


Abstract
In bacterial expression systems, translation initiation is usually the rate limiting and the least predictable stage of protein synthesis. Efficiency of a translation initiation site can vary dramatically depending on the sequence context. This is why many standard expression vectors provide very poor expression levels of some genes. This notion persuaded us to develop an artificial genetic selection protocol, which allows one to find for a given target gene an individual efficient ribosome binding site from a random pool. In order to create Darwinian pressure necessary for the genetic selection, we designed a system based on translational coupling, in which microorganism survival in the presence of antibiotic depends on expression of the target gene, while putting no special requirements on this gene. Using this system we obtained superproducing constructs for the human protein RACK1 (receptor for activated C kinase).
INTRODUCTION
Bacterial expression systems, in spite of some limitations in their applicability, remain an important domain in recombinant protein production, because of the robustness and low cost of these systems. This widely used method for preparing eukaryotic proteins includes the synthesis of an intronless gene encoding the target protein and its insertion into a bacterial vector containing the regulatory elements necessary for correct and efficient transcription and translation as a separate polypeptide or as part of a fusion protein. Modern expression vectors based usually on bacteriophage T7 RNA polymerase-driven transcription reliably provide robust transcription of the target gene and the rate limiting step in protein synthesis is most likely to be translation, especially its initiation.
The relatively simple translational apparatus of bacteria, requiring only a short Shine–Dalgarno (SD) sequence several nucleotides upstream of the start codon, however, is very sensitive to the sequence context (1,2), including the coding sequence of the target gene. A single nucleotide substitution in the translation initiation region not affecting the SD sequence, initiation codon or length of the spacer may cause a 20- to 40-fold increase in translation efficiency (3). High efficiency of translation is provided by a fragile balance of two conditions: the ribosomal 30S subunit should readily bind to the translation initiation region, yet should be able to easily move on to start elongation. Binding is mainly driven by the Watson–Crick base pairing of rRNA with the SD sequence. An extended SD sequence (>5 nt) might be beneficial for translation if there is a competing secondary structure or some spatial hindrance of the translation initiation region, however, it compromises further progression of the ribosome. Therefore, the key to a high translational efficiency is an optimal sequence context providing a high binding rate with low affinity for the 30S subunit. It is most likely the secondary structure that is crucial for fulfilling these conditions, however, oligo(A) linker, for example, which should not potentially support strong secondary structures, was found not to be optimal for maximum translation initiation (4). What is beyond doubt is that the optimal sequence context depends on the sequence of the coding region. This creates a major hurdle for designing versatile expression vectors other than fusion protein vectors with a constant proximal coding part. Ideally, the translation initiation region ought to be fitted individually for every target gene (5), but there is no rational basis for such ‘fitting’.
Genetic selection is a powerful combinatorial approach, which allows one to select feature sequences out of a random pool in an artificially designed selection system (6). Projected to our problem, this would mean randomization of the translation initiation region of the target gene and selection of the clones most efficiently expressing this gene. In this hypothetical system microorganism survival should depend on expression of the target gene, an exogenous gene of our interest. In order to provide such dependence, we decided to introduce a second gene (an antibiotic resistance marker) downstream of the target gene. The marker was to be translationaly coupled with the upstream gene (generating, however, a separate polypeptide), so that the higher the translation of the target gene, the higher the translation of the marker, resulting in a higher antibiotic resistance (Fig. 1A).
Figure 1.

Schematic representation of the genetic selection system. (A) A plasmid construct with a two-cistron T7 promoter-driven expression cassette, containing the translation conjugation site from MS2 phage capable of reinitiating translation of the Tet resistance gene when the ribosomes are delivered into this site after termination of translation of the target gene. The construct also contains an ampicillin resistance marker for propagation of the plasmid independently of T7 polymerase. (B) A randomized fragment can be introduced into this plasmid through a BstXI site. (C) Sequencing pattern of the supercoiled plasmid pool recovered after transforming bacteria with the ligation mixture described in (B). Note that the sequencing pattern becomes gibberish for the 20 nt of the random insert and then resumes the dominant readable sequence from the same point where it was broken, indicating that the majority of the individual plasmids in this pool have inserts of the correct length.
