

Abstract
A uniform extracellular stimulus triggers cell-specific patterns of Ca2+ signals, even in genetically identical cell populations. However, the underlying mechanism that generates the cell-to-cell variability remains unknown. We monitored cytosolic inositol 1,4,5-trisphosphate (IP3) concentration changes using a fluorescent IP3 sensor in single HeLa cells showing different patterns of histamine-induced Ca2+ oscillations in terms of the time constant of Ca2+ spike amplitude decay and the Ca2+ oscillation frequency. HeLa cells stimulated with histamine exhibited a considerable variation in the temporal pattern of Ca2+ signals and we found that there were cell-specific IP3 dynamics depending on the patterns of Ca2+ signals. RT-PCR and western blot analyses showed that phospholipase C (PLC)-β1, -β3, -β4, -γ1, -δ3 and -ε were expressed at relatively high levels in HeLa cells. Small interfering RNA-mediated silencing of PLC isozymes revealed that PLC-β1 and PLC-β4 were specifically involved in the histamine-induced IP3 increases in HeLa cells. Modulation of IP3 dynamics by knockdown or overexpression of the isozymes PLC-β1 and PLC-β4 resulted in specific changes in the characteristics of Ca2+ oscillations, such as the time constant of the temporal changes in the Ca2+ spike amplitude and the Ca2+ oscillation frequency, within the range of the cell-to-cell variability found in wild-type cell populations. These findings indicate that the heterogeneity in the process of IP3 production, rather than IP3-induced Ca2+ release, can cause cell-to-cell variability in the patterns of Ca2+ signals and that PLC-β1 and PLC-β4 contribute to generate cell-specific Ca2+ signals evoked by G protein-coupled receptor stimulation.
Introduction
Many extracellular stimuli trigger increases in the cytosolic concentration of Ca2+ ([Ca2+]) that control a wide range of physiological processes, including fertilization, proliferation, development, learning and memory, contraction, and secretion [1], [2]. In a multitude of cell types, released Ca2+ causes oscillatory changes in [Ca2+], and the frequency of Ca2+ oscillations is correlated with the stimulus intensity [3]–[5], while the time course of an individual Ca2+ spike depends on the type of receptor stimulated [6], [7]. The versatility of Ca2+ signals is considered to be dependent on the spatiotemporal patterns of intracellular Ca2+ signals [8].
Extracellular stimuli, such as growth factors, hormones, and neurotransmitters, activate an intracellular effector molecule, phosphoinositide-specific PLC, that catalyzes the conversion of phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol 1,4,5-trisphosphate (IP3) and diacylglycerol [9]. IP3 acts as a second messenger in many cell types, where its primary effect is to trigger the release of Ca2+ ions from intracellular stores and generate cytosolic Ca2+ signals. The forms of the Ca2+ signals vary from cell to cell, even in clonal populations of genetically identical cells, while individual cells show characteristic and reproducible Ca2+ responses in terms of amplitude, frequency, and shape of the transient as well as the latency of onset of the initial Ca2+ rise [5]. Therefore, Ca2+ signals have been referred to as cell “fingerprints”.
Real-time monitoring of cytosolic IP3 in living cells is pivotal for understanding the mechanism that underlies the generation of cell-specific patterns of Ca2+ signals. In a previous study, we monitored the cytosolic IP3 concentration ([IP3]) changes during agonist-evoked Ca2+ oscillations in HeLa cells using a genetically-encoded fluorescent IP3 sensor IP3R-based IP3 sensor 1 (IRIS-1) [10]. The observed IP3 dynamics were unexpectedly complex because [IP3] did not return to its basal level during the intervals between Ca2+ spikes, and IP3 gradually accumulated in the cytosol with little or no fluctuation during cytosolic Ca2+ oscillations [10]. Manipulation of the cytosolic Ca2+ increases during agonist application indicated the presence of both Ca2+-dependent and Ca2+-independent components for IP3 generation in this cell type [10]. The molecular natures of these components and, more fundamentally, the roles of IP3 dynamics in the generation of the cell-specific patterns of Ca2+ signals remain elusive.
There are six families of mammalian PLC enzymes (PLC-β, -γ, -δ, -ε, -ζ, and -η) consisting of 13 isoforms in humans [11], [12]. PLC-β is activated by the Gα and Gβγ subunits of heterotrimeric G proteins downstream of G protein-coupled receptors (GPCRs). PLC-γ is activated through receptor and non-receptor tyrosine kinases. PLC-δ is activated by cytosolic Ca2+ and the high-molecular-weight G protein Gh. PLC-ε is regulated directly by small GTPases from the Ras family, and its enzymatic activity can be stimulated by subunits of heterotrimeric G proteins and small GTPases from the Rho family. PLC-ζ was identified as a sperm-specific PLC, and shows extremely high Ca2+ sensitivity for its enzymatic activity compared with the other PLC isoforms. PLC-η is suggested to be activated through GPCR stimulation. Although various tissues express multiple PLC isozymes, the effects of the individual PLC isozymes on the generation of Ca2+ signals are poorly understood.
In this study, we measured the IP3 dynamics using IRIS-1 in cells showing different patterns of Ca2+ signals within a clonal population of genetically identical HeLa cells stimulated with histamine. The IP3 dynamics were characteristically different depending on the patterns of cellular Ca2+ signals, suggesting that the heterogeneity in IP3 production contributes to the cell-to-cell variability observed in Ca2+ signals. We combined an RNAi technique with the real-time IP3 imaging technique to identify the PLC isozymes involved in the generation of IP3 production in this cell type, and found that at least two isozymes in the PLC-β family, PLC-β1 and PLC-β4, are involved in histamine-induced IP3 increases in HeLa cells. RNAi-mediated silencing and overexpression experiments for PLC-β1 and PLC-β4 revealed that these isozymes differentially contribute to the determination of the characteristic parameters of Ca2+ signals, i.e. the Ca2+ oscillation frequency and the time constant of Ca2+ spike amplitude decay. These findings provide important clues toward understanding the molecular mechanism that underlies the generation of cell-specific patterns of Ca2+ signals in genetically identical cell populations.
Results
Reproducible Cell-specific Patterns of [Ca2+] and [IP3] Changes in HeLa Cells Stimulated with 3 µM Histamine
To examine whether the cell-specific patterns of Ca2+ oscillations were accompanied by characteristic IP3 dynamics in individual cells, we performed simultaneous monitoring of [Ca2+] and [IP3] at the single-cell level. Indo-5F was loaded into IRIS-1-expressing HeLa cells and the cells were stimulated twice with 3 µM histamine with an interval of approximately 20 min. Consistent with previous studies [5], [13]–[15], there was marked intercellular heterogeneity in the Ca2+ dynamics, which varied in the pattern (sustained oscillations or damped oscillations) and the frequency of Ca2+ oscillations (for more details, see below), even within the same microscopic field of view (Fig. 1A). These cell-specific Ca2+ patterns were reproducible upon repetitive histamine additions (Fig. 1B), as observed in different cell types [5], [13]–[15], suggesting that individual HeLa cells have characteristic Ca2+ dynamics. As shown in Fig. 1B, the patterns of IP3 dynamics also differed between cells. In cells showing sustained Ca2+ oscillations (e.g. cell 1 in Fig. 1B), IP3 was maintained at relatively low levels during the period of stimulation, and fluctuations in [IP3] that synchronized with Ca2+ spikes were observed with almost constant amplitudes. In cells showing damped Ca2+ oscillations (e.g. cell 2 in Fig. 1B), the initial increase in [IP3] was relatively high and the IP3 level gradually increased during the period of stimulation. [IP3] fluctuations that synchronized with Ca2+ spikes were also observed in cells showing damped Ca2+ oscillations, but the amplitude of these [IP3] fluctuations gradually decreased depending on the amplitude of the Ca2+ spikes. In both cell types, the IP3 dynamics were reproducible (Fig. 1B). These findings indicate that the IP3 dynamics are characteristic of individual cells, and that the IP3 dynamics may be involved in the determination of cell-specific Ca2+ responses.
Figure 1. Reproducible cell-specific patterns in Ca2+ and IP3 responses in HeLa cells stimulated with histamine.
