B interface identified for BphDLB400(16). Of interest here are the distinctions in sequence and structure that mediate assembly of the BphDLB400, HsaD, and MphC tetramers through additional interfaces we label as tetrameric interfaces; in this context, tetrameric refers to the A-B’ and A-A’ interfaces(16) that build the tetramer and does not necessarily imply interactions between four monomers at the same interface.
Figure 4.
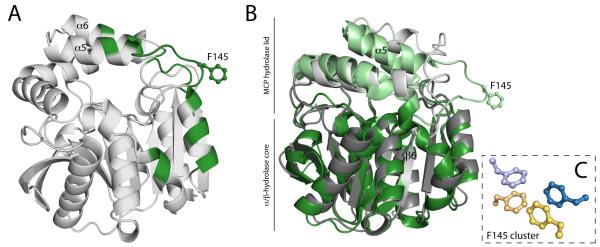
(A) Ribbon drawings of the BphDLB400 protomer (PDB ID: 2OG1, chain A). The regions that are involved either the A-B’ and/or A-A’ tetrameric interfaces, which include the NC-loop and lid domain helices, are colored green. (B) Superposition of the DxnB2 S105A:3-Cl HOPDA binary complex and BphDLB400 demonstrating differences in the NC-loop. (C) The apolar phenylalanine cluster conserved among MCP hydrolase tetramers. The cluster is comprised of the F145 residue from each protomer within the tetrameric assembly. The dimer pairs (A-B pairs) are color coded blue and yellow.
Prior to the NC-loop, a conserved LMG sequence (DxnB2 127-9) completes the β6 strand in all known structures. The glycine residue, at position i, has a left-handed α conformation, which creates a sharp turn as the backbone departs from the core. The next highly conserved sequence positions are a pair of branched non-polar residues at positions j and j+3 (DxnB2 146 and 149); the segment between i and j encompasses 12-17 residues in the known structures. In all structures except that of CarC (wherein these residues are disordered), the side chains of residues j and j+3 project from a helical segment of the lid domain into the NP-substrate binding subsite. Residues between positions i and j are prominent at the tetrameric interfaces, and the sequences and structures of BphDLB400, HsaD, and MhpC present features distinguishing them from the dimeric MCP hydrolases.
In particular, the tetrameric MCP hydrolases of known crystal structure all share an F-X-P-X-P motif that begins at position i+9 and contributes to the tetrameric interface that associates a pair of lid domains, the A-B’ interface. In fact, Fi+9 makes a visually striking contribution to both tetrameric interfaces as illustrated in Figure 4A and 4C. Thus Fi+9 lies at the apex of a loop that protrudes away from the hydrolase and lid domains, and Fi+9 residues from all four monomers cluster to form a buried apolar core at the junction of the A-B’ and A-A’ interfaces (Figure 4C). Residues immediately preceding Fi+9 as well as residues in the linker and the helix (α10) that follows the triad Asp also contribute to tetramer formation, but they do not belong to sequence motifs that distinguish dimers and tetramers. BphDLB400, HsaD, and MhpC belong to the same class, class I, on the basis of amino acid sequences(10). The sequences of other homologs in this class (such as PcbD from Pseudomonas stain sp. DJ12 and Ro09014 from Rhodococcus jostii RHA1) show variations at the first position of the F-X-P-X-P motif. However, the oligomeric states are not known to us.
In contrast to the protruding structure observed for the tetramers, the first few residues of the NC-loop in DxnB2 associate with helix α10 of the hydrolase core domain as well as residues just prior to helix α9 via several hydrogen bonds (Figure 5A). The NC-loop of CumD is five residues shorter than that of DxnB2 and, while it follows a similar path, is not involved in the same suite of H-bonding interactions. However, CumD and several other class II enzymes share an F-P-X-P-Q-X4-D motif that disrupts helix α8 in CumD and places the Gln side chain from this motif in position to hydrogen bond with the backbone carbonyl and amide of NC-loop i+6 and i+8 residues, respectively.