Conjugated translation is a natural phenomenon used by microorganisms and their parasites. There are several mechanisms by which the translation of a downstream gene on a polycistronic mRNA is regulated by translation of the upstream gene (7). The mechanism which appeared most suitable for our system is the ‘ribosome delivery mechanism’, which has been shown to conjugate translation of the coat protein gene and the lysis protein gene in bacteriophage MS2 (8). In this example, the downstream gene possesses a very weak ribosome binding site virtually incapable of independent translation initiation. However, such a structure can accomplish initiation of translation when ribosomes are delivered to this site from the upstream gene. This mechanism has two advantages for our genetic selection system: (i) the efficiency of translational conjugation is not dependent on the primary structure of the upstream gene, which makes the construct versatile with respect to the target gene; (ii) the efficiency of translation initiation of the downstream gene is largely reduced compared to the upstream gene, which would mean for our system limited expression of the selection marker under overexpression of the target gene.
Using these features we constructed a genetic selection system which allowed us to select an efficient clone producing recombinant human protein receptor for activated C kinase (RACK1).
MATERIALS AND METHODS
Plasmid construction
pGCT vector. The 72 nt linker (top strand 5′-GGGTTAACTTTACCATAACAGTGGATAACATATGTAGGTACCTTTAAAACTGGTAACCCAATTGTACCTGCAGAG; restriction sites are in bold, the translational enhancer is italicized and the MS2 reinitiation site is underlined) was constructed from overlapping oligonucleotides representing fragments of the top and the bottom strands of the desired segment. This was done using a combination of Klenow enzyme fill-in reactions and PCR. This duplex, after treatment with PstI, was cloned into the SmaI and PstI sites of plasmid pGEM1 (Promega), resulting in plasmid pGC. The tetracycline resistance gene (Tet) was PCR amplified from plasmid pBR322 (Pharmacia) with the primers TetUS (5′-CCGCAATTGAAATCTAACAATGCGCTC) and TetDS (5′-CAGGTACCTCAGGTCGAGGTGGCCCGGC). The PCR product was cloned into the PstI and MfeI sites of pGC, resulting in plasmid pGCT.
pGCT-RACK vector. RACK1 cDNA was PCR amplified from a human fetal brain cDNA library (Clontech) using the primers RACK-US (5′-TGACCGAGCAAATGACCCTTCGT) and RACK-DS (5′-CCGGTACCTTAGCGTGTACCAATAGTCACC). The PCR product was trimmed with Klenow enzyme and cleaved with KpnI. The vector pGCT was cleaved with NdeI, trimmed with Klenow enzyme, then cleaved with KpnI and ligated with the fragment, resulting in plasmid pGCT-RACK.
Randomization of the translation initiation site
Plasmid pGCT-RACK (10 µg) was cleaved with NdeI and BstXI and purified from the small fragment by means of a High Pure PCR Product Purification Kit (Roche). This vector was ligated with 100 pmol of the randomized oligonucleotide N20 (5′-TGTTANNNNNNNNNNNNNNNNNNNNCCACTGTT) in a 70 µl reaction mixture for 14 h at 4°C. The mixture was purified using a High Pure PCR Product Purification Kit to remove non-ligated oligo and treated with NdeI restriction enzyme to cleave any dimerized vector. The second strand of the randomization fragment was synthesized by fill-in reaction using Klenow enzyme. The mixture was purified with a High Pure PCR Product Purification Kit and intramolecular blunt-end ligation was carried out (16 h at room temperature). The ligation mixture was used for transformation of Escherichia coli XL1-blue strain with 0.2 µg of the starting vector material per 100 µl of competent cells (transformation efficiency for supercoiled plasmid DNA was ∼5 × 106 clones/µg).
All colonies (∼50 000) were pooled by washing plates with LB broth and plasmid DNA prepared from this pool pGCT-RACK-N20 using a Qiagen Tip-500 ion exchange column.
Genetic selection
Competent cells (efficiency ∼104 colonies/µg) of E.coli strain BL21(DE3) containing an IPTG-inducible gene for T7 RNA polymerase were transformed with the pGCT-RACK-N20 plasmid pool. Transformants (∼20 000) were grown on LB-agar [50 µg/ml Ampicillin (Ap)] Petri plates, washed off the plates with LB medium, pelleted, then resuspended in 10 ml of LB medium containing 50% glycerol, aliquoted and frozen.