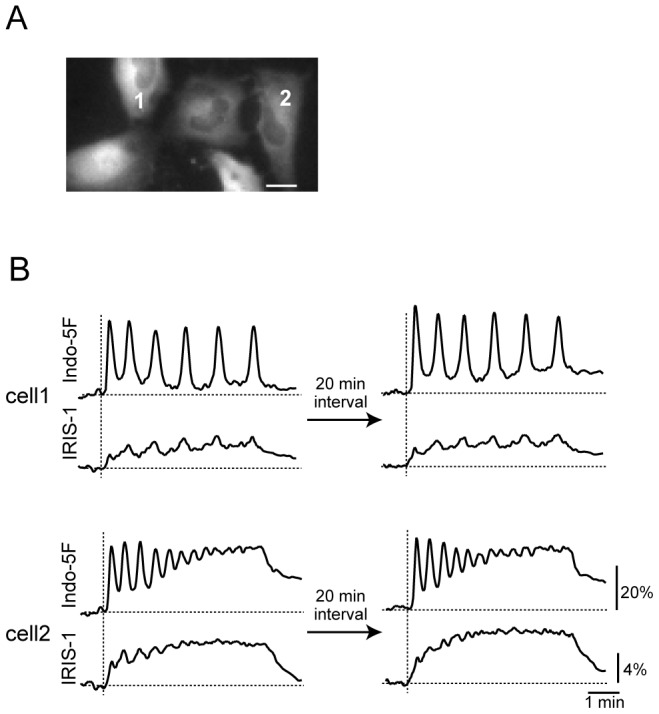
(A) Venus fluorescence image of IRIS-1-expressing HeLa cells before histamine stimulation. Bar, 20 µm. (B) Changes in 460–510 nm emission of Indo-5F signals (F/F0, where F0 is the basal level of F; top) and ECFP/Venus emission ratio of IRIS-1 signals (ΔR/R0; ΔR was defined as R – R0, where R0 is the basal level of R; bottom) after repeated additions of 3 µM histamine with a 20-min interval in two different cells within the same field of view shown in (A). The horizontal broken lines indicate the baseline levels of the IRIS-1 and Indo-5F signals. The vertical broken lines indicate the onsets of stimulation.
[Ca2+] and [IP3] Dynamics Observed in Cells Stimulated with Different Concentrations of Histamine
As shown in Fig. 1, there were two prominent types of cells depending on their temporal patterns of [Ca2+] changes after stimulation with 3 µM histamine. To investigate whether these differences arose from heterogeneity in the histamine sensitivity of the cells, as proposed by Rooney et al. [15], or from other components in the signal transduction pathway, we measured the [Ca2+] and [IP3] changes in the same cells stimulated with three different concentrations of histamine. Representative results are shown in Fig. 2A (cells showing sustained Ca2+ oscillations at 3 µM histamine) and Fig. 2B (cells showing damped Ca2+ oscillations at 3 µM histamine). To analyze the results quantitatively, we measured the time constants for the progressive decrease in the amplitude of Ca2+ spikes (Fig. S1A). Sustained oscillations had larger time constants of Ca2+ spike amplitude decay, while damped oscillations had smaller time constants of Ca2+ spike amplitude decay. Fig. 2C shows the relationships between the time constants and the histamine concentrations applied. There were large cell-to-cell varieties for all three histamine concentrations examined, and the inverse time constants varied from less than 0.001 s−1 (stable sustained oscillations) to 0.3 s−1 (rapidly damped oscillations). There was no obvious dependence of the time constant on the histamine concentration, and individual cells showed similar time constants irrespective of the histamine concentration (Fig. 2C). These findings suggest that a difference in histamine sensitivity is not the primary mechanism for the generation of different patterns of Ca2+ signals and that the time constants are characteristic of individual cells. Because there were two peaks in the histogram of the time constants observed in cells stimulated with 3 µM histamine (Fig. S1B), we classified the cells into two groups, namely S-cells (cells showing sustained oscillations: red in Fig. 2C) and D-cells (cells showing damped oscillations: blue in Fig. 2C), depending on the time constants of Ca2+ amplitude decay observed at 3 µM histamine with a threshold of 0.0056 s−1.
Figure 2. Classification of cells depending on the time constants of peak amplitude decay of Ca2+ spikes.

Representative traces of Indo-5F signal changes (F/F0; top) and IRIS-1 signal changes (ΔR/R0; bottom) in cells showing sustained Ca2+ oscillations (S-cell) (A) and damped oscillations (D-cell) (B). Three different concentrations of histamine (1, 3, and 10 µM) were sequentially applied to the same cells with an interval of 20 min. (C) Relationships between the histamine concentrations and the inverse time constants of Ca2+ amplitude decay in S-cells (red) and D-cells (blue). (D) Relationships between the histamine concentrations and the Ca2+ oscillation frequencies observed in S-cells (red) and D-cells (blue). (E–G) Comparisons of the Ca2+ oscillation frequencies between S-cells (red) and D-cells (blue). The histamine concentrations were 1 µM (E), 3 µM (F), and 10 µM (G). (H–J) Comparisons of integrated IRIS-1 signals between S-cells (red) and D-cells (blue). The histamine concentrations were 1 µM (H), 3 µM (I), and 10 µM (J).
Fig. 2D shows the relationships between the histamine concentrations and the Ca2+ oscillation frequencies in S-cells (red) and D-cells (blue). The frequency increased depending on the concentration of histamine, with slopes of 2.2±1.1 mHz/µM histamine (mean ± SD, n = 14) for S-cells and 1.2±1.0 mHz/µM histamine (mean ± SD, n = 23) for D-cells. There were large cell-to-cell variabilities in the Ca2+ oscillation frequencies, but the frequencies in S-cells tended to be smaller than those in D-cells (Fig. 2D–G). We detected significant differences (P<0.05, Student's t-test) in the Ca2+ oscillation frequencies between S-cells and D-cells following stimulation with 1 µM histamine (mean ± SD: 20.5±10.3 mHz, n = 11, for S-cells vs. 31.1±10.6 mHz, n = 11, for D-cells) and 3 µM histamine (mean ± SD: 25.0±6.9 mHz, n = 13, for S-cells vs. 32.5±11.2 mHz, n = 23, for D-cells) (Fig. 2E and F). These findings indicate the presence of a correlation between the interval of Ca2+ spikes and the time dependence of the change in the amplitude of Ca2+ spikes.
Representative traces showed that the dependence of the Ca2+ spike amplitude on the histamine concentration differed between S-cells and D-cells (Fig. 2A and B). We quantified the peak amplitudes of the first Ca2+ spike following stimulation with three different concentrations of histamine (Fig. S2A and B). The amplitude of the first spike of S-cells remained almost constant irrespective of the histamine concentration applied, while that of D-cells was dependent on the histamine concentration (Fig. S2B). These results suggest that the histamine concentration-dependence of the amplitude of the first Ca2+ spikes is tightly coupled to the rate of changes in the amplitude of the following Ca2+ spikes in HeLa cells.
Fig. 2H–J shows histograms of the integrals of the IP3 signals during stimulation with the three different concentrations of histamine. As shown in Fig. S2C, the mean values of the integrated IP3 signals were dependent on the histamine concentration in both S-cells and D-cells. The IP3 productions in S-cells were significantly smaller than those in D-cells under all three conditions, suggesting that the amount of IP3 production and/or IP3 dynamics should be involved in the formation of the cell-specific patterns of [Ca2+] changes. To understand the relationship between IP3 dynamics and Ca2+ dynamics more precisely, we investigated the changes in Ca2+ dynamics by modifying IP3 production in the following analyses.