Figure 5.

(A) Ball-and-stick representation of the polar interactions between the side chain and backbone atoms of the poorly conserved loop (white) extending from β6 and the α/β-hydrolase core (grey) of DxnB2. (B) Combined ribbon drawings and ball-and-stick representation of the structural superposition of three representatives from each MCP hydrolase class: CumD (blue), DxnB2 (white) and BphDLB400 (green). The lid domains and relevant residues contributing to the structural divergence are shown and numbered. Possible polar interactions are drawn as dashed lines.
In general, the beginning and end of the lid show greatest variability among the known MCP hydrolase crystal structures (Figure 5B). Thus in many pair-wise or multi-protein comparisons it can be difficult to structurally align residues from the NC-loop and α5 and/or residues within α8 and the segment that links α8 to α9. In addition, the NC-loop and α5 are prone to disorder and are sensitive to the occupancy of ligands in the active site. These uncertainties should be considered in evaluating sequence alignments, including those based on structures (Figure S3). By contrast, the segment of the lid spanning helices α6 and α7, residues 155-186 of DxnB2 and residues 164-196 of BphDLB400, is more readily superposed and aligned.
DxnB2 WT and S105A active sites
As described for BphDLB400(16), the DxnB2 substrate binding site can be divided into polar (P) and non-polar (NP) subsites that merge at the locations of the catalytic serine, Ser105, and oxyanion hole residues, Met106 and Gly34. The NP-subsite is largely formed by the side chains of residues from both the core domain and the lid. Because of disorder in the NC-loop and α5 (see above) and perhaps the absence of substrate, the NP-subsite is not fully defined by the electron density of the S105A structure. Thus Leu146 and Val149 from α5, which correspond to residues in contact with the substrates in structures of BphDLB400 and HsaD(15, 16, 18), are not included in the model. These residues are present in the model for the WT structure, and all others predicted by sequence alignment to be involved in substrate binding in the NP site are present in both structures. The P site residues are well-defined in both structures.
The structure of the DxnB2 WT active site includes a sulfate ion that binds at the heart of the active site within hydrogen bonding distance of the catalytic serine, the backbone amides of the oxyanion hole, the triad histidine (His255), Trp256, and Asn104 (Figure 6A). The influence of the sulfate is extended by bound water molecules. For example, two water molecules form a bridge between the sulfate and conserved P-site Arg180, and one of these also hydrogen bonds with Asn43. Similarly two additional water molecules contribute to the interactions of the sulfate with Asn104 and Ser105. Despite the presence of the sulfate, the catalytic triad residues are within H-bonding distances of each other: the Ser105 Oγ is 2.9 Å from the His255 Nε2, and the His255 Nε1 is 2.6 Å and 3.2 Å from the Asp227 carboxylate oxygens.
Figure 6.

(A) Ball-and-stick representation of the DxnB2 (white) and BphDLB400 (PDB ID: 2OG1 chain A, green) active site (DxnB2 numbering). For simplicity, only the polar contacts among the catalytic triad residues or involving Arg180 are drawn as dashed lines. The sulfate and water molecules (salmon) that occupy the DxnB2 active site are also shown. (B) Ball-and-stick representation of the stable 3-Cl HOPDA binding mode observed in the crystalline DxnB2 S105A complex. The 2Fo – Fc map (green) for the substrate is shown, contoured at 1σ. Polar contacts ≤ 3.4 Å are drawn as dashed lines.
The ordered active site residues in the DxnB2 S105A model generally superpose on their WT counterparts with high fidelity and well within the expected positional error with the exception of a slight difference in the side chain conformation of Met229 (see below). The final model for S105A does not include a multi-atom ligand or waters bound near the catalytic residues. Nevertheless, it remains possible that the active site is organized by a poorly ordered ligand inasmuch as the final difference map includes a non-definitive density feature above 0.15 e/Å3 and large enough to accommodate an aspartate side chain. At one end, the density predicts an atom within 3.0 Å of the oxyanion hole amides. At the other end, the density extends to within non-bonded contact distance of the side chain of Met229, which is modeled into relatively poor electron density. Thus the presence of some multi-atom ligand of variable structure could explain the slightly different conformation and non-definitive density of Met229; disorder in α5 could also contribute. Malonate is the prime candidate for the poorly ordered ligand because it is present at high concentration in the crystals.
