

Abstract
The coordinate regulation of HLA class II (HLA-II) is controlled by the class II transactivator, CIITA, and is crucial for the development of anti-tumor immunity. HLA-II in breast carcinoma is associated with increased IFN-γ levels, reduced expression of the estrogen receptor (ER) and reduced age at diagnosis. Here, we tested the hypothesis that estradiol (E2) and ERα signaling contribute to the regulation of IFN-γ inducible HLA-II in breast cancer cells. Using a panel of established ER− and ER+ breast cancer cell lines, we showed that E2 attenuated HLA-DR in two ER+ lines (MCF-7 and BT-474), but not in T47D, while it augmented expression in ER− lines, SK-BR-3 and MDA-MB-231. To further study the mechanism(s), we used paired transfectants: ERα+ MC2 (MDA-MB-231 c10A transfected with the wild type ERα gene) and ERα− VC5 (MDA-MB-231 c10A transfected with the empty vector), treated or not with E2 and IFN-γ. HLA-II and CIITA were severely reduced in MC2 compared to VC5 and were further exacerbated by E2 treatment. Reduced expression occurred at the level of the IFN-γ inducible CIITA promoter IV. The anti-estrogen ICI 182,780 and gene silencing with ESR1 siRNA reversed the E2 inhibitory effects, signifying an antagonistic role for activated ERα on CIITA pIV activity. Moreover, STAT1 signaling, necessary for CIITA pIV activation, and selected STAT1 regulated genes were variably downregulated by E2 in transfected and endogenous ERα positive breast cancer cells, whereas STAT1 signaling was noticeably augmented in ERα− breast cancer cells. Collectively, these results imply immune escape mechanisms in ERα+ breast cancer may be facilitated through an ERα suppressive mechanism on IFN-γ signaling.
Introduction
Antigen presentation by major histocompatibility complex (MHC) class II molecules (MHC-II), known as HLA-II (HLA-DR, -DP, -DQ) in humans and co-chaperones HLA-DM and the invariant chain (Ii) are important for the development of adaptive immune responses including anti-tumor immunity [1]–[4]. Typically, HLA-II expression is limited to professional antigen presenting cells (pAPC), but is induced by IFN-γ on most cell types including those derived from cancer [5], [6]. HLA-DR positive tumor cells have been described in several malignancies, such as melanoma [7], colon [8], [9] and breast [10]–[12], but the underlying mechanisms are likely diverse. The number of HLA-II positive tumor cells in breast cancer is directly associated with tumor infiltrating immune cells and levels of IFN-γ [12]–[14], but other cytokines, hormones, growth factors and oncogenes are also implicated in regulating HLA-II expression [15]–[20].
HLA-II expression is controlled at the transcription level by a highly conserved regulatory module, located in the promoter of genes encoding the α- and β-chains of all HLA-II molecules and in the gene encoding the Ii co-chaperone [21]–[26]. This regulatory module forms a platform for the class II transactivator (CIITA), a non-DNA binding protein, which acts as a transcriptional integrator by connecting transcription factors, bound to the MHC-II promoter with components of the general transcriptional machinery [23], [27]–[30]. The central role of CIITA is evident from lack of constitutive or IFN-γ inducible HLA-II in bare lymphocyte syndrome [31], [32].
CIITA expression is controlled by three distinct promoters: promoter I (pI) for constitutive expression in dendritic cells; promoter III (pIII), for constitutive expression in B cells; promoter IV (pIV) for IFN-γ inducible expression [21], [26], [33]. This promoter system is crucial for controlling CIITA messenger RNA (mRNA) and protein levels, and they, in turn, regulate HLA-II expression. The molecular regulation of CIITA pIV is intricately linked to the classical IFN-γ signaling pathway. IFN-γ, binds to IFN-γ receptors (IFNGR) on the cell surface, resulting in autophosphorylation of Janus kinase 2 (JAK2) and JAK1, followed by phosphorylation, dimerization and nuclear translocation of signal transducer and activator of transcription 1 (STAT1) [34], [35]. Phosphorylated STAT1 (pSTAT1) binds to IFN-activated sites (GAS) in the promoter of target genes including the IFN-regulatory factor 1 (IRF1), thus stimulating its expression. IRF1 binds cooperatively with IRF2 to its associated IRF element (IRF-E) in CIITA pIV, and concomitant pSTAT1 binding to GAS in CIITA pIV results in transcriptional activation of CIITA [33], [36]. Moreover, signaling pathways such as mitogen activated protein kinases (MAPK) and PI3K/Akt that are frequently activated in breast cancer cells [37] modulate expression of IRF1 and STAT1 [38]–[40], further impacting the levels of IFN-γ inducible CIITA and subsequent HLA-II expression on tumor cells.
Previously, we showed that HLA-II (HLA-DR, HLA-DM and Ii) was discordantly expressed on tumor cells in human breast cancer tissues [12]. Furthermore, tumor cell expression of HLA-DR and Ii, but not HLA-DM, correlated with reduced expression of estrogen receptors (ER) and reduced age at diagnosis. Importantly, tumors with coordinate expression of HLA-DR, Ii and HLA-DM had the highest IFN-γ mRNA levels and correlated with increased patient survival [12]. Undoubtedly, the mechanisms governing tumor cell expression of HLA-II in breast carcinoma are likely multifaceted, involving IFN-γ secreted by infiltrating immune cells [12], circulating and tumor-associated estrogens [41] and activation of growth factor and hormone receptor pathways in the tumor cells [42], [43]. Estradiol and anti-estrogens, tamoxifen and fulvestrant or ICI 180,720 (ICI), were shown to modulate IFN-γ inducible MHC-II in various cell types [17], [19], [44], [45] through mechanisms not involving ligand activation of the estrogen receptor (ER) pathway.