PLC Isozymes Expressed in HeLa Cells and their Specific Knockdown Using Small Interfering RNAs (siRNAs)
In a previous study [10], we found that there are multiple components of IP3 production in HeLa cells stimulated with histamine (see below). To identify the PLC isozymes involved in the generation of IP3 dynamics in histamine-stimulated HeLa cells, total RNA was isolated from HeLa cells and subjected to RT-PCR analyses with specific primer pairs (Table 1). We found that PLC-β1, -β3, -β4, -γ1, -δ3 and -ε were expressed at relatively high levels among the 13 PLC genes (Fig. 3A). All of the isozymes were successfully detected by western blotting with isozyme-specific antibodies (Fig. 3B). In the western blot analyses, two spliced variants of PLC-β1 and PLC-β4 were detected (Fig. 3B). Next, we examined the effects of specific knockdown of the PLC isozymes using siRNAs. As shown in Fig. 3C, the expressions of the endogenous PLC isozymes, except for PLC-ε, were markedly reduced by two different siRNAs for each isozyme. We confirmed that the PLC isozyme-specific knockdown did not affect the expression levels of the other PLC isozymes in HeLa cells (Fig. S3).
Table 1. Specific primer sequences for RT-PCR analyses.
| Target gene | RT-PCR primer sequencesa (5′ to 3′) | Product size (bp) |
| PLC-β1 | F TGTCTCAGCCCCTTTCTCAC | 111 |
| R ACAGGAGCACTTGGCGATAC | ||
| PLC-β2 | F GAGGACTTCCCAGAACCTGTC | 116 |
| R TCCTTCGTCATGTAGGGTTTG | ||
| PLC-β3 | F AAGTCCTTCGACCCCTTCAC | 110 |
| R TAGATGCCCACCTTCCTGTC | ||
| PLC-β4 | F CCGAACTCGCATGGTTATG | 114 |
| R TCATACACAGCTATTCTCAAGACAG | ||
| PLC-γ1 | F CGACTGGATTCCTCCAACTAC | 108 |
| R GGCCTGGTTCATCTGCATAG | ||
| PLC-γ2 | F TCTGCGCTTTGTGGTTTATG | 109 |
| R CAGAGGAACGGACCTGAATC | ||
| PLC-δ1 | F CCAGCAGCACTGAAGCCTAC | 114 |
| R TGAAAGTATAGCCGTGGTAGATG | ||
| PLC-δ3 | F GGCCTATGTTAGGGCCTTTG | 110 |
| R TCTTGGAGGTGAGGGTATGG | ||
| PLC-δ4 | F CTGTCGTTTACCACGGACAC | 117 |
| R TCTCCAGGGACAAGATGACTG | ||
| PLC-ε | F GAAAACACCAGCGATCTTCAG | 107 |
| R TGCCTCTGGTTGTCCGATAG | ||
| PLC-ζ | F TCGGTGCAGTTATATTCATGTG | 117 |
| R TCTTCTCTGTGCGTGATAATTC | ||
| PLC-η1 | F GGAAGGGGAAGTTTCTGATG | 112 |
| R CCTTATGAAAGATTCCACCTGATG | ||
| PLC-η2 | F GGACATCATCGAGCAGTTTG | 119 |
| R GGTGCTCAGGGTTGAAGATG | ||
| β-actin | F TCGTGCGTGACATTAAGGAG | 110 |
| R GTCAGGCAGCTCGTAGCTCT |
F and R denote foreword and reverse primers, respectively.
Figure 3. Identification of PLC isozymes expressed in HeLa cells and their specific knockdown using siRNAs.
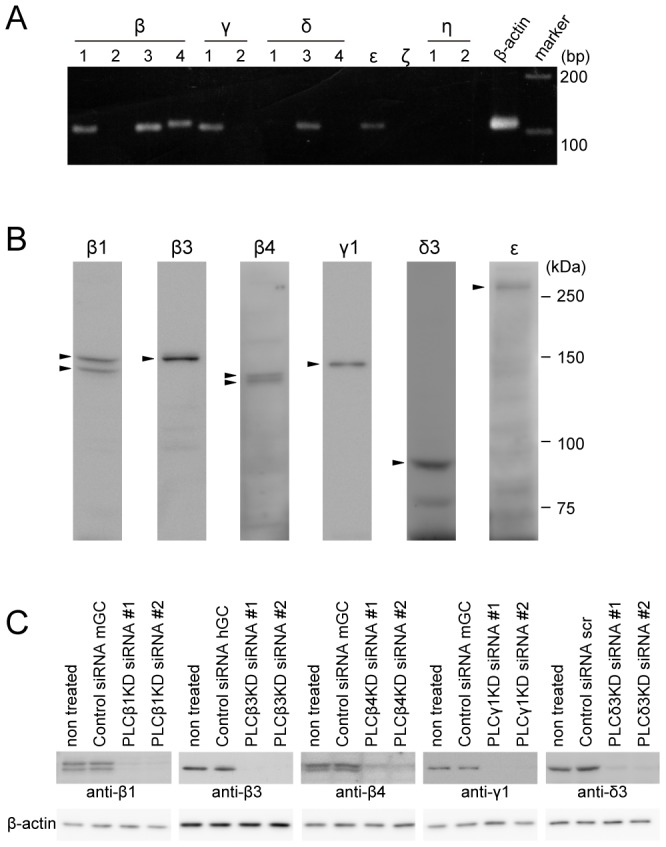
(A) Results of RT-PCR analyses using total RNA isolated from HeLa cells. (B) Western blotting analyses of endogenous PLC isozymes expressed in HeLa cells using isozyme-specific antibodies. Molecular mass markers are shown on the right. (C) Western blotting analyses of total lysates prepared from HeLa cells treated with PLC isozyme-specific siRNAs. The antibodies used are shown at the bottom. The results are representative of at least three independent experiments.
Identification of PLC Isozymes Involved In Histamine-induced [IP3] Increases in HeLa Cells
There are multiple components in the IP3 increases observed in HeLa cells [10]: (1) a fast IP3 increase in response to histamine receptor stimulation without cytosolic Ca2+ increase (Fig. 4A, left); (2) a slow IP3 increase in response to cytosolic Ca2+ increase alone (Fig. 4A, middle); and (3) a complex [IP3] change composed with a fast transient (1st component) and a slow gradual increase (2nd component) in response to the combination of histamine receptor stimulation and cytosolic Ca2+ increase (Fig. 4A, right). The presence of cytosolic Ca2+ enhanced the rate of the histamine-induced IP3 increase and also elicited a delayed negative feedback (Fig. 4B). We attempted to identify the PLC isozymes contributing to each component by measuring the IP3 dynamics in PLC isozyme-specific knockdown cells. To exclude the effects of Ca2+ released through IP3 receptor stimulation on the [IP3] changes, the cells were pretreated with thapsigargin to deplete intracellular Ca2+ stores, and cytosolic [Ca2+] was increased using the capacitive Ca2+ entry pathway activated by Ca2+ store depletion, using previously described methods [10]. We used the maximal IRIS-1 signal change or the increasing rate of the IRIS-1 signal as an index of PLC activity in the following experiments because these values were not affected by the IRIS-1 expression level, the resting level of [IP3], or the increasing rate of [Ca2+] or total amount of [Ca2+] increase mediated by capacitive Ca2+ entry (Fig. S4).
Figure 4. Identification of PLC isozymes involved in IP3 generation in HeLa cells stimulated with histamine.

(A) Representative traces of IRIS-1 signal changes (ΔR/R0; top) and Indo-5F signal changes (F/F0; bottom) observed in thapsigargin-treated HeLa cells. The horizontal broken lines indicate the baseline levels. (B) Traces of the mean ± SD IRIS-1 signal changes (ΔR/R0) observed in thapsigargin-treated HeLa cells after addition of 3 µM histamine (blue circles) or 3 µM histamine plus 2 mM Ca2+ (green circles). (C–F) Effects of PLC isozyme knockdown on the IP3 increase evoked by 3 µM histamine alone (C), the IP3 increase evoked by 2 mM Ca2+ alone (D), the first component of the IP3 increase evoked by 3 µM histamine plus 2 mM Ca2+, and the second component of the IP3 increase evoked by 3 µM histamine plus 2 mM Ca2+. Data are shown as means ± SD. The numbers of cells measured are shown in parentheses. Statistical analyses were performed by one-way ANOVA followed by Scheffe’s multiple comparison test. *P<0.05, **P<0.01, vs. the values in control siRNA-transfected cells.