A 100 µl aliquot from this stock was used to inoculate 10 ml of LB medium (50 µg/ml Ap) and 0.05 mM IPTG was added to induce T7 RNA polymerase synthesis and expression of the recombinant construct. After 3–6 h of shaking at 37°C, the LB volume was increased to 50 ml (50 µg/ml Ap, 0.05 mM IPTG) and Tet was added (5 µg/ml) to start the selection. After overnight incubation the culture was pelleted and plasmid DNA purified. This DNA was separated on a preparative agarose gel alongside plasmid pGCT-RACK and the full-length fraction of the pool was purified from the gel using a Getsorb gel extraction kit (Genomed). This pool was reintroduced into BL21(DE3) cells and separate colonies were taken through the same procedure to select individual clones resistant to 5 µg/ml Tet.
Clone analysis for RACK1 expression
Fresh colonies of transformants from the agarose purified pool were inoculated into separate test tubes containing 2 ml of LB medium (50 µg/ml Ap), incubated with shaking for ∼3 h, 0.5 mM IPTG was added and the tubes were incubated overnight at 37°C. The cells were then pelleted, resuspended in 800 µl of loading buffer, heated for 10 min at 96°C and 10 µl was loaded on a gel (10%).
Immunoblotting
Homogenates were separated by denaturing SDS–PAGE (Bio-Rad), blocked (1 h, 3% non-fat dried milk, 2% w/v BSA, 0.2% Tween-20 in Tris-buffered saline) and blotted. Immunodetection was done with mouse monoclonal antibody against RACK1 (R20620; Transduction Laboratories, Lexington, KY) diluted 1:2500. Secondary antibodies were horseradish peroxidase-conjugated goat anti-mouse (1:10 000) (Jackson Laboratory). Chemiluminescent detection was with an enhanced chemiluminescence kit (Amersham Pharmacia Life Science, Uppsala, Sweden), as instructed.
RESULTS AND DISCUSSION
To fulfill the idea of a genetic selection system based on conjugated translation of the target gene with an antibiotic resistance gene, we created a plasmid vector derived from plasmid pGEM1 (Promega), designated pGCT (Fig. 1A), containing the Tet resistance gene preceded by a linker with the translation initiation site from the MS2 lysis gene (the downstream gene), as well as restriction sites for cloning of the target gene and for introducing a randomized fragment upstream of the target gene. We also introduced a translational enhancer ε (9,10) upstream of the randomization site. As expected, this plasmid conferred no resistance to Tet because of the weakness of the Tet ribosome binding site. At this stage we obtained the basic construct containing all the necessary elements for picking up a target gene and conducting the cloning and selection procedure to create a super-producing clone. We have chosen to clone the RACK1 (11) cDNA as the target gene. This was cloned into the NdeI and KpnI sites of the construct to create a plasmid pGCT-RACK.
To create a positive control for our system we cloned a synthetic duplex containing a 5 nt SD sequence in the place of the ribosome binding site of pGCT-RACK to create pGCT-RACK-SD, with the following translation initiation site: GTGGAT CCA GGAGAT AACATATG (SD sequence in bold, start codon underlined). This site conferred resistance to 1 but not to 5 µg/ml Tet and showed moderate expression of the RACK1 gene (Fig. 2B, lane 1), supporting potential utility of the system.
Figure 2.
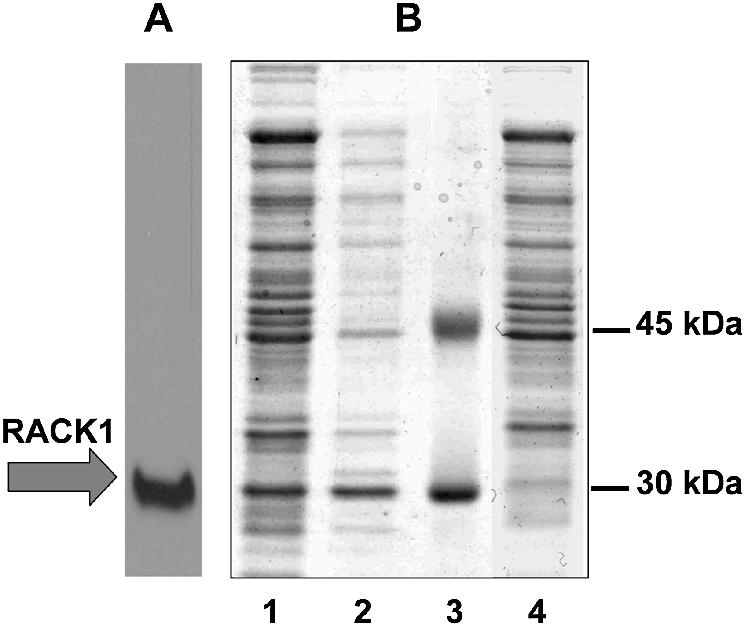
Electrophoretic analysis of homogenates from individual clones retrieved from the genetic selection procedure. (A) Western blot analysis of the homogenate from lane 1 with antibodies against RACK1; a strong signal co-localizes with the expected RACK1 band [the molecular weight of RACK1 is 35.1 kDA, but its electrophoretic mobility corresponds to ∼30 kDa (12)]. (B) Crude homogenates obtained from the non-selected control ribosome binding site pGCT-RACK-SD construct (lane 1), the selected clone (lane 2) and the negative control (empty vehicle) construct (lane 4) induced with 0.5 mM IPTG. Lane 3, molecular weight marker.