The location and interactions of conserved P-subsite residue Arg180 are unexpectedly and remarkably different in DxnB2 compared to the architecture observed in BphDLB400 and HsaD. In the latter, the guanidinium groups of the corresponding Arg residues are positioned to interact with the substrate’s carboxylate group and with residues at the apex of the connector between β3 and α1 (DxnB2 residues 35 and 36). In DxnB2, Arg180 interacts with residues from the α1 helix; the shortest contact, 3.2 Å, is between Nη2 and the carbonyl O of Ser42. Relative to BphDLB400, the guandinium group of Arg180 is displaced by ~3.5 Å. The orientation of Arg180 is shared between the structures of the WT and S105A variant, and therefore cannot be attributed to the presence of sulfate in the former.
DxnB2 S105A:3-Cl HOPDA complex
To define DxnB2-specific interactions with recalcitrant substrates, S105A crystals were incubated with 3-Cl HOPDA resulting in a structure of the binary complex at a resolution of 2.0 Å. Similar to the S105A structure in the absence of HOPDA, electron density was poorly defined for residues 140 through 151. Although the model includes these residues, only the course of the backbone is clear from the density. The electron density for the 3-Cl HOPDA ligand was also incomplete in early maps such that the distal atoms of the phenyl moiety (CB3, CB4 and CB6) and the C1-carboxylate could not be confidently positioned. Nevertheless, the density clearly indicated that the C3-Cl and C2-O atoms are in a cis-conformation around the C2-C3 bond and that the carbon atoms of the dienoate moiety are coplanar (Figure 6B). Ultimately, the 3-Cl HOPDA molecule was modeled as a (2Z,4E)-2-oxido-6-oxo- isomer based on the available electron density and the yellow color of the crystals, which is indicative of a dianionic species (3-Cl HOPDA pKa2 = 6.1(8)). Other isomers of HOPDA may be consistent with the electron density. In line with the spectrophotometric data, the binding mode of 3-Cl HOPDA differed significantly from those observed in BphDLB400 S112A:HOPDA and DxnB2 S105A:HOPDA or 5,8-diF HOPDA binary complexes, which have been correlated to ESred(11, 15).
Although 3-Cl HOPDA binds to DxnB2 S105A in a planar conformation with the C6-O in the oxyanion hole, as previously observed for BphDLB400, enzyme-substrate interactions in the P-subsite differ markedly. In particular, the guanidinium group of Arg180 does not interact with the substrate’s carboxylate group and its C2-O atom as in the BphDLB400 complex. Although it appears that the Arg180 side chain could be placed in an appropriate location to bind the carboxylate, it maintains the same slightly removed location and conformation as in the unliganded structures. Interactions of the carboxylate with Trp256 Nε2, Asn104 Nδ2, and a water molecule bridging to Arg180 replace the direct HOPDA:Arg190 interactions observed in the BphDLB400 complex.
The interactions of the 3-Cl substituent also vary between the complexes. In BphDLB400, the Cl binding pocket includes atoms within 4.4 Å from five residues, and it is capped by side chains from a well-ordered and close-packed α5 helix, which supplies a 3.7 Å contact with the side chain of Leu156 that is backed by the side chain of a second-shell residue Ile153 (Figure 7A). By contrast, the DxnB2 pocket includes only three side chain interactions within 4.4 Å (Figure 7B). In addition, α5 is poorly ordered, as noted above, and the Cα atoms of the residues corresponding to Leu156 and Ile153 in BphDLB400, DxnB2 Val149 and Leu146, are 1.4 Å and 3.1 Å further removed from the chlorine atom.
Figure 7.