Figure 4C shows the results of PLC isozyme knockdown on the [IP3] increase in response to histamine receptor stimulation alone. Among the five isozymes examined, only PLC-β4 knockdown caused significant impairments of the [IP3] increase irrespective of the siRNA used. The maximal IRIS-1 change was significantly decreased by 46% (siRNA#1) and 71% (siRNA#2) compared with the mean change with the control siRNAs mGC and hGC (Fig. 4C). These findings indicate that PLC-β4 is selectively activated downstream of histamine receptors in the absence of [Ca2+] elevation in HeLa cells.
Figure 4D shows the results of PLC isozyme knockdown on the [IP3] increase in response to cytosolic Ca2+ increase alone, which was elicited by the application of extracellular 2 mM CaCl2 whithout histamine. In thapsigargin-treated HeLa cells, store-operated Ca2+ influx channels on the plasma membrane are activated by the depletion of intracellular Ca2+ stores and the addition of extracellular Ca2+ immediately elicites [Ca2+] increases through the Ca2+ influx channels [10]. Because knockdown of any single PLC isozyme alone did not significantly decrease the amount of the IP3 increase (Fig. 4D), multiple PLC isozymes might be activated in response to [Ca2+] elevation. A significant increase in [IP3] was observed in cells treated with PLCβ4KD siRNA#2 (Fig. 4D), even though the treatment with this siRNA did not change the expression levels of other PLC isozymes (Fig. S3). Because the other siRNA designed for PLC-β4, PLCβ4KD siRNA#1, did not increase [IP3] significantly (Fig. 4D), the increase in [IP3] may be caused by some side-effects of PLCβ4KD siRNA#2. PLC-δ3 knockdown cells using two different siRNAs showed significant increases in [IP3] evoked by Ca2+ increase (Fig. 4D). The treatments with these two siRNAs did not change the expression level of other PLC isozymes (Fig. S3). PLC-δ3 may possess some inhibitory effects on the Ca2+-dependent IP3 production in HeLa cells.
Figure 4E (1st component) and 4F (2nd component) show the results of PLC isozyme knockdown on the [IP3] increases in response to the combination of histamine receptor stimulation and Ca2+ increase. Both the 1st and 2nd components of the [IP3] increase after addition of 3 µM histamine plus 2 mM Ca2+ were impaired by knockdown of PLC-β1 or PLC-β4. The increasing rate of the IRIS-1 signal of the 1st component was significantly reduced by 48% (PLCβ1KD siRNA#1), 38% (PLCβ1KD siRNA#2), 47% (PLCβ4KD siRNA#1), and 51% (PLCβ4KD siRNA#2) compared with the mean value for the control siRNAs mGC and hGC (Fig. 4E). The maximal IRIS-1 signal change of the 2nd component was significantly reduced by 33% (PLCβ1KD siRNA#1), 40% (PLCβ1KD siRNA#2), 32% (PLCβ4KD siRNA#1), and 34% (PLCβ4KD siRNA#2) compared with the mean value for the control siRNAs mGC and hGC (Fig. 4F). These findings indicate that both PLC-β1 and PLC-β4 are activated by histamine stimulation accompanied by [Ca2+] elevation and contribute to the generation of the 1st and 2nd components. Delayed inhibition of IP3 production after the 1st component of the [IP3] increase was also detected in PLC-β1 and PLC-β4 knockdown cells (Fig. S5), suggesting that this inhibitory effect was mediated by components upstream of PLC (GPCR and/or G proteins) or by both PLC-β1 and PLC-β4 to the same degree.
Roles of PLC-β1 and PLC-β4 in the Generation of Ca2+ Oscillations
We examined how PLC-β1 and PLC-β4 contribute to the generation of Ca2+ dynamics by measuring [Ca2+] in PLC-β1 and PLC-β4 knockdown cells stimulated with 3 µM histamine. We found that specific knockdown of PLC-β1 resulted in alterations of both Ca2+ dynamics and IP3 dynamics (Fig. 5A). One of the obvious differences was that sustained Ca2+ oscillations were hardly detected in PLC-β1 knockdown cells (Fig. 5B). The mean inverse time constants of Ca2+ spike amplitude decay (0.0290±0.0263 s−1 for siRNA#1 and 0.0247±0.0114 s−1 for siRNA#2) were larger than that in control cells (0.0146±0.0093 s−1, n = 45) and the fraction of cells showing damped oscillations was increased (Fig. 5B). In contrast, the mean Ca2+ oscillation frequencies in PLC-β1 knockdown cells were similar to that in control cells (37.1±8.6 mHz) (Fig. 5C). The integrals of the IP3 signals were significantly reduced in PLC-β1 knockdown cells (Fig. 5D).
Figure 5. Effects of PLC-β1 or PLC-β4 knockdown on Ca2+ and IP3 dynamics evoked by histamine stimulation.

(A) Representative traces of Indo-5F signal changes (F/F0; top) and IRIS-1 signal changes (ΔR/R0; bottom) in siRNA-treated cells stimulated with 3 µM histamine. The siRNAs used are shown on the left. The horizontal broken lines indicate the baseline levels of the IRIS-1 and Indo-5F signals. The vertical broken lines indicate the onsets of stimulation. Stacked histograms of the inverse time constants for exponential decay of the Ca2+ oscillation amplitude (B), Ca2+ oscillation frequencies (C), and integrated IP3 signals (D) of cells treated with control siRNA (top row), PLCβ1KD siRNAs (second row), and PLCβ4KD siRNAs (third row). The results for the two siRNAs are shown in different colors. The means ± SD are shown at the bottom. The numbers of cells measured are shown in parentheses. Statistical analyses were performed by one-way ANOVA followed by Scheffe’s multiple comparison test. **P<0.01, vs. the values in control siRNA mGC-transfected cells.
When the expression of PLC-β4 was selectively reduced, different phenotypes were observed (Fig. 5A). In PLC-β4 knockdown cells, sustained Ca2+ oscillations with small inverse time constants (less than 10−3 s−1) were detected in some cells, but the overall mean inverse time constants did not differ significantly from that in control cells (Fig. 5A and B). The mean frequencies of Ca2+ oscillations in PLC-β4 knockdown cells (21.1±7.2 mHz for siRNA#1 and 17.0±10.5 mHz for siRNA#2) were significantly smaller than that in control cells (37.1±8.6 mHz) (Fig. 5C). The integrals of the IP3 signals were also reduced (Fig. 5D), similar to the case for PLC-β1 knockdown cells. These findings indicate that knockdown of PLC-β1 increases the time constant of Ca2+ spike amplitude decay, while knockdown of PLC-β4 decreases the Ca2+ oscillation frequency in HeLa cells stimulated with histamine. It is important to note that the integrals of the IP3 signals were similarly reduced in both PLC-β1 and PLC-β4 knockdown cells (Fig. 5D), while the temporal patterns of IP3 dynamics were characteristically different between these cells (Fig. 5A). In PLC-β1 knockdown cells, fluctuation of the IP3 signals was hardly detected during Ca2+ oscillations, while fluctuations of [IP3] synchronized with Ca2+ spikes were obviously detected in PLC-β4 knockdown cells (Fig. 5A). These findings suggest that the temporal pattern of IP3 dynamics, but not the absolute IP3 concentration, is important for determining the pattern of [Ca2+] changes, such as sustained oscillations and damped oscillations.