In this study, using established human ER− and ER+ breast cancer cell lines (BCCL) and an ERα-transfected BCCL, we investigated the specific and combined effects of estradiol (E2) and ERα on HLA-II regulation. We found IFN-γ inducible HLA-II expression was modulated by E2-ER activation at the level of the CIITA pIV. Furthermore, E2-treatment of ERα+ BCCL and ERα− BCCL differentially affected various components of the IFN-γ signaling pathway that are required for transactivation of CIITA pIV.
Results
Estradiol differentially modulates HLA-DR expression in breast cancer cell lines
Stemming from our previous finding that HLA-II expression in breast carcinoma tissues correlates with increased IFN-γ mRNA, reduced age at diagnosis and reduced ER levels [12] we questioned whether E2, in the absence or presence of its cognate receptor ERα, modulates HLA-DR expression in established ER− and ER+ BCCL, treated or not with IFN-γ for 96 hours. Analysis of ER− BCCL using flow cytometry (Figure 1A & 1B) revealed low basal expression of HLA-DR in MDA-MB-231, but not in SK-BR-3 while IFN-γ induced strong expression in both cell lines. E2-treatment augmented IFN-γ inducible HLA-DR, although this was significant for only SK-BR-3 (Figure 1B). These results, confirmed by Western blot analysis of cell lysates (Figure 1C & 1D), suggest E2 may modulate HLA-DR expression in ER− breast cancer through an ERα independent mechanism [46].
Figure 1. E2 differentially modulates inducible HLA-DR expression in ERα+ and ERα− breast cancer cell lines.

MDA-MB-231, SK-BR-3, MCF-7, BT-474, and T47D were cultured in E2-depleted media, treated with vehicle (ethanol) or E2 (10−9 M) and stimulated or not with IFN-γ (100 U/ml) for 96 hours. (A & E) HLA-DR cell surface expression (L243) was analyzed by flow cytometry: grey line, isotype control; black line, constitutive expression; shaded histogram, IFN-γ induced expression. (B & F) Bar graphs represent the MFI (mean florescence intensity) ± SEM for HLA-DR expression of three independent experiments. (C & G) Western blot analysis was performed on cytoplasmic and nuclear extracts for ERα expression (HC-20) and on cytoplasmic extracts for HLA-DRα (TAL 1B5). Protein loading controls included α-tubulin (B-7) and P84 (5E10) for cytoplasmic and nuclear proteins, respectively. (D & H) Bar graphs show the ratio of band intensity for HLA-DRα, normalized to the α-tubulin band intensity and represent the mean ± SEM of three independent experiments (*p<0.05, **p<0.01, ***p<0.001).
Since the least HLA-DR in human breast carcinoma tissues occurred in ER+ tumors [12] we hypothesized that E2-activation of the ERα pathway inhibits HLA-DR expression. Analysis of ER+ BCCL, treated as described above, revealed a variable pattern of IFN-γ inducible HLA-DR expression with amounts that were barely detectable, moderate and abundant in BT-474, MCF-7, and T47D, respectively (Figure 1E & 1F). Constitutive HLA-DR was detected at the cell surface in only T47D (Figure 1E). Furthermore, E2 treatment significantly reduced HLA-DR in MCF-7 and BT-474, but not in T47D (Figure 1E). Similar results were obtained from Western blot analysis of cell lysates (Figure 1G & 1H). Notably, ERα levels were not altered by IFN-γ but E2 treatment increased the amount in the nucleus, indicating ligand activation of the ERα pathway (Figure. 1G). Taken together these data suggest that E2-inhibition of HLA-II expression in ERα+ BCCL is mediated through activation of ligand-dependent ERα pathway.
Transfection of ESR1 in an ER- cell line diminishes IFN-γ inducible HLA-II proteins
To further explore the role of ERα on IFN-γ inducible HLA-DR, we used two stably transfected cell lines, derived from MDA-MB-231 clone 10A [47], [48]: MC2 expresses wild type ERα and VC5 expresses the empty vector. Since MDA-MB-231 clone 10A was selected for negative expression of ERα and ERβ [47], the transfected pair is a suitable model to assess ERα mediated effects on HLA-II without interference from other ERs including GPR30, reported to be deficient in MDA-MB-231 [48], [49]. The cells, treated and analyzed for HLA-DR expression as described above, revealed significantly reduced cell surface HLA-DR in MC2, as compared to VC5 and MDA-MB-231 clone 10A (Figure 2A & 2B). Moreover, E2-treatment greatly diminished HLA-DR in MC2 but not in VC5 and MDA-MB-231 clone 10A. These results were confirmed by Western blot analysis of cell extracts (Figure 2C). Again, HLA-DR protein in the ERα+ MC2 was severely reduced and exacerbated by E2, whereas MDA-MB-231 clone10A and VC5 expressed abundant HLA-DR in the presence and absence of E2. As the only known difference between MC2 and VC5 is the expression of ERα, these results further implicate ERα in negatively regulating HLA-DR expression.
Figure 2. IFN-γ inducible HLA-DR is down regulated in the ERα+ transfected breast cancer cell line, MC2.