Protein surface representation of the hydrophobic pocket that stabilizes the planar 3-Cl HOPDA binding mode in (A) DxnB2 S105A and (B) BphDLB400 S112A (PDB ID: 2RHT). Leu156 of the BphDLB400 lid domain is the positional equivalent of Val149 in DxnB2, which is further removed from the MCP bound in the dimeric homolog. Other DxnB2 lid domain residues illustrated include Ile162 (BphDLB400 Met171) and Leu166. The remaining residues that help to form the hydrophobic pocket are part of the α/β-hydrolase core domain.
DISCUSSION
The single turnover data together with the proposed catalytic mechanism of MCP hydrolases indicate that the higher specificity of DxnB2 for 3-Cl HOPDAs (kcat/Km) versus BphD is due to higher rate of ESred formation in the former. More specifically, the hydrolysis of HOPDAs by DxnB2(11) and BphDLB400(14-16) is defined by three observable steps for which the rates have been determined: (i) ESred formation (> 500 s−1); (ii) ESred decay (50 to 70 s−1), which is coupled to acylation/HPD formation and limited by the ketonization reaction; and (iii) deacylation (kcat ~0.4 to 6.5 s−1). In contrast, the hydrolysis of 3-Cl HOPDAs by these enzymes is characterized by just two observable processes (Table 1): (i) formation of an alternative, presumably non-productive, binding mode named ESe in BphDLB400(14) and characterized by a hypo- and/or hypsochromically-shifted absorption spectrum in DxnB2; and (ii) ESe decay, which is coupled to HPD formation and corresponds to kcat (Table 1). The failure to detect ESred during 3-Cl HOPDA cleavage indicates that it is consumed at a rate that precludes detection. Thus, the similarity between the observed rates of HPD formation and kcat values indicate that ESred formation is rate-limiting to catalysis(10). For 3-Cl HOPDA, this rate (1/τ2) is at least 3000-fold slower than for HOPDA in DxnB2(17). However, formation of ESred from 3-Cl HOPDA in DxnB2 (0.152 s−1) is 20-fold faster than in BphDLB400 (0.0077 s−1), accounting for the increased specificity of the former for recalcitrant PCB metabolites. Interestingly, ESred formation during turnover of 3,9,11-triCl HOPDA was only ~3-fold slower than 3-Cl HOPDA. This small reduction is in line with the general observation that ring-substituents are minor contributors to the substrate specificity of BphDLB400 and DxnB2(8, 10).
The structural analyses of the MCP hydrolases further support a significant contribution of the NC-loop to catalysis. This loop, which joins the core and lid domains, has been proposed to contribute to catalysis in several other α/β-hydrolase families including the lipases(35), haloalkane dehalogenases(36) and epoxide hydrolases(37). Furthermore, substitution of the NC-loop was required to convert an esterase into an epoxide hydrolase(38). In the MCP hydrolases, domain-swapping experiments between BphD (BphDLA4) and MfphA from Dyella ginsengisoli LA-4 further established that the identity of the lid domain is a strong determinant of substrate specificity(39), and a single amino acid substitution located in the NC-loop of BphDLA4 reduced kcat/Km for HOPDA by nearly 300-fold(40). Molecular dynamics simulations have also been used to argue the relevance of NC-loop flexibility in the haloalkane dehalogenases(36) and significant α5 helix motions were observed in the Candida antarctica lipase B (CALB)(41). Interestingly, while the CALB and MCP hydrolase lid domains are distinct, the first helix of each, which follows the NC-loop, occupy very similar three-dimensional space in relation to the α/β-hydrolase core. Indeed, the sum of the available data for the α/β-hydrolases sets a precedent for the involvement of the NC-loop in substrate binding and catalysis by the MCP hydrolases.