To confirm the effects of PLC-β1 or PLC-β4 knockdown on the generation of Ca2+ dynamics, we examined cells transfected with PLCβ1- or PLCβ4-IRES-mRFP constructs (Fig. 6A) and subjected mRFP-positive cells to single-cell imaging of [IP3] and [Ca2+]. Overexpression of PLC-β1 or PLC-β4 led to different patterns of Ca2+ dynamics (Fig. 6B). PLC-β1-overexpressing cells showed sustained Ca2+ oscillations more frequently than control cells transfected with the IRES-mRFP construct (Fig. 6B), and the mean inverse time constant of Ca2+ spike amplitude decay (0.00349±0.00456 s−1, n = 26) was significantly smaller than that in control cells (0.0137±0.0100 s−1, n = 19) (Fig. 6C). In contrast, PLC-β4-overexpressing cells showed Ca2+ oscillations with higher frequency (mean ± SD, 56.8±11.6 mHz) than that in control cells (mean ± SD, 42.5±4.5 mHz) (Fig. 6D). These findings are consistent with those in the RNAi-mediated silencing experiments when we make two assumptions as follows. First, histamine-induced IP3 production is mainly operated by PLC-β1 and PLC-β4 in HeLa cells. Therefore, PLC-β4 alone works in PLC-β1 knockdown cells and PLC-β1 alone works in PLC-β4 knockdown cells. No compensation by the other PLC isozymes exists in knockdown cells, as supported by the data shown in Fig. S3. Second, overexpressed PLC-β isozyme has a dominant activity, even though the other type of endogenous PLC-β is expressed in the transfected cells. According to these assumptions, we can conclude that PLC-β1 activity is involved in determination of the time-dependence of Ca2+ spike amplitude decay, while PLC-β4 activity is involved in determination of the Ca2+ oscillation frequency in HeLa cells. As previously described in some reports [16], [17], the overexpression of PLC-βs alone without G proteins did not cause a marked increase in the IP3 production (Fig. 6E). Instead, the integral of the IP3 signals was significantly decreased in PLC-β4-overexpressing cells to 65% of the value in control cells suggesting that an excess amount of PLC-β4 would function inhibitory for the IP3 production. The exact mechanism of this phenomenon is not clear, but one possibility is that GTP hydrolysis by Gαq would be promoted due to an excess amount of PLC-β4 because PLC-βs are GTPase activating proteins for Gαq [12]. When we compared cells showing almost the same integral amounts of IP3 production, a phenotypic difference in the patterns of [Ca2+] changes was still observed between PLC-β1-overexpressing cells and PLC-β4-overexpressing cells (Fig. 6B). Taken together, these findings provide further support for the notions that a constant threshold of [IP3] for driving Ca2+ spikes does not exist, and that a temporal pattern of IP3 dynamics is critical for the determination of Ca2+ signaling patterns.
Figure 6. Effects of PLC-β1 or PLC-β4 overexpression on Ca2+ and IP3 dynamics evoked by histamine stimulation.

(A) Western blotting analyses of cell lysates prepared from HeLa cells transfected with PLC-β1-IRES-mRFP (lane 2) or PLC-β4-IRES-mRFP (lane 4). Non-transfected cells were used as controls (lanes 1 and 3). (B) Representative traces of Indo-5F signal changes (F/F0; top) and IRIS-1 signal changes (ΔR/R0; bottom) in transfected cells stimulated with 3 µM histamine. The plasmid DNAs used to transfect the cells are shown on the left. The horizontal broken lines indicate the baseline levels of IRIS-1 and Indo-5F signals. The vertical broken lines indicate the onsets of stimulation. (B–D) Histograms for the inverse time constants for exponential decay of the Ca2+ oscillation amplitude (B), Ca2+ oscillation frequencies (C), and integrated IP3 signals (D) in cells expressing mRFP (top row), PLC-β1 and mRFP (second row), and PLC-β4 and mRFP (third row). The means ± SD are shown at the bottom. The numbers of cells measured are shown in parentheses. Statistical analyses were performed by one-way ANOVA followed by Scheffe’s multiple comparison test. **P<0.01, vs. the values in IRES-mRFP transfected cells.
Discussion
During the past decade, it has become increasingly evident that genetically identical cells can exhibit variability in their cellular responses, even in identical environments. Underlying much of this variability is the stochasticity in gene expression that can produce unique proteomes in individual cells, and this cell population heterogeneity is an important factor in processes ranging from stem cell differentiation to chemotherapy resistance [18], [19]. In the present study, we found that individual HeLa cells exhibited characteristic and highly reproducible patterns of Ca2+ changes in response to bath applications of histamine, even in the same microscopic field of view, as reported in other cell lines [5], [13]–[15]. We quantified the patterns of Ca2+ signals by the Ca2+ oscillation frequencies and the time constants of Ca2+ spike amplitude decay, and found that a difference in histamine sensitivity could not account for the cell-to-cell variability in Ca2+ signals. A remarkable finding in this study is that the IP3 dynamics are characteristically different depending on the patterns of Ca2+ signals (Fig. 1), suggesting that the cell-to-cell variety in Ca2+ signals originates from the upstream of the IP3 generation process in the signal transduction cascade in this cell type.
Identification of PLC Isozymes Activated in HeLa Cells Stimulated with Histamine
HeLa cells, an epithelial cell line derived from a carcinoma of the human uterine cervix, express the histamine H1 receptor [20]. Ten Gα proteins among the 16 mammalian Gα proteins and four Gβ proteins among five Gβ proteins have been detected in HeLa cells [21]. RT-PCR analyses positively identified transcripts for all 12 Gγ genes in HeLa cells [21]. In this study, we found that, among the 13 PLC isozyme genes, six (PLC-β1, -β3, -β4, -γ1, -δ3, and -ε) are expressed in HeLa cells. RNAi-mediated silencing of the expressions of the PLC genes showed that at least two isozymes in the PLC-β family, PLC-β1 and PLC-β4, are actually involved in the generation of IP3 production evoked by histamine stimulation. Among the three PLC-β isozymes identified in HeLa cells, specific knockdown of PLC-β3 did not induce a significant decrease in the histamine-induced IP3 production. Recently, Adjobo-Hermans et al. showed that PLC-β1 and PLC-β4 are enriched at the plasma membrane [22], while PLC-β2 and PLC-β3 are mainly localized in the cytosol in resting HeLa cells following expression of GFP-fused PLC-β constructs. Our findings suggest that the intracellular localization of PLC-β isozymes is a critical factor for the selection of PLC-β isozymes activated downstream of the G protein-coupled histamine H1 receptor.
A Possible Mechanism that Underlies the Generation of Cell-specific Patterns of Histamine-induced Ca2+ Signals in HeLa Cells
The RNAi-mediated silencing experiments and PLC isozyme overexpression experiments provide coherent evidence that PLC-β1 activity is involved in determination of the time constant of Ca2+ spike amplitude decay during Ca2+ oscillations, while PLC-β4 activity is involved in determination of the Ca2+ oscillation frequency in HeLa cells stimulated with histamine. These findings suggest that the relative amounts of PLC-β1 and PLC-β4 have a large impact on the pattern of histamine-induced Ca2+ signals. We hypothesize that stochastic variations in the amounts of PLC-β1 and PLC-β4 expressed in HeLa cells are involved in the generation of the non-genetic cell-to-cell variability in agonist-induced Ca2+ signals (Fig. 7). According to this hypothesis, low-frequency sustained Ca2+ oscillations tended to be observed in cells in which PLC-β1 was dominant, while high-frequency damped oscillations tended to be observed in cells in which PLC-β4 was dominant (Fig. 7). The former may correspond to the S-cells shown in Fig. 2A, while the latter may correspond to the D-cells shown in Fig. 2B. Ambler et al. described that sister cells in a BC3H-1 clonal cell population displayed unique temporal responses in histamine-induced Ca2+ signals immediately following cell division [13], and proposed that the linkage of receptor occupancy to Ca2+ elevation is a functionally unique property for individual cells that can be influenced by epigenetic factors. Quantification of the individual PLC isozymes and rapid manipulation of their expression and/or intracellular localization in single HeLa cells will be useful to verify our hypothesis in future studies.
Figure 7. A possible mechanism that underlies the generation of cell-specific patterns of histamine-induced Ca2+ signals.

The diagrams show the typical IP3 and Ca2+ dynamics observed in HeLa cells stimulated with histamine. Low-frequency sustained Ca2+ oscillations tend to be observed in cells in which PLC-β1 is dominant (left), while high-frequency damped oscillations tend to be observed in cells in which PLC-β4 is dominant (right).