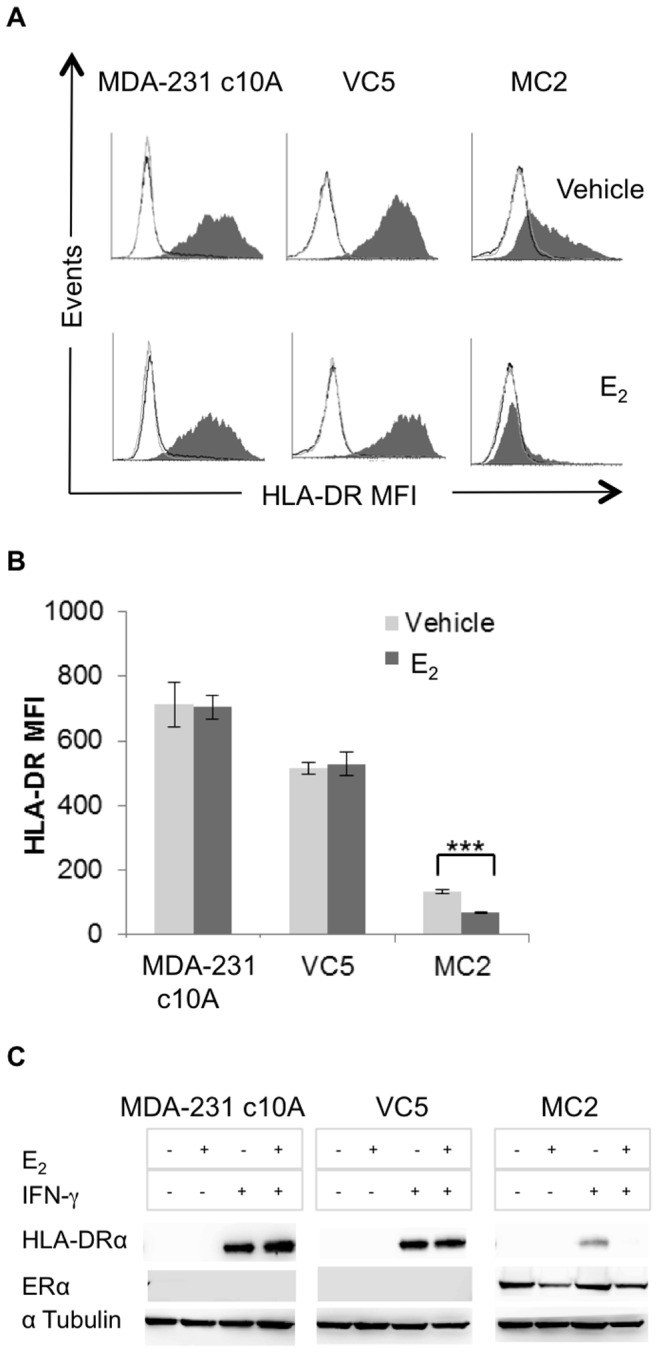
MDA-MB-231 clone 10A (MDA-231 c10A), VC5 (MDA-231 c10A, transfected with the empty plasmid vector) and MC2 (MDA-231 c10A, transfected with wild type ESR1) were cultured in E2-depleted medium and stimulated or not with IFN-γ (100 U/ml) for 96 hours. (A) HLA-DR cell surface expression (L243) was analyzed by flow cytometry: grey line, isotype control; black line, constitutive expression; shaded histogram, IFN-γ induced expression. (B) Bar graphs represent the MFI ± SEM for HLA-DR expression of three independent experiments (***p<0.001). (C) Western blot analysis was performed on whole cell lysates for HLA-DRα (TAL 1B5) and ERα (HC-20).
Although HLA-II genes are coordinately regulated [25], we found most breast cancer lesions with HLA-DR+ tumor cells do not have detectable HLA-DM expression [12]. We reasoned that if ERα and its activation by E2 coordinately down regulates HLA-II, then blocking ER signaling with ICI, a selective anti-estrogen that degrades ER, should reverse the inhibition. To test this hypothesis, MC2 and VC5 were pretreated with 10−6 M ICI in the presence or absence of 10−9 M E2. Following stimulation with IFN-γ for 96 hours, HLA-DR, -DM and Ii were analyzed by flow cytometry and Western blot. HLA-DR, -DM and Ii expression levels were significantly reduced in MC2 compared to VC5 (Figure 3A–3C), while E2-treatment further diminished HLA-II expression in MC2, but not in VC5. Although ICI-treatment, alone or with E2, did not restore HLA-II in MC2 to VC5 levels, it clearly reversed the E2-inhibitory effect on HLA-II expression. Western blot analysis (Figure 3D–3G) and immunocytochemistry (data not shown) confirmed the reduced expression of HLA-DR, -DM and Ii in MC2 and the involvement of ERα signaling in the inhibitory effect of E2 on HLA-II expression.
Figure 3. Coordinate downregulation of IFN-γ inducible HLA-II expression by E2 is reversed by ICI-mediated degradation of ERα in MC2 cells.

VC5 and MC2 cells were cultured in E2-depleted media, treated with vehicle (ethanol), E2 (10−9 M) or/and ICI (10−6 M) followed by stimulation with IFN-γ (100 U/ml) for 96 hours. HLA-II expression was analyzed by surface flow cytometry using (A) anti-DR, (L243), and intracellular flow cytometry using (B) anti-DM (Map.DM1) and (C) anti-Ii (LN2). Bar graphs represent the MFI ± SEM of three independent experiments. (*p<0.05, **p<0.01). (D) Western blot analysis was performed on whole cell extracts using for HLA-DRα (TAL 1B5), HLA-DM (TAL18.1) and Ii (LN2); GAPDH (Ab8245) is the protein loading control. Bar graphs show the ratio of band intensities, normalized to GAPDH band intensities and represent the mean ± SEM ratio of three independent experiments: (E) HLA-DRα/GAPDH (F) HLA-DM/GAPDH, and (G) Ii/GAPDH (* p<0.05, ** p<0.01).
Activation of the ERα signaling pathway impedes CIITA expression
Since HLA-II expression is coordinately regulated by CIITA, we predicted that ERα interfered with CIITA expression in ERα-expressing MC2. MC2 and VC5 were pretreated with E2 and/or ICI, as described above, followed by addition of IFN-γ for 24 hours. Western blot analysis of nuclear and cytoplasmic extracts showed inducible CIITA expression in MC2 was about 70% of VC5 levels (Figure 4A & 4B). E2-treatment further reduced CIITA in MC2 while increasing the amount of nuclear ERα; in contrast, ICI reversed the inhibitory effect of E2 on CIITA expression, coincident with ICI-mediated reduced ER levels (Fig 4A Lanes 7 and 8). These results indicated that E2 inhibits HLA-II expression by downregulating CIITA expression.
Figure 4. E2-ERα signaling down regulates CIITA protein and mRNA expression in ER+ BCCL.