In order to introduce a random fragment upstream of the target gene in our system, we developed a new technique based on the use of a restriction enzyme generating 3′-protruding non-palindromic ends, BstXI (Fig. 1B). These ends were to serve as a template for ligation of the randomized oligonucleotide and a primer for building the second strand of the oligonucleotide. A single-stranded oligonucleotide containing a 20 nt random fragment was ligated to the BstXI terminus of the plasmid linearized with NdeI and BstXI. The second strand was filled-in by polymerase. Then the plasmid was recircularized by intramolecular blunt-end ligation. The ligation mixture was used to transform E.coli and a randomized pool of supercoiled plasmids containing ∼50 000 individual sequences was obtained using ampicillin as a selection marker. Restriction analysis of individual clones from this pool (not shown) and sequencing of the pool (Fig. 1C) indicated that ∼70% of the plasmids contained an insert.
This pool did not cover the sequence space of the 20 nt long randomization (420 sequences). However, it represented several hundred sequences satisfying the minimal requirement for the translation initiation site, a 4 nt SD sequence situated 6–9 nt upstream of the initiation codon. Therefore, we considered it wide enough for our selection purposes.
The pool was taken into the selection procedure. To this end it was introduced into a bacterial strain containing an inducible gene for T7 RNA polymerase, in which the target gene cassette could be expressed. First, the colonies were grown on ampicillin without induction of the T7 cassette, colonies were then pooled together and an aliquot of this pool was used to inoculate liquid medium. This was first incubated in the presence of ampicillin and IPTG, the inducer of T7 RNA polymerase synthesis, to allow synthesis of the target gene and tet resistance protein. In order to avoid compromised growth of clones super-producing the target gene, we used the lowest concentration of the inducer (0.05 mM) which could confer further growth on tetracycline. After 3 h, tetracycline (5 µg/ml) was added to start the genetic selection. The Tet-resistant plasmid pool was purified from overnight cultures and analyzed by agarose gel. Most of the pool comprised plasmids which had lost the target gene and expressed the tetracycline resistance gene directly from the upstream translation initiation site. However, a fraction of the pool had the correct length, indicative of both genes being present in the cassette. This fraction was purified from the agarose gel and reintroduced into bacteria. Separate clones from this transformation were incubated first with Ap and IPTG and then with Tet (1 or 5 µg/ml). All 10 clones taken for analysis, but not the negative control, were able to grow on 1 µg/ml Tet, but only three of them grew on 5 µg/ml. When these three clones were analyzed for recombinant RACK1 production (using a standard IPTG concentration of 0.3 mM), they proved to be very efficient producers of RACK1 (Fig. 2B, lane 2), compared to the control construct pGCT-RACK-SD (Fig. 2B, lane 1), with the yield of recombinant protein being >30% of total protein. Western blot analysis of one of the homogenates confirmed the presence of RACK1 in the 30 kDa major band (Fig. 2A).
Sequencing of the selected ribosome binding sites revealed that all three clones were identical, the ribosome binding site being TAA AAA GGA AAC CCT ATG (ATG start codon). The selected sequence was shorter then the expected insert and lacked the NdeI restriction site, but retained the correct start codon. It contained a classical 4 nt SD sequence at 6 nt distance from the start codon and appeared to be AT-rich, except for a stretch of three cytidines close to the start codon. The fact that we found only one sequence in the final pool is most likely not due to the low number of sequences tested, but rather to the property that a selection from a relatively large pool will converge to a single variant. This phenomenon has been frequently observed in in vitro selection studies (13). The reason for such convergence is that in the initial pool, every sequence is represented by a tiny fraction, and a great deal of exponential amplification occurs throughout the selection; therefore, even a very small advantage of one of the sequences in growth (or other selection–amplification process) may result in strong dominance in the final pool.