Regulatory Factors for PLC-β Activity
Comparisons of the functional differences between PLC-β1 and PLC-β4 should provide clues toward understanding the mechanistic basis for the generation of cell-specific patterns of Ca2+ signals. The RNAi-mediated silencing experiments revealed that PLC-β1 only became activated when the histamine stimulation was accompanied by [Ca2+] increases in thapsigargin-treated cells, whereas PLC-β4 was activated by histamine stimulation with or without [Ca2+] elevation. These findings are consistent with previous biochemical measurements showing that purified PLC-β1 exhibits Ca2+-dependent PIP2 hydrolysis activity with a maximal activation at 100–220 nM [Ca2+] [23], while the activity of purified PLC-β4 is less sensitive to Ca2+ and the PLC-β4 activity in the absence of Ca2+ is higher than those of the other PLC-βs [24]. Because feedback regulation by cytosolic Ca2+ must be involved in the Ca2+ signal formation, the difference in Ca2+ sensitivity for the PIP2 hydrolysis activity between PLC-β1 and PLC-β4 should be critical in determining the pattern of cell-specific Ca2+ signals.
PLC-β isozymes are regulated by heterotrimeric G proteins, whose activities are controlled by the balance of the rate of GTP binding, which causes activation, and the rate of GTP hydrolysis, which terminates activation [25], [26]. Because Gα subunits show remarkably slow rates of basal guanine nucleotide exchange and a low rate of GTP hydrolysis [27], [28], the GTPase catalytic cycle is regulated by GPCRs, which accelerate GTP binding, and by GTPase-activating proteins (GAPs), which accelerate hydrolysis. PLC-βs are GAPs for Gαq [29] and accelerate the deactivation limb of the cycle. In the continued presence of an agonist, it has been proposed that activated GPCR stays bound to Gαq through multiple cycles of GTP binding and hydrolysis because the rate of dissociation of the GPCR from GTP-bound Gαq is much slower than that of the PLC-β-promoted return of the G protein to the GDP-bound state. This phenomenon has been referred to as a kinetic scaffold [27], [28], [30], and the GAP activities of the PLC-β isozymes may influence the temporal pattern of the activation of each isozyme independently. Recently, it was shown that the intracellular localization of PLC-β1 is regulated by PKC phosphorylation [31] and by translin-associated factor X binding [32]. In addition to the PIP2 hydrolysis activity, examination of the differences between PLC-β1 and PLC-β4 in terms of these regulatory factors in living cells will be pivotal for understanding the molecular basis of their differential roles in the generation of Ca2+ signals.
Materials and Methods
Materials
Anti-PLC-β1, anti-PLC-β3, anti-PLC-β4, and anti-PLC-γ1 rabbit polyclonal antibodies were obtained from Santa Cruz Biotechnology Inc. A mouse monoclonal antibody against β-actin was purchased from Sigma-Aldrich. A mouse monoclonal antibody against PLC-δ3 [33] was used. Mouse PLC-β1 cDNA (U85712) and mouse PLC-β4 cDNA (NM_013829) were subcloned and inserted into pBluescriptII KS(+). A rabbit polyclonal antibody against PLC-ε [34] was a generous gift from Dr. T. Kataoka (Kobe University, Kobe, Japan). Two different predesigned siRNA duplexes targeting each of PLC-β1, PLC-β3, PLC-β4, PLC-γ1, and PLC-ε (Stealth™ RNAi) were purchased from Invitrogen. Two Stealth™ RNAi Negative Control Medium GC Duplexes (mGCs) and two Stealth™ RNAi Negative Control High GC Duplexes (hGCs) (both from Invitrogen) were used as negative controls. For knockdown of PLC-δ3, two original siRNAs were synthesized (ST Pharm Co. Ltd.). The sequence of PLCδ3KD siRNA#1 (Table 2) was scrambled to minimize the sequence homology with any known vertebrate transcripts and used to synthesize a negative control siRNA. The nucleotide sequences of the siRNAs used in this study are shown in Table 2.
Table 2. siRNA sequences.
| Name | Sequences (5′ to 3′) | GC%a | Positionb |
| Control siRNA mGC | GGUAGGUGAGUGUACAGACGCAAUA | 48 | |
| Control siRNA mGC | CAGGACGUCUAGCUGUAGUCGCCAU | 56 | |
| PLCβ1KD siRNA #1 | GCUACUGGAUCUCAGCCUUGUCAAA | 48 | 186 |
| PLCβ1KD siRNA #2 | GCCCUCGACCUGAAAUUGAUAACAU | 44 | 659 |
| PLCβ3KD siRNA #1 | GGCUUCACUUCGCAUUGCAGCCUUU | 52 | 2372 |
| PLCβ3KD siRNA #2 | ACCCGAGACUCAACGAAGUGCUGUA | 52 | 766 |
| PLCβ4KD siRNA #1 | GCUGCCAGAUGGUUUCACUGAACUA | 48 | 1928 |
| PLCβ4KD siRNA #2 | GCAUGGUUAUGAAUAAUGGACUCAA | 36 | 2222 |
| PLCγ1KD siRNA #1 | CCCUGCGCUGUAAUGAGUUUGAGAU | 48 | 1928 |
| PLCγ1KD siRNA #2 | CCUUGUUGACCUCAUCAGCUACUAU | 44 | 2199 |
| PLCεKD siRNA #1 | CGCCACCCUCCAAAGGACUUCAAUA | 52 | 1362 |
| PLCεKD siRNA #2 | CAGCGAUCUUCAGCCUGACCUAGAU | 52 | 3792 |
| Control siRNA scr | GGCAUAGCCAGAUGGUAGAGAUAAGAA | 44 | |
| PLCδ3KD siRNA #1 | AGAUGAGCUUCAAGGAGAUCAAGAGAG | 44 | 602 |
| PLCδ3KD siRNA #2 | UGGUCAACGUGGACAUGAACGACAUAG | 48 | 638 |
The GC% column indicates the GC content of each siRNA sequence.
The numbers in the position column denote where the sequence is located in the mRNA of the target gene (counted from the first nucleotide of the start codon) of human origin.
Plasmid Construction
The plasmid IRES-mRFP was prepared from IRES2-AcGFP1 (Clontech Laboratories Inc.) by replacing the AcGFP1 cDNA with an mRFP cDNA. A full-length mouse PLC-β1 cDNA was obtained by PCR from mouse PLCβ1-pBluescriptII KS(+), and cloned into the EcoRI and BamHI sites of IRES-mRFP. A full-length mouse PLC-β4 cDNA was obtained from mouse PLCβ4-pBluescriptII KS(+) by XhoI digestion, and inserted into IRES-mRFP.
Cell Culture
HeLa cells were maintained in DMEM (Nacalai Tesque) supplemented with 10% FBS at 37°C under 5% CO2.
RT-PCR
Total RNA was extracted from HeLa cells using a TRIzol Reagent Kit (Invitrogen), and reverse-transcribed using random primers and SuperScriptII reverse transcriptase (Invitrogen) according to the manufacturer’s protocol. cDNA fragments for the 13 PLC genes were amplified by PCR using specific primer sets (Table 1). The primers were designed using Primer 3 software to fullfil the following requirements: (1) the PCR products should contain an exon–exon junction to prevent amplification of any contaminating genomic DNA; (2) all known splicing variants should be detected; (3) the PCR products should be approximately 100 bp; and (4) the primers should have almost the same Tm values to maintain uniform amplification efficiency. Primers for β-actin were designed under similar criteria and used as an internal control. The PCR fragments were analyzed by 2% agarose gel electrophoresis.
Western Blotting
Total HeLa cell lysates were prepared by direct lysis with SDS sample buffer (62.5 mM Tris-HCl pH 6.8, 10% glycerol, 2% SDS, 5% β-mercaptoethanol, 0.05% bromophenol blue) and subjected to electrophoresis. The separated proteins were transferred onto PVDF membranes (Millipore), and the PLC isozymes and β-actin were probed with specific primary antibodies. After treatment with HRP-conjugated anti-rabbit or anti-mouse secondary antibodies (GE Healthcare), the antibody-bound proteins were detected with chemiluminescence reagents (ECL: GE Healthcare) and a LAS4000mini system (Fujifilm).
siRNA Transfection
Fresh culture medium (0.8 ml) was supplied to HeLa cells cultured in 35-mm dishes at 30 min prior to transfection. Next, 1 µl of 20 µM siRNA solution and 1 µl of LipofectAMINE 2000 (Invitrogen) were independently diluted in 100 µl of Opti-MEM (Invitrogen), and the two mixtures were combined and incubated for 20 min at room temperature. The mixed solution was added to the cells to give a final concentration of 20 nM siRNA. After incubation for 8–12 h in a CO2 chamber, the siRNA solution was removed by replacement with 1.5 ml of fresh culture medium.