VC5 and MC2 cells were cultured in E2-depleted media, treated with vehicle (ethanol), E2 (10−9 M) or/and ICI (10−6 M) and stimulated or not with IFN-γ (100 U/ml) for 24 and 4 hours, for CIITA protein and mRNA expression, respectively. (A) Western blot analysis was performed on cytoplasmic and nuclear extracts for CIITA (antiserum #21) and ERα (HC-20). (B) Cytoplasmic CIITA and nuclear CIITA were normalized to GAPDH and P84 respectively; bar graphs represent the mean ± SEM ratio of three independent experiments (**p<0.01). (C) CIITA mRNA was relatively quantified by real time PCR using Taqman gene expression assay. GAPDH was used as an endogenous control and the data were expressed relative to a control B cell line (RAJI). Bar graphs represent the mean ± SEM of three replicate assays (**p<0.01).
To further determine the inhibitory effect of E2 on CTIIA gene expression, VC5 and MC2 cells were pretreated with E2 and/or ICI for 1 hour and then stimulated with and without IFN-γ for 4 hours, an optimal time for CIITA mRNA expression [50]. CIITA transcription was induced in both VC5 and MC2, but the induction of CIITA mRNA in MC2 was about half in VC5 (Figure 4C). E2 further decreased CIITA mRNA in MC2, while ICI reversed the E2-mediated effect on CIITA.
To confirm the above results, we silenced the ERα transgene in MC2 using ESR1 siRNA and then treated with E2 or vehicle control followed by IFN-γ stimulation for 24 hours. VC5, treated in the same way, was used as a control. Western blot analysis of cell lysates showed ERα was greatly reduced in MC2 transfected with ESR1 siRNA, but not with scrambled siRNA (Figure 5A). Similar to the ICI-mediated effects, ESR1 siRNA clearly reversed the E2-mediated inhibition observed in the scrambled siRNA transfectants. E2 increased CIITA in the ER− VC5, whether transfected with scrambled or ESR1 siRNA. Analysis of CIITA transcripts using real time PCR on siRNA-treated cells (Figure 5B), revealed equivalent levels of CIITA transcripts in ESR1 and scrambled siRNA transfectants; again, ESR1-siRNA abolished the inhibitory effect of E2 on constitutive and induced CIITA transcripts. These results suggest a mechanism whereby E2-activated ER interferes with CIITA transcription induced by IFN-γ in breast cancer cells.
Figure 5. Silencing ERα with ESR1 siRNA reversed the inhibitory effect of E2 on CIITA expression.
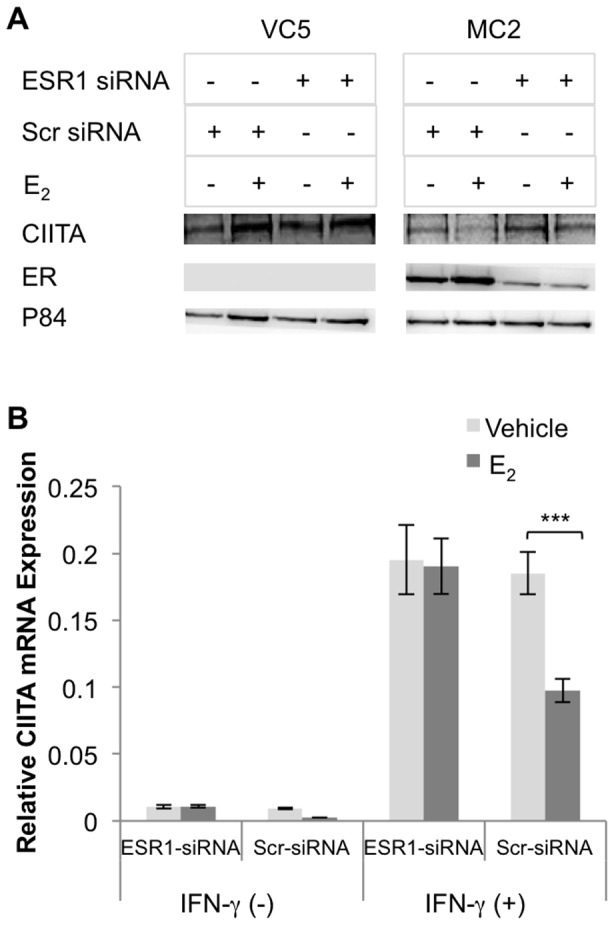
(A) ERα was silenced (ESR1 siRNA) or not (scrambled siRNA) in MC2; VC5 served as an ERα negative cell control. Cells were treated with vehicle (ethanol) or E2 (10−9 M) and stimulated or not with IFN-γ (100 U/ml) for 24 hours. Nuclear lysates were prepared and probed for CIITA (anti-serum #21), ERα (HC-20), and p84. Each figure represents one of three individual experiments. (B) ESR1 siRNA and scrambled siRNA transfected MC2 cells were treated with either vehicle (ethanol) or E2 (10−9 M) followed by stimulation with or without IFN-γ (100 U/ml) for 4 hours and CIITA mRNA was relatively quantified by real time PCR using Taqman gene expression assay. GAPDH was used as an endogenous control and the data were expressed relative to a control B cell line (RAJI). Bar graphs represent the mean ± SEM of three replicate assays (*** p<0.001).
E2 activated ERα inhibits CIITA promoter IV activity
Since IFN-γ inducible HLA-II expression requires activation of CIITA pIV [33], we hypothesized that E2 activation of ERα interferes with CIITA pIV activity. We transfected VC5 and MC2 with a CIITA pIV luciferase construct and treated the cells with E2 and/or ICI, followed by stimulation or not with IFN-γ for 12 hours. E2-treatment further reduced both basal and IFN-γ induced CIITA pIV activity in MC2, while ICI reversed the inhibitory effect of E2 in MC2 cells (Figure 6). Treatment with ICI and/or E2 did not significantly affect constitutive or IFN-γ inducible CIITA pIV activity in VC5.
Figure 6. E2-ERα signaling pathway interferes with CIITA pIV activity in MC2.