The identification of the NC-loop as a determinant of MCP hydrolase oligomeric state provides a further example of the ability of the α/β-hydrolase core to accommodate family-specific insertions to determine substrate and chemical specificity(42, 43). Thus, comparison of the seven structurally characterized MCP hydrolases representing three different oligomeric states revealed high structural variation for the NC-loop. The NC-loop residues appear to be most highly conserved among the class I enzymes, and to at least partially mediate tetramer formation. In particular, the NC-loop contributes to two distinct tetrameric interfaces and the phenylalanine residue of its F-X-P-X-P motif lies at the core of the biological assembly. Moreover, differences in the NC-loop are propagated to the lid domain architecture: the position of and interactions involving the first helix of the MCP hydrolase lid, α5, are not conserved between the phylogenetically distinct classes. Each class possesses a distinguishing set of intra-lid interactions, which are likely to mitigate substrate specificity.
The variable disorder in helix α5 in crystal structures of DxnB2 suggests that this region has increased mobility relative to other MCP hydrolases with the exception of CarC. Such movement is probably restricted in the tetrameric enzymes based on the location the tetrameric interfaces. Interestingly, superposition of three DxnB2 S105A:HOPDA binary complexes reveals large difference in the position of helix α5, which varies up to ~6 Å based on placement of the Cα atoms (Figure 8). The position of the α5 helix drastically affects the form of the NP-subsite. Specifically, helix α5 carries Leu146 and Val149, which are further removed from the substrate in crystal structures of DxnB2 than in BphDLB400. Accordingly, the comparatively limited restraint placed by the DxnB2 NP-site on the substrate is also likely to contribute to the relative increase in hydrolytic activity towards recalcitrant PCB metabolites. Thus, the possible mobility of this helix may account for the increased accessibility to ESred in DxnB2 versus BphDLB400.
Figure 8.

Comparison of DxnB2 S105A binary complexes reveals differences in the lid domains. The three structures include S105A in complex with 3-Cl HOPDA (green), HOPDA (white) and 5,8-diF HOPDA (grey).
The 3-Cl HOPDA binding mode observed in DxnB2 suggests that significant conformational changes must occur in order to reach a productive substrate-binding mode. Many of these changes have been highlighted by studies in BphDLB400(14). The DxnB2 S105A:3-Cl HOPDA complex is particularly intriguing since in addition to the distance observed between the NP-subsite and the substrate, it is also missing a number of polar contacts at the P-subsite. Thus, neither Asn43 nor Arg180 contact the dienoate moiety in the DxnB2 S105A:3-Cl HOPDA binary complex. These contrast with the direct interactions observed in other crystalline complexes that were correlated to ESred(11, 15), and those that potentially form in the BphDLB400 S112A:3-Cl HOPDA complex(14). These observed differences in stable, planar ESe complexes may also contribute to the ability of DxnB2 to form ESred faster than BphDLB400.
The high structural variance of the NC-loop contributes to the ability of DxnB2 to overcome the stabilization of 3-Cl HOPDAs in non-productive, planar dienoate binding modes. Moreover, the NC-loop is prominent at MCP hydrolase tetrameric interfaces suggesting that it at least partially stabilizes the oligomeric state of class I family members. The attributes of this loop have implications for both environmental bioremediation and the design of therapeutics. Replacement of BphDLB400 (or other bona fide BphD enzymes) by DxnB2 in the Bph pathway may alleviate the metabolic bottleneck caused by 3-Cl HOPDAs. The dimeric MCP hydrolases could also represent better starting points for future enzyme engineering efforts. The plasticity of the NC-loop should also be considered in designing inhibitors of HsaD, which appears to be required for survival of Mycobacterium tuberculosis in macrophages(44). Finally, while the results further establish the catalytic adaptability of the α/β-hydrolase superfamily and highlight the influence of lid architecture within a specific enzyme family, the correlation of loop and lid movements to catalysis is relevant to a broader understanding of enzyme dynamics(45).
Supplementary Material
ACKNOWLEDGMENT
We acknowledge Dr. Michael Murphy and Marek Kobylarz for access and technical assistance SEC-MALS instrument, respectively.