Imaging
For imaging of siRNA-treated cells, HeLa cells at 36–42 h after siRNA transfection were transfected with plasmids containing the IRIS-1 cDNA using a transfection reagent (TransIT; Mirus). The cells were subjected to imaging at 24–30 h after the IRIS-1 transfection. For imaging of cells expressing recombinant PLC-βs, HeLa cells were transfected with PLCβ1-IRES-mRFP, PLCβ4-IRES-mRFP, or IRES-mRFP plasmids using the TransIT transfection reagent at 20–36 h before IRIS-1 transfection. After loading the cells with 5 µM Indo-5F AM (Dojindo), imaging was performed under a constant flow (2 ml/min) of balanced salt solution (20 mM Hepes pH 7.4, 115 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM glucose) as an imaging medium at 37°C. For experiments with artificial control of [Ca2+], cells were pretreated with 1 µM thapsigargin in the absence of extracellular Ca2+ to deplete the cytosolic Ca2+ stores by blocking sarcoplasmic or endoplasmic reticulum Ca2+-ATPase pumps. Imaging was performed at 37°C using an inverted microscope (IX-71; Olympus) with a cooled charge-coupled device camera (ORCA-ER; Hamamatsu Photonics) and a 40× (NA 1.35) objective. A 330–348-nm excitation filter and 460–510-nm emission filter and a 425–445-nm excitation filter and a pair of 460–510-nm (cyan) and 525–565-nm (yellow) emission filters were used for the fluorochromes Indo-5F and IRIS-1, respectively. An emission splitter (W-view; Hamamatsu Photonics) was used with a fast light source exchanger (DG-4; Sutter Instrument Co.). Sequential excitation of IRIS-1 and Indo-5F was performed using a 450-nm dichroic mirror and two excitation filters (425–445-nm filter for IRIS-1 and 330–348-nm filter for Indo-5F). Dual emission at 460–490 nm (for IRIS-1 and Indo-5F) and >520 nm (for IRIS-1) was split with a 460–490-nm filter, a long-path 520-nm barrier filter, and two 505-nm dichroic mirrors equipped in W-view. Images were acquired at 0.5 or 1 Hz for measurements of Ca2+ oscillations in intact cells and at 0.17, 0.25, or 0.5 Hz for measurements in thapsigargin-treated cells. Image acquisition was performed with MetaFluor software (Molecular Devices). Spectral analyses of Ca2+ oscillations were performed as described previously [35].
Supporting Information
Histogram of the time constants for exponential decay of the Ca2+ spike amplitude. (A) Ca2+ spike amplitudes were defined as shown in the left scheme. The time constants were estimated by fitting the amplitudes with a single exponential function, as shown on the right. (B) Histogram of the time constants of Ca2+ spike amplitude decay observed in HeLa cells stimulated with 3 µM histamine. The arrow indicates the threshold for dividing the cells into S-cells and D-cells.
(PDF)
Difference in histamine concentration-dependence of the amplitude of the first Ca2+ spike and the IP3 production between S-cells and D-cells. (A) Relationships between the histamine concentrations and the peak amplitudes of the first increase in Indo-5F signals (F/F0) in S-cells (red) and D-cells (blue). (B) Mean values of the Indo-5F signal changes shown in (A). Statistical analyses were performed by one-way ANOVA followed by Scheffe’s multiple comparison test. *P<0.05, **P<0.01. NS: not significant. (C) Relationships between the histamine concentration and the integrated IP3 signals observed in S-cells (red) and D-cells (blue). Data represent means ± SD. Statistical analyses were performed using Student’s t-test. *P<0.05.
(PDF)
PLC isozyme-specific knockdown does not affect the expressions of other PLC isozymes. Western blotting analyses of total lysates prepared from HeLa cells treated with PLC isozyme-specific siRNAs. The isozyme-specific antibodies used for the western blotting analyses are shown on the left. The data are representative of at least two independent experiments.
(PDF)
Validation of the IRIS-1 signal changes observed in thapsigargin-treated HeLa cells. (A and B) The maximal IRIS-1 signal changes after addition of 3 µM histamine (A) and 2 mM Ca2+ (B) were plotted against the basal CFP intensity of IRIS-1 (top), resting IRIS-1 C/Y ratio (middle), and integrated value of Indo-5F change for 10 min (bottom). (C) The initial rate of IRIS-1 signal change (1st component) after addition of 3 µM histamine plus 2 mM Ca2+ was plotted against the basal CFP intensity of IRIS-1 (top), resting IRIS-1 C/Y ratio (middle), and initial rate of Indo-5F change (bottom). (D) The maximal IRIS-1 signal changes after addition of 3 µM histamine plus 2 mM Ca2+ (2nd component) were plotted against the basal CFP intensity of IRIS-1 (top), resting IRIS-1 C/Y ratio (middle), and integrated value of Indo-5F change for 10 min (bottom).
(PDF)
[IP3] changes after histamine stimulation plus [Ca2+] elevation in thapsigargin-treated PLC-β1 and PLC-β4 knockdown cells. Representative traces of IRIS-1 signal changes (ΔR/R0) after addition of 3 µM histamine (horizontal open bars) plus 2 mM Ca2+ (horizontal filled bars) observed in thapsigargin-treated HeLa cells transfected with control siRNA mGC (left), PLCβ1KD siRNAs (middle), or PLCβ4KD siRNAs (bottom).
(PDF)
Acknowledgments
We thank Drs. Takafumi Inoue, Haruka Yamazaki, Masahiro Enomoto, and Hideki Nakamura for fruitful discussions.