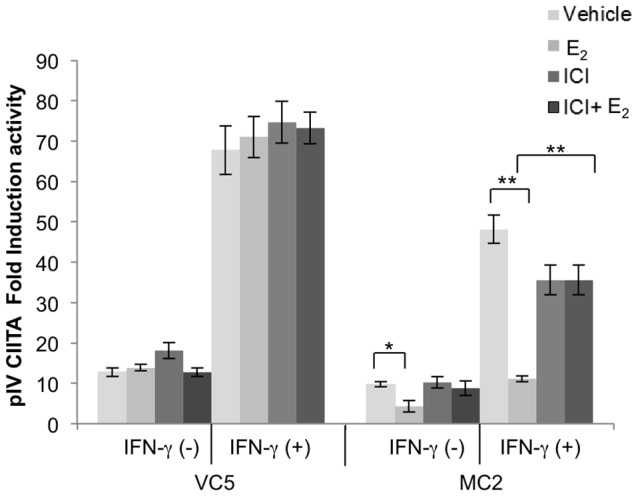
VC5 and MC2 cells were cultured in E2-depleted media followed by transfection with CIITA pIV luciferase constructs. On the following day, cells were treated with vehicle (ethanol), E2 (10−9 M) and/or ICI (10−6 M), and stimulated or not with IFN-γ (100 U/ml) for 12 hours. Data are expressed as fold induction over the PGL2 Basic empty plasmid after controlling for transfection efficiency using cells dual transfected with GFP (Green Florescent Protein). The effect of ERα on the transcription activation of CIITA PIV was determined from relative luciferase activities in transfected MC2. Error bars represent the mean ± SEM of three independent experiments (**p<0.01).
To determine whether E2 directly regulates CIITA pIV activity, we searched for presence of ERE sites using three different computer software programs (http://tfbind.hgc.jp/, http://alggen.1si.upc.es/ and http://www.cbrc.jp/index.eng.html) and identified four putative ERE sites in CIITA pIV (Figure 7A, bold letters in boxes). Sites 1 to 3 are upstream of the STAT1 and IRF1 binding sites. Site 4 is downstream of these sites and precedes the start codon. To determine if either of these sites serves as an ERα repressor of CIITA transcription, three deletion mutant constructs (Site 1/2 deletion mutant, Site 3/4 deletion mutant and Site 1–4 deletion mutant) were created (Figure 7A, open boxes). VC5 and MC2, transfected with one of the mutant CIITA pIV constructs, were pretreated with E2 or vehicle control and then stimulated with IFN-γ for 12 hours, followed by measurement of luciferase activity (Figure 7B, left panel). All three deletion constructs demonstrated significantly reduced IFN-γ stimulated CIITA pIV activity in E2-treated MC2, similar to that observed in MC2 transfected with the wild type CIITA pIV plasmid. By comparison CIITA pIV activity was similar in E2 or vehicle treated VC5 cells whether transfected with wild type or deletion constructs. Intriguingly, constructs Del 3 & 4 and Del 1–4 resulted in dramatic and significant loss of CIITA pIV activity in both cell lines, suggesting there may be other or overlapping sites in CIITA pIV that interact with currently unknown transcription factors for a fully active promoter. Alternatively, the deletion of these sites may have led to the creation of a novel site that has an inhibitory effect on CIITA pIV activity. Importantly, these results do not support the hypothesis that diminished CIITA pIV activity in MC2 treated with E2 occurs via ERE sites in the proximal region of CIITA pIV.
Figure 7. Mutation of putative ERE sites in CIITA pIV does not enhance CIITA pIV activation in MC2.

(A) CIITA pIV nucleotide sequence from −346 to +50 with the GAS and IRF1 binding sites (shaded hexagon) and the predicted ERE (clear rectangles) were identified using online transcription factor prediction software, (http://tfbind.hgc.jp/, http://alggen.lsi.upc.es/ and http://www.cbrc.jp/index.eng.html). Site directed mutagenesis was used to perform deletion of the predicted ERE. (B) VC5 and MC2 were transfected with CIITA pIV constructs, then treated with vehicle (ethanol) or E2 (10−9 M) and stimulated with IFN-γ (100 U/ml) for 12 hours, followed by determination of luciferase activity. Bar graphs represent the mean ± SEM of three independent experiments (**p<0.01, ***p<0.001).
E2-ERα interferes with STAT1 signaling in ERα transfected MC2 cells
To explore whether STAT1 signaling, necessary for activation of CIITA pIV, is adversely affected by ERα activation, we transfected the 8 X GAS luciferase plasmid in VC5 and MC2, followed by treatment, or not, with E2 and/or IFN-γ for 6 hours. Compared to VC5, STAT1 signally was clearly reduced in MC2 (Figure 8A & 8B); moreover, E2 significantly reduced basal and induced GAS promoter activity by about 44% and 40%, respectively, in MC2 (Figure 8B). Although E2 increased basal GAS promoter activity by about 28% in VC5, this was not significant; E2 had no effect on induced activity (Figure 8A).
Figure 8. GAS promoter activity, STAT1 activation and IRF1 expression were reduced in MC2 as compared to VC5.

(A) VC5 and (B) MC2 were cultured in E2-depleted media and transfected with 8 X GAS binding sequence construct, then treated with vehicle (ethanol), E2 (10−9 M) and stimulated or not with IFN-γ (100 U/ml) for 6 hours. Firefly luciferase activities in samples were normalized to Renilla luciferase activities in the same samples and expressed as fold induction over the un-stimulated mock. Error bars represent the mean ± SEM of three independent experiments (*p<0.05, ** p<0.01). (C) VC5 and MC2 were stimulated with IFN-γ (100 U/ml) for 15 minutes, STAT1 activation was detected using STAT1 Phospho-Tyrosine701 and Phospho-Serine 727 antibodies. (D) VC5 and MC2 were treated or not with E2 (10−9 M) for 4 hours, followed by stimulation with IFN-γ (100 U/ml) for 15 minutes, STAT1 activation was detected using STAT1 Phospho-Tyrosine701. (E) Western blot analysis of whole cell lysates, prepared from VC5 and MC2 stimulated with IFN-γ (100 U/ml) for 96 hours, for IRF1 (BD-20) expression. Error bars represent the mean ± SEM of three independent experiments (*p<0.05, *** p<0.001).