FUNDING STATEMENT This research was supported by a Discovery grant from the Natural Sciences and Engineering Research Council of Canada to L.D.E., and A.C.R. is an NSERC PGS-D scholar. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. Some of the diffraction data were obtained using GM/CA at the APS facilities. GM/CA at the APS has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104). Use of BioCARS Sector 14 was supported by grants from the National Center for Research Resources (5P41RR007707) and the National Institute of General Medical Sciences (8P41GM103543) from the National Institutes of Health.
ABBREVIATIONS
- Bph
biphenyl
- HOPDA
2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoic acid
- HPD
2-hydroxy-2,4-pentadienoic acid
- MCP
meta-cleavage product
- PCB
polychlorinated biphenyl
Footnotes
Supporting Information. HPLC analysis of reaction products, SEC-MALS and sequence alignment. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
REFERENCES
- 1.Faroon OM, Keith LS, Smith-Simon C, De Rose CT. Polychlorinated Biphenyls: Human Health Aspects. World Health Organization; Geneva: 2003. [Google Scholar]
- 2.Agency for Toxic Substances and Disease Registry . Toxicological Profile for Polychlorinated Biphenyls (PCBs) U.S. Department of Human Health and Human Services; Atlanta, GA: 2000. [PubMed] [Google Scholar]
- 3.Lunt D, Evans WC. The microbial metabolism of biphenyl. Biochem J. 1970;118:54P–55P. doi: 10.1042/bj1180054pb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmed M, Focht DD. Degradation of polychlorinated biphenyls by two species of Achromobacter. Can J Microbiol. 1973;19:47–52. doi: 10.1139/m73-007. [DOI] [PubMed] [Google Scholar]
- 5.Furukawa K. Biochemical and genetic bases of microbial degradation of polychlorinated biphenyls (PCBs) J Gen Appl Microbiol. 2000;46:283–296. doi: 10.2323/jgam.46.283. [DOI] [PubMed] [Google Scholar]
- 6.Bedard DL, Haberl ML. Influence of Chlorine Substitution Pattern on the Degradation of Polychlorinated Biphenyls by Eight Bacterial Strains. Microbial Ecology. 1990;20:87–102. doi: 10.1007/BF02543870. [DOI] [PubMed] [Google Scholar]
- 7.Furukawa K, Tomizuka N, Kamibayashi A. Effect of chlorine substitution on the bacterial metabolism of various polychlorinated biphenyls. Appl Environ Microbiol. 1979;38:301–310. doi: 10.1128/aem.38.2.301-310.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seah SY, Labbe G, Nerdinger S, Johnson MR, Snieckus V, Eltis LD. Identification of a serine hydrolase as a key determinant in the microbial degradation of polychlorinated biphenyls. J Biol Chem. 2000;275:15701–15708. doi: 10.1074/jbc.275.21.15701. [DOI] [PubMed] [Google Scholar]
- 9.Seah SY, Labbe G, Kaschabek SR, Reifenrath F, Reineke W, Eltis LD. Comparative specificities of two evolutionarily divergent hydrolases involved in microbial degradation of polychlorinated biphenyls. J Bacteriol. 2001;183:1511–1516. doi: 10.1128/JB.183.5.1511-1516.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seah SY, Ke J, Denis G, Horsman GP, Fortin PD, Whiting CJ, Eltis LD. Characterization of a C-C bond hydrolase from Sphingomonas wittichii RW1 with novel specificities towards polychlorinated biphenyl metabolites. J Bacteriol. 2007;189:4038–4045. doi: 10.1128/JB.01950-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruzzini AC, Bhowmik S, Yam KC, Ghosh S, Bolin JT, Eltis LD. 2013. unpublished.