There was a chance that the improvement in protein yield observed in the selected clone was due to changes which had occurred in other sites of the plasmid during the selection procedure, affecting replication or transcription efficiency. In order to rule out this possibility, we have excised a BstXI–KpnI fragment (including RACK1 with the translation initiation site) from the selected clone and reintroduced this fragment into plasmid pGCT, which had not been through the selection process. The resulting plasmid showed virtually the same efficiency of recombinant protein expression as the previously selected clone (data not shown).
In the case of RACK1, the described system proved instrumental in creating an efficient bacterial producer of this protein in a small-scale genetic selection experiment. As this system does not impose any special requirements on the target gene, we believe that it can be used to optimize expression of other genes as well. The scale of the selection can be readily increased by orders of magnitude, if needed, by using commercially available competent cells with high transformation efficiencies.
Acknowledgments
ACKNOWLEDGEMENT
We are thankful to Prof. Edwin Southern of Oxford University for reading and commenting on the manuscript.
REFERENCES
- 1.Jacques N. and Dreyfus,M. (1990) Translation initiation in Escherichia coli: old and new questions. Mol. Microbiol., 4, 1063–1067. [DOI] [PubMed] [Google Scholar]
- 2.Fuchs E. (1999) The translation initiation signal in E. coli and its control. Genet. Eng. (N Y), 21, 15–35. [DOI] [PubMed] [Google Scholar]
- 3.Buell G., Schulz,M.F., Selzer,G., Chollet,A., Movva,N.R., Semon,D., Escanez,S. and Kawashima,E. (1985) Optimizing the expression in E. coli of a synthetic gene encoding Somatomedin-C (IGF-I). Nucleic Acids Res., 13, 1923–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalboge H., Carlsen,S., Jensen,E.B., Christensen,T. and Dahl,H.H. (1988) Expression of recombinant growth hormone in Escherichia coli: effect of the region between the Shine-Dalgarno sequence and the ATG initiation codon. DNA, 7, 399–405. [DOI] [PubMed] [Google Scholar]
- 5.Bucheler U.S., Werner,D. and Schirmer,R.H. (1992) Generating compatible translation initiation regions for heterologous gene expression in Escherichia coli by exhaustive periShine-Dalgarno mutagenesis. Human glutathione reductase cDNA as a model. Nucleic Acids Res., 20, 3127–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hilvert D. (2000) Genetic selection as a tool in mechanistic enzymology and protein design. Ernst Schering Res. Found. Workshop, 32, 253–268. [DOI] [PubMed] [Google Scholar]
- 7.Andre A., Puca,A., Sansone,F., Brandi,A., Antico,G. and Calogero,R.A. (2000) Reinitiation of protein synthesis in Escherichia coli can be induced by mRNA cis-elements unrelated to canonical translation initiation signals. FEBS Lett., 468, 73–78. [DOI] [PubMed] [Google Scholar]
- 8.Adhin M.R. and van Duin,J. (1990) Scanning model for translational reinitiation in Eubacteria. J. Mol. Biol., 213, 811–818. [DOI] [PubMed] [Google Scholar]
- 9.Lehmeier B. and Amann,E. (1992) Tac promoter vectors incorporating the bacteriophage T7 gene 10 translational enhancer sequence for improved expression of cloned genes in Escherichia coli. J. Biotechnol., 23, 153–165. [DOI] [PubMed] [Google Scholar]
- 10.O’Connor M. and Dahlberg,A.E. (2001) Enhancement of translation by the epsilon element is independent of the sequence of the 460 region of 16S rRNA. Nucleic Acids Res., 29, 1420–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ron D., Chen,C.H., Caldwell,J., Jamieson,L., Orr,E. and Mochly-Rosen,D. (1994) Cloning of an intracellular receptor for protein kinase C: a homolog of the β subunit of G proteins. Proc. Natl Acad. Sci. USA, 91, 839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Birikh K.R., Sklan,E.H., Shoham,S. and Soreq,H. (2003) Interaction of “readthrough” acetylcholinesterase with RACK1 and PKSβII correlates with intensified fear-induced conflict behavior. Proc. Natl Acad. Sci. USA, 100, 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lozupone C., Changayil,S., Majerfeld,I. and Yarus,M. (2003) Selection of the simplest RNA that binds isoleucine. RNA, 9, 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]