To test whether reduced GAS activity in MC2 was the result of reduced pSTAT1, we performed Western blot analysis on lysates from cells treated or not with IFN-γ for 15 minutes. As shown in Figure 8C, total STAT1 and pSTAT1 at tyrosine (Y) 701 and serine (S) 727 were reduced in MC2, compared to VC5. Similar results were observed in an experiment in which cells were also treated with E2 for 4 hours, followed by IFN-γ treatment for 15 minutes; moreover, E2 did not alter levels of phosphorylated or total STAT1 in MC2 or in VC5 (Figure 8D). We next examined IRF1 expression, also essential for CIITA pIV activation, in MC2 and VC5, treated with E2 and stimulated with IFN-γ for 96 hours (Figure 8E). We found IRF1 levels were significantly decreased in MC2, compared to VC5, that E2-treatment had only a trivial effect on IRF1 in MC2, whereas it significantly increased the levels in VC5. Collectively, these results show that ectopic expression of ERα and, moreover, its activation by E2 attenuates STAT1 signaling, however, E2 has only a marginal inhibitory effect on IRF1 levels in MC2. These findings imply that attenuation of CIITA pIV and subsequent reduced HLA-II expression in ERα positive breast cancer may be due to defects in STAT1 regulation.
E2 differentially affects IFN-γ signaling in established ERα+ and ERα− breast cancer cells
To ensure that attenuated STAT1 signaling in MC2 was not merely a peculiarity of the transfected model, we further analyzed GAS promoter activity in endogenously ERα+ BCCL: MCF-7, BT-474 and T47D and ERα− BCCL: MDA-MB-231 and SK-BR-3. E2 significantly decreased IFN-γ induced GAS activity in MCF-7 and BT-474, (Figure 9A & 9B) but not in T47D (Figure 9C). To further confirm the inhibitory effect of E2 on IFN-γ signaling in BCCLs, other than HLA-DR (Figure 1), we conducted Western blot analysis of IFN-γ inducible proteins. These included STAT1, IRF1, IRF9, a member of the IRF family of transcription factors that is not implicated in CIITA expression [51], and gamma-interferon-inducible lysosomal thiol reductase (GILT), a STAT1 regulated but CIITA-independent protein, that is important for antigen processing [52] Basal and IFN-γ inducible STAT1 levels were not substantially altered by E2 in either cell line (Figure 9D–9F); however, STAT1 regulated proteins, IRF1, IRF9 and GILT were differentially modulated in E2-treated MCF-7 and BT-474 (Fig 9D & 9E).
Figure 9. E2 differentially down regulates IFN-γ signaling and IFN-γ induced proteins in endogenous ER+ breast cancer cell lines.

(A) MCF-7, (B) BT-474, (C) T47D, (G) MDA-MD-231, and (H) SK-BR-3 were cultured in E2-depleted media, transfected with 8 X GAS binding sequence construct, then treated with vehicle (ethanol), E2 (10−9 M) and stimulated or not with IFN-γ (100 U/ml) for 6 hours. Firefly luciferase activities in samples were normalized to Renilla luciferase activities in the same samples and expressed as fold induction over the un-stimulated mock. (D) MCF-7, (E) BT-474, (F) T47D, (I) MDA-MB-231 and (J) SK-BR-3 were cultured in E2-depleted media, treated with vehicle (ethanol), or E2 (10−9 M) and stimulated or not with IFN-γ (100 U/ml) for 96 hours. Western blot analysis of cytoplasmic extracts was performed for expression of IFN-γ inducible proteins: STAT1 (06-501), IRF1 (BD-20), IRF9 (C-20), GILT (T-18). Each figure represents one of three independent experiments.
In contrast to the E2-inhibitory effect on GAS promoter activity in the ERα+ lines, E2 noticeably enhanced GAS promoter activity in ERα− BCCL, MDA-MB-231 and SK-BR-3 (Figure 9G & 9H). Furthermore, E2-treatment augmented expression of IRF1 and GILT in MDA-MB-231 cells, and of STAT1 in SK-BR-3 (Figure 9I & 9J). Taken together, the results suggest that E2 differentially modulates the IFN-γ and HLA-II pathways in ERα+ and ERα− BCCL.
Discussion
We previously reported the frequency of HLA-II positive tumor cells in ER+ breast carcinomas is decreased, compared to ER− tumors from younger women [12]. As estrogen levels are high in breast carcinoma tissues, irrespective of age and menopausal status[41], we hypothesized a negative role for estrogen-activated ERα in HLA-II regulation in breast cancer cells. Herein, we provided experimental evidence that ERα and E2-activated ERα attenuate HLA-II expression in BCCL. Using paired ERα (MC2) and vector (VC5) transfected MDA-MB-231 clone 10A cells we showed: i) E2-treatment coordinately decreased IFN-γ inducible HLA-II and CIITA in ERα+ MC2 but not in ERα− VC5; ii) reduction of ERα by ICI or siRNA reversed the E2-inhibitory effect on HLA-II expression, CIITA pIV activity and transcriptional activation of CIITA in MC2; iii) E2-activated ERα adversely affected IFN-γ induced transcription as shown by GAS reporter assay and expression levels of IFN-γ inducible proteins. Importantly, similar results were observed in the ERα+ BCCL, MCF-7 and BT-474, in which GAS activity, STAT1 regulated genes and HLA-DR were down regulated by E2; by contrast, E2 augmented GAS activity and expression of STAT1 regulated genes in the ERα− BCCL, MDA-MB-231 and SK-BR-3.