- 12.Lam WW, Bugg TD. Purification, characterization, and stereochemical analysis of a C-C hydrolase: 2-hydroxy-6-keto-nona-2,4-diene-1,9-dioic acid 5,6-hydrolase. Biochemistry. 1997;36:12242–12251. doi: 10.1021/bi971115r. [DOI] [PubMed] [Google Scholar]
- 13.Li C, Li JJ, Montgomery MG, Wood SP, Bugg TD. Catalytic role for arginine 188 in the C-C hydrolase catalytic mechanism for Escherichia coli MhpC and Burkholderia xenovorans LB400 BphD. Biochemistry. 2006;45:12470–12479. doi: 10.1021/bi061253t. [DOI] [PubMed] [Google Scholar]
- 14.Bhowmik S, Horsman GP, Bolin JT, Eltis LD. The molecular basis for inhibition of BphD, a C-C bond hydrolase involved in polychlorinated biphenyls degradation: large 3-substituents prevent tautomerization. J Biol Chem. 2007;282:36377–36385. doi: 10.1074/jbc.M707035200. [DOI] [PubMed] [Google Scholar]
- 15.Horsman GP, Bhowmik S, Seah SY, Kumar P, Bolin JT, Eltis LD. The tautomeric half-reaction of BphD, a C-C bond hydrolase. Kinetic and structural evidence supporting a key role for histidine 265 of the catalytic triad. J Biol Chem. 2007;282:19894–19904. doi: 10.1074/jbc.M702237200. [DOI] [PubMed] [Google Scholar]
- 16.Horsman GP, Ke J, Dai S, Seah SY, Bolin JT, Eltis LD. Kinetic and structural insight into the mechanism of BphD, a C-C bond hydrolase from the biphenyl degradation pathway. Biochemistry. 2006;45:11071–11086. doi: 10.1021/bi0611098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruzzini AC, Horsman GP, Eltis LD. The catalytic serine of meta-cleavage product hydrolases is activated differently for C-O bond cleavage than for C-C bond cleavage. Biochemistry. 2012;51:5831–5840. doi: 10.1021/bi300663r. [DOI] [PubMed] [Google Scholar]
- 18.Lack NA, Yam KC, Lowe ED, Horsman GP, Owen RL, Sim E, Eltis LD. Characterization of a carbon-carbon hydrolase from Mycobacterium tuberculosis involved in cholesterol metabolism. J Biol Chem. 2010;285:434–443. doi: 10.1074/jbc.M109.058081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otwinowski Z, Minor W. Methods in Enzymology. Academic Press; New York: 1997. Processing of X-ray Diffraction Data Collected in Oscillation Mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 20.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Habe H, Morii K, Fushinobu S, Nam JW, Ayabe Y, Yoshida T, Wakagi T, Yamane H, Nojiri H, Omori T. Crystal structure of a histidine-tagged serine hydrolase involved in the carbazole degradation (CarC enzyme) Biochem Biophys Res Commun. 2003;303:631–639. doi: 10.1016/s0006-291x(03)00375-9. [DOI] [PubMed] [Google Scholar]
- 22.Vagin A, Teplyakov A. An approach to multi-copy search in molecular replacement. Acta Crystallogr D Biol Crystallogr. 2000;56:1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- 23.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 24.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 26.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 27.Russell RB, Barton GJ. Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins. 1992;14:309–323. doi: 10.1002/prot.340140216. [DOI] [PubMed] [Google Scholar]
- 28.Roberts E, Eargle J, Wright D, Luthey-Schulten Z. MultiSeq: unifying sequence and structure data for evolutionary analysis. BMC Bioinformatics. 2006;7:382. doi: 10.1186/1471-2105-7-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 30.Nandhagopal N, Senda T, Hatta T, Yamada A, Masai E, Fukuda M, Mitsui Y. Three-dimensional structure of microbial 2-hydroxyl-6-oxo-phenylhexa-2,4-dienoic acid (HPDA) hydrolase (BphD enzyme) from Rhodococcus sp. strain RHA1, in the PCB degradation pathway. Proceedings of the Japan Academy, Ser. B. 1997;73:154. [Google Scholar]