Funding Statement
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan to T. Matsu-ura (22770227), T. Michikawa (20370054), and K. Mikoshiba (2022007). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Clapham DE (1995) Calcium signaling. Cell 80: 259–268. [DOI] [PubMed] [Google Scholar]
- 2. Berridge MJ, Lipp P, Bootman MD (2000) The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1: 11–21. [DOI] [PubMed] [Google Scholar]
- 3. Woods NM, Cuthbertson KS, Cobbold PH (1986) Repetitive transient rises in cytoplasmic free calcium in hormone-stimulated hepatocytes. Nature 319: 600–602. [DOI] [PubMed] [Google Scholar]
- 4. Berridge MJ (1988) Inositol trisphosphate-induced membrane potential oscillations in Xenopus oocytes. J Physiol (Lond) 403: 589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prentki M, Glennon MC, Thomas AP, Morris RL, Matschinsky FM, et al. (1988) Cell-specific patterns of oscillating free Ca2+ in carbamylcholine-stimulated insulinoma cells. J Biol Chem 263: 11044–11047. [PubMed] [Google Scholar]
- 6. Cobbold PH, Sanchez-Bueno A, Dixon CJ (1991) The hepatocyte calcium oscillator. Cell Calcium 12: 87–95. [DOI] [PubMed] [Google Scholar]
- 7. Thomas AP, Renard DC, Rooney TA (1991) Spatial and temporal organization of calcium signalling in hepatocytes. Cell Calcium 12: 111–126. [DOI] [PubMed] [Google Scholar]
- 8. Berridge MJ (2012) Calcium signalling remodelling and disease. Biochemical Society Transactions 40: 297–309. [DOI] [PubMed] [Google Scholar]
- 9. Berridge MJ (2009) Inositol trisphosphate and calcium signalling mechanisms. Biochimica et Biophysica Acta 1793: 933–940. [DOI] [PubMed] [Google Scholar]
- 10. Matsu-ura T, Michikawa T, Inoue T, Miyawaki A, Yoshida M, et al. (2006) Cytosolic inositol 1,4,5-trisphosphate dynamics during intracellular calcium oscillations in living cells. J Cell Biol 173: 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bunney TD, Katan M (2011) PLC regulation: emerging pictures for molecular mechanisms. Trends Biochem Sci 36: 88–96. [DOI] [PubMed] [Google Scholar]
- 12. Suh PG, Park JI, Manzoli L, Cocco L, Peak JC, et al. (2008) Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep 41: 415–434. [DOI] [PubMed] [Google Scholar]
- 13. Ambler SK, Poenie M, Tsien RY, Taylor P (1988) Agonist-stimulated oscillations and cycling of intracellular free calcium in individual cultured muscle cells. J Biol Chem 263: 1952–1959. [PubMed] [Google Scholar]
- 14. Kuchtey J, Fewtrell C (1996) Subcloning the RBL-2H3 mucosal mast cell line reduces Ca2+ response heterogeneity at the single-cell level. J Cell Physiol 166: 643–652. [DOI] [PubMed] [Google Scholar]
- 15. Rooney TA, Sass EJ, Thomas AP (1989) Characterization of cytosolic calcium oscillations induced by phenylephrine and vasopressin in single fura-2-loaded hepatocytes. J Biol Chem 264: 17131–17141. [PubMed] [Google Scholar]
- 16. Kim MJ, Min DS, Ryu SH, Suh PG (1998) A cytosolic, galphaq- and betagamma-insensitive splice variant of phospholipase C-beta4. J Biol Chem 273: 3618–3624. [DOI] [PubMed] [Google Scholar]
- 17. Wu DQ, Lee CH, Rhee SG, Simon MI (1992) Activation of phospholipase C by the alpha subunits of the Gq and G11 proteins in transfected Cos-7 cells. J Biol Chem 267: 1811–1817. [PubMed] [Google Scholar]
- 18. Niepel M, Spencer SL, Sorger PK (2009) Non-genetic cell-to-cell variability and the consequences for pharmacology. Curr Opin Chem Biol 13: 556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK (2009) Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459: 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hazama A, Yada T, Okada Y (1985) HeLa cells have histamine H1-receptors which mediate activation of the K+ conductance. Biochim Biophys Acta 845: 249–253. [DOI] [PubMed] [Google Scholar]
- 21. Krumins AM, Gilman AG (2006) Targeted knockdown of G protein subunits selectively prevents receptor-mediated modulation of effectors and reveals complex changes in non-targeted signaling proteins. J Biol Chem 281: 10250–10262. [DOI] [PubMed] [Google Scholar]
- 22. Adjobo-Hermans MJ, Crosby KC, Putyrski M, Bhageloe A, van Weeren L, et al. (2012) PLCbeta isoforms differ in their subcellular location and their CT-domain dependent interaction with Galphaq. Cell Signal 25: 255–263. [DOI] [PubMed] [Google Scholar]
- 23. Blank JL, Ross AH, Exton JH (1991) Purification and characterization of two G-proteins that activate the beta 1 isozyme of phosphoinositide-specific phospholipase C. Identification as members of the Gq class. J Biol Chem 266: 18206–18216. [PubMed] [Google Scholar]
- 24. Min DS, Kim DM, Lee YH, Seo J, Suh PG, et al. (1993) Purification of a novel phospholipase C isozyme from bovine cerebellum. J Biol Chem 268: 12207–12212. [PubMed] [Google Scholar]
- 25. Morris AJ, Malbon CC (1999) Physiological regulation of G protein-linked signaling. Physiol Rev 79: 1373–1430. [DOI] [PubMed] [Google Scholar]
- 26. Wettschureck N, Offermanns S (2005) Mammalian G proteins and their cell type specific functions. Physiol Rev 85: 1159–1204. [DOI] [PubMed] [Google Scholar]
- 27. Sprang SR (1997) G protein mechanisms: insights from structural analysis. Annu Rev Biochem 66: 639–678. [DOI] [PubMed] [Google Scholar]
- 28. Ross EM, Wilkie TM (2000) GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem 69: 795–827. [DOI] [PubMed] [Google Scholar]
- 29. Berstein G, Blank JL, Jhon DY, Exton JH, Rhee SG, et al. (1992) Phospholipase C-beta 1 is a GTPase-activating protein for Gq/11, its physiologic regulator. Cell 70: 411–418. [DOI] [PubMed] [Google Scholar]
- 30. Waldo GL, Ricks TK, Hicks SN, Cheever ML, Kawano T, et al. (2010) Kinetic scaffolding mediated by a phospholipase C-beta and Gq signaling complex. Science 330: 974–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aisiku O, Dowal L, Scarlata S (2011) Protein kinase C phosphorylation of PLCbeta1 regulates its cellular localization. Arch Biochem Biophys 509: 186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aisiku OR, Runnels LW, Scarlata S (2010) Identification of a novel binding partner of phospholipase cbeta1: translin-associated factor X. PLoS One. 5: e15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakamura Y, Hamada Y, Fujiwara T, Enomoto H, Hiroe T, et al. (2005) Phospholipase C-delta1 and -delta3 are essential in the trophoblast for placental development. Mol Cell Biol 25: 10979–10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu D, Tadano M, Edamatsu H, Masago-Toda M, Yamawaki-Kataoka Y, et al. (2003) Neuronal lineage-specific induction of phospholipase Cepsilon expression in the developing mouse brain. Eur J Neurosci 17: 1571–1580. [DOI] [PubMed] [Google Scholar]
- 35. Uhlen P (2004) Spectral analysis of calcium oscillations. Sci STKE 2004: pl15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Histogram of the time constants for exponential decay of the Ca2+ spike amplitude. (A) Ca2+ spike amplitudes were defined as shown in the left scheme. The time constants were estimated by fitting the amplitudes with a single exponential function, as shown on the right. (B) Histogram of the time constants of Ca2+ spike amplitude decay observed in HeLa cells stimulated with 3 µM histamine. The arrow indicates the threshold for dividing the cells into S-cells and D-cells.
(PDF)
Difference in histamine concentration-dependence of the amplitude of the first Ca2+ spike and the IP3 production between S-cells and D-cells. (A) Relationships between the histamine concentrations and the peak amplitudes of the first increase in Indo-5F signals (F/F0) in S-cells (red) and D-cells (blue). (B) Mean values of the Indo-5F signal changes shown in (A). Statistical analyses were performed by one-way ANOVA followed by Scheffe’s multiple comparison test. *P<0.05, **P<0.01. NS: not significant. (C) Relationships between the histamine concentration and the integrated IP3 signals observed in S-cells (red) and D-cells (blue). Data represent means ± SD. Statistical analyses were performed using Student’s t-test. *P<0.05.
(PDF)
PLC isozyme-specific knockdown does not affect the expressions of other PLC isozymes. Western blotting analyses of total lysates prepared from HeLa cells treated with PLC isozyme-specific siRNAs. The isozyme-specific antibodies used for the western blotting analyses are shown on the left. The data are representative of at least two independent experiments.
(PDF)
Validation of the IRIS-1 signal changes observed in thapsigargin-treated HeLa cells. (A and B) The maximal IRIS-1 signal changes after addition of 3 µM histamine (A) and 2 mM Ca2+ (B) were plotted against the basal CFP intensity of IRIS-1 (top), resting IRIS-1 C/Y ratio (middle), and integrated value of Indo-5F change for 10 min (bottom). (C) The initial rate of IRIS-1 signal change (1st component) after addition of 3 µM histamine plus 2 mM Ca2+ was plotted against the basal CFP intensity of IRIS-1 (top), resting IRIS-1 C/Y ratio (middle), and initial rate of Indo-5F change (bottom). (D) The maximal IRIS-1 signal changes after addition of 3 µM histamine plus 2 mM Ca2+ (2nd component) were plotted against the basal CFP intensity of IRIS-1 (top), resting IRIS-1 C/Y ratio (middle), and integrated value of Indo-5F change for 10 min (bottom).
(PDF)
[IP3] changes after histamine stimulation plus [Ca2+] elevation in thapsigargin-treated PLC-β1 and PLC-β4 knockdown cells. Representative traces of IRIS-1 signal changes (ΔR/R0) after addition of 3 µM histamine (horizontal open bars) plus 2 mM Ca2+ (horizontal filled bars) observed in thapsigargin-treated HeLa cells transfected with control siRNA mGC (left), PLCβ1KD siRNAs (middle), or PLCβ4KD siRNAs (bottom).
(PDF)