Overall our data support a negative role for E2-ERα signaling in the regulation of HLA-II in breast cancer cells, but cell-specific differences are evident. For example, E2 treatment attenuated HLA-DR in MCF-7 and BT-474, but not in T47D. This finding is compatible with an older study in which BCCL, cultured in E2-sufficient medium, exhibited a hierarchy of IFN-γ inducible HLA-DR levels with T47D>MCF-7>BT-474 [6]. Differential HLA-II in these cells is not surprising, given that ER+ BCCL, although expressing many of the same genes associated with a luminal subtype, will differ in expression of many other genes [53], which may or may not be regulated by E2. Multiple factors including the ratio and localization of ERα and ERβ receptors, levels of coactivators and corepressors, cell surface receptors such as GPR30 and EGFR and cross-talk with other signaling pathways determine which genes are up or down regulated [54]. E2-activated ERβ inhibits recruitment of ERα to ERE in target genes, thus, suppressing ERα regulated gene expression [55]. Furthermore, activation of the ERβ2 isoform results in ERβ2/ERα heterodimers that are targeted for proteasomal degradation [56]. It is noteworthy, then, that E2 increases ERβ in T47D but not in MCF-7 or BT-474 [57] and the ER β:α ratio in T47D is reported to be greater than in MCF-7[53], [58] thus, suggesting that cell-specific differences in ER subtypes and other receptors may underlie differential HLA expression in breast cancer.
The most convincing evidence that activated ERα modulates HLA-II and CIITA expression came from our experiments using the transfected ERα+ line, MC2. Since MC2 and its ERα− vector control, VC5, are derived from MDA-MB-231 clone 10A, which is negative for both ERα and ERβ[47], it should be a valid model to directly assess the effect of activated ERα on the HLA-II pathway. Our finding, that E2 attenuation of HLA-II and CIITA in MC2 could be reversed by knockdown of ERα in MC2 with ICI (Figures 3D and 4A) or siRNA (Figures 5A), provides compelling evidence that the classical ERα signaling pathway interferes with CIITA regulation. However, we were puzzled that even without adding E2, HLA-II and CIITA were reduced in MC2 and that knockdown of ERα by ICI and siRNA did not restore CIITA activity in MC2 to VC5 levels. Although we used phenol red free medium and E2-depleted FBS, there might still be a minimum level of E2 in the culture medium, which is sufficient to activate ERα and suppress CIITA activity. Furthermore, the incomplete depletion of ERα by ICI or siRNA (Figures 3D, 4A & 5A), may also explain why HLA-II and CIITA expression were not completely restored.
Identification of putative ERE binding sites in the proximal region of CIITA pIV (Figure 7A) led us to explore a direct role for ERα as a suppressor of CIITA pIV activation. Although mutagenesis of these sites did not reverse the inhibitory effect of ERα or E2-activated ERα on CIITA pIV activity (Figure 7B), the experiments do not completely exclude direct ERα suppression of CIITA activity as there may be other unidentified ERE sites in either the proximal or distal region of CIITA pIV through which this effect is mediated. Alternatively, ERα may indirectly suppress CIITA pIV activation through interacting with another factor such as AP1 or NFKβ that may bind CIITA pIV [21], or by interacting with factors such as CREB, SRC-1 and CBP/p300 [59] that interact with the regulatory elements of CIITA pIV and HLA-II promoters [23], [60], [61]. This remains to be further studied.
Although others have shown an E2 inhibitory effect on MHC class II expression [17], [19], [44], [45], the described mechanisms were not CIITA dependent. Tzortzakaki et al (2003) reported E2-inhibition of IFN-γ inducible HLA-DR in both MCF-7 and T47D, whereby the mechanism involved sequestering the steroid receptor co-activator 1 (SRC-1) away from the HLA-DRA promoter by the E2-activated ER [17]. Our study did not assess cofactors, but similarly, we found E2-inhibition of DR expression and DRA promoter activity with only slightly reduced CIITA in MCF-7 (Figure 1 and data not shown). However, our results for T47D conflict with theirs, as we found no E2 inhibition of HLA-DR in this cell line. This could be due to differences in the amounts of E2, as their study used 3–4 log fold more than ours. Higher than physiological concentrations of E2 were also used to show an E2 inhibitory effect on murine MHC-II that did not involve reduced CIITA[45]. Here the E2 inhibitory effect was mediated through reduced association of the histone acetylation transferase, CBP, with the MHC-II promoter. Since CBP is required for acetylation of histones 3 and 4 in the MHC-II promoter, this resulted in decreased transcription of MHC-II. Intriguingly, the cell lines in this study expressed both ER subtypes, which bound to the MHC I-Eβ promoter, but as neither ICI nor tamoxifen reversed the E2 inhibitory effect on MHC-II promoter, they concluded the mechanism was ER-independent. Subsequently, they showed the E2 inhibitory effect on CBP was mediated through E2 activation of JNK MAPK pathway [45]. Although these studies are not directly comparable to ours, they do suggest additional factors may have contributed to E2-inhibition of HLA-DR. However, the underlying mechanisms for E2-ERα inhibition of CIITA transactivation and STAT1 signaling in breast cancer are likely to be more diverse and complex.
Studies investigating deficient CIITA and MHC class II expression in various cancer cell lines have identified epigenetic modifications that result in transcriptional silencing [61], [62]. These include histone deacetylation of the CIITA pIV in squamous cell carcinomas [63] and rhabdomyosarcomas [64], and hypermethylation of the CpG islands in CIITA pIV colon and gastric carcinoma lines. Hypermethylation and recruitment of dysregulated methyltransferases were hypothesized as mechanisms for defective CIITA and HLA-II expression in metastatic breast cancer [65], [66], but these studies were based on a presumed breast cancer cell line MDA-MB-435. This cell line and its metastatic variants have a controversial history [67], as there is strong evidence that they originated from a melanoma cell line [68]. However, it is conceivable that epigenetic modifications are implicated in the E2-liganded ERα deleterious effect on CIITA pIV, as numerous epigenetic modifications have been described in breast cancer that include silencing of ERα in the MDA-MB-231 cell line and downregulation of tumor suppressor genes [69]–[73].