- 31.Fushinobu S, Saku T, Hidaka M, Jun SY, Nojiri H, Yamane H, Shoun H, Omori T, Wakagi T. Crystal structures of a meta-cleavage product hydrolase from Pseudomonas fluorescens IP01 (CumD) complexed with cleavage products. Protein Sci. 2002;11:2184–2195. doi: 10.1110/ps.0209602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lack N, Lowe ED, Liu J, Eltis LD, Noble ME, Sim E, Westwood IM. Structure of HsaD, a steroid-degrading hydrolase, from Mycobacterium tuberculosis. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:2–7. doi: 10.1107/S1744309107065931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunn G, Montgomery MG, Mohammed F, Coker A, Cooper JB, Robertson T, Garcia JL, Bugg TD, Wood SP. The structure of the C-C bond hydrolase MhpC provides insights into its catalytic mechanism. J Mol Biol. 2005;346:253–265. doi: 10.1016/j.jmb.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 34.Barth S, Fischer M, Schmid RD, Pleiss J. The database of epoxide hydrolases and haloalkane dehalogenases: one structure, many functions. Bioinformatics. 2004;20:2845–2847. doi: 10.1093/bioinformatics/bth284. [DOI] [PubMed] [Google Scholar]
- 35.Li B, Yang G, Wu L, Feng Y. Role of the NC-loop in catalytic activity and stability in lipase from Fervidobacterium changbaicum. PLoS One. 2012;7:e46881. doi: 10.1371/journal.pone.0046881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otyepka M, Damborsky J. Functionally relevant motions of haloalkane dehalogenases occur in the specificity-modulating cap domains. Protein Sci. 2002;11:1206–1217. doi: 10.1110/ps3830102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barth S, Fischer M, Schmid RD, Pleiss J. Sequence and structure of epoxide hydrolases: a systematic analysis. Proteins. 2004;55:846–855. doi: 10.1002/prot.20013. [DOI] [PubMed] [Google Scholar]
- 38.Jochens H, Stiba K, Savile C, Fujii R, Yu JG, Gerassenkov T, Kazlauskas RJ, Bornscheuer UT. Converting an esterase into an epoxide hydrolase. Angew Chem Int Ed Engl. 2009;48:3532–3535. doi: 10.1002/anie.200806276. [DOI] [PubMed] [Google Scholar]
- 39.Zhou H, Qu Y, Shen E, Kong C, Zhang X, Ma Q, Zhou J. Tuning the substrate selectivity of meta-cleavage product hydrolase by domain swapping. Appl Microbiol Biotechnol. 2012 doi: 10.1007/s00253-012-4405-5. [DOI] [PubMed] [Google Scholar]
- 40.Zhou H, Qu Y, Kong C, Shen E, Wang J, Zhang X, Ma Q, Zhou J. The key role of a non-active-site residue Met148 on the catalytic efficiency of meta-cleavage product hydrolase BphD. Appl Microbiol Biotechnol. 2013 doi: 10.1007/s00253-013-4814-0. [DOI] [PubMed] [Google Scholar]
- 41.Skjot M, De Maria L, Chatterjee R, Svendsen A, Patkar SA, Ostergaard PR, Brask J. Understanding the plasticity of the alpha/beta hydrolase fold: lid swapping on the Candida antarctica lipase B results in chimeras with interesting biocatalytic properties. Chembiochem. 2009;10:520–527. doi: 10.1002/cbic.200800668. [DOI] [PubMed] [Google Scholar]
- 42.Holmquist M. Alpha/Beta-hydrolase fold enzymes: structures, functions and mechanisms. Curr Protein Pept Sci. 2000;1:209–235. doi: 10.2174/1389203003381405. [DOI] [PubMed] [Google Scholar]
- 43.Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, et al. The alpha/beta hydrolase fold. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 44.Rengarajan J, Bloom BR, Rubin EJ. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci U S A. 2005;102:8327–8332. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nashine VC, Hammes-Schiffer S, Benkovic SJ. Coupled motions in enzyme catalysis. Curr Opin Chem Biol. 2010;14:644–651. doi: 10.1016/j.cbpa.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.