In our study the E2 mediated downregulation of CIITA pIV and HLA-II expression in the ERα+ BCCL appears likely due to aberrant STAT1 signaling with reduced expression of IRF1 or reduced ability to bind the CIITA promoter. Others have shown that STAT1 and IRF1 are aberrantly expressed in some ER+ breast cancer tissues and cell lines [74]–[77] and both have tumor suppressor properties. Chan et al (2012) reported significantly decreased STAT1 in human neoplastic tissue of ER+ breast tumors and showed that knocking out STAT1 in a mouse model correlated with the development of ER+PR+ luminal A adenocarcinoma [77]. Intriguingly, the reduced phosphorylation of STAT1 and reduced levels of total STAT1 in MC2, compared to VC5 (Figure 8C), whether treated or not with E2 (Figure 8D) implies that ERα somehow negatively regulates STAT1 activation and signaling. We speculate this could occur via direct interaction of ERα with STAT1, possibly interfering with dimerization and nuclear translocation or indirectly by interfering with STAT1 promoter activation. Whatever the mechanism, aberrant STAT1 signaling is likely to result in reduced IRF1 levels and subsequently reduced CIITA activation. However, as ICI treatment of MC2 did not substantially increase STAT1 levels (data not shown), nor completely degrade ERα, more studies are required to test this concept.
A potential explanation for the dramatic reduction of CIITA pIV activity in MC2 is decreased IRF1 (Figure 8D), which is essential for IFN-γ inducible CIITA transcriptional activation and HLA-II expression [50], [78], [79]. Furthermore, E2 diminished IRF1 in MCF-7 and dramatically reduced its expression in BT-474, a cell line that expresses insignificant amounts of HLA-DR in the presence and absence of E2 (Figures 1 & 9). In contrast, ERα− lines appear to have an intact IFN-γ signaling pathway that is not inhibited by E2. We did not investigate mechanisms underlying E2-mediated increase in GAS and STAT1 activity, but others have shown a dependency on SRC kinase activity [80]. Furthermore, E2 also activates other pathways such as MAPK and PI3K pathways that interact with the JAK-STAT1 pathway [40], [81], [82].
In conclusion, our results show that HLA-II expression is regulated differently by estrogen in ER− and ER+ breast cancer cells. To our knowledge this report is the first to show that activation of ERα by its ligand E2, results in downregulation of CIITA pIV activity. Although the mechanism is not fully elucidated, the data suggest that the dysregulation occurs at the level of STAT1 activation. Such a mechanism would explain the HLA-DR negative tumor cells in breast carcinomas despite infiltrating T-cells and high levels of IFN-γ and has further implications for tumor immune escape.
Materials and Methods
Cells
Breast cancer cell lines, obtained from ATCC, included: ERα+ (MCF-7, T47D, and BT-474) and ERα− (SK-BR-3, MDA-MB-231 (MDA-231). Cells were grown in Iscove's Modified Dulbecco's Medium (IMDM) (Gibco) supplemented with 10% heat inactivated fetal bovine serum (FBS) (Gibco), 2 mM L-glutamine, antibiotic-antimycotic mixture (100 units/ml penicillin G sodium, 100 µg/ml streptomycin sulfate, and 0.25 µg/ml amphotericin B as Fungizone®), all from Invitrogen. MDA-MB-231 clone 10A and two stably-transfected lines, MC2 (MDA-MB-231 clone 10A transfected with ESR1 (NM_000125) and VC5 (MDA-MB-231 clone 10A transfected with an empty vector) were generous gifts from Dr. Craig Jordan. Cells were grown in phenol red free minimum essential medium (MEM) (Invitrogen) supplemented with 5% charcoal/dextran heat inactivated FBS (CD FBS) (Hyclone), MEM non-essential amino acids, 6 ng/ml recombinant human insulin, 2 mM L-glutamine and antibiotic-antimycotic mixture (all from Invitrogen). MC2 and VC5 were maintained under selective conditions with G418, 5 µg/ml (Sigma). For experiments cell lines were detached with 0.25% trypsin (Invitrogen) and plated at 3×105 cells/well in 6-well plates or 2×104 cells/well in 96-well plates. After 24 hours medium was replaced with fresh medium containing 10−9 M E2 and/or 10−6 M ICI (Sigma) or vehicle control (ethanol) and left un-stimulated or stimulated with IFN-γ, 100 units/ml (BD Biosciences) for the indicated times depending on the experiment.
Antibodies
Expression of HLA-II and CIITA was determined as follows: HLA-DR conformers, clone L243 [83] ATCC, purified IgG2a from supernatant diluted to 2.4 µg/ml for flow cytometry (FC) or 10 ng/ml for Western blot analysis; HLA-DRα, mouse IgG1 (clone Tal 1B5, Abcam, 40 ng/ml, IB); Ii, mouse IgG1 (clone LN2, BD Biosciences, 5 µg/ml, FC or 200 ng/ml, IB); HLA-DM, mouse IgG1 (clone MaP.DM1, BD Biosciences, 10 µg/ml, FC and clone TAL18.1, Abcam, 40 ng/ml, IB); CIITA (rabbit antiserum # 21, diluted 1/4000), prepared in Dr. Viktor Steimle's laboratory [84]. Other antibodies used for Western blotting included anti-ERα, rabbit IgG (HC-20, Santa Cruz Biotechnology, 500 ng/ml); STAT1, rabbit IgG (06-501, Upstate Biotechnology, 200 ng/ml); STAT1 Phospho-Tyrosine701 and Phospho-Serine 727, both rabbit IgG (GenScript, 500 ng/ml); ISGF-3γ p48 (IRF9), rabbit IgG (C-20, Santa Cruz Biotechnology, 400 ng/ml); IRF1, mouse IgG1 (clone BD-20, BD Biosciences, 125 ng/ml); GILT, goat polyclonal IgG (T-18, Santa Cruz Biotechnology, 250 ng/ml). Isotype-matched nonspecific monoclonal antibodies (mAbs) included: IgG2a (clone NSG2a) from a local source and IgG1 (clone MOPC-21, BD Biosciences). Housekeeping proteins were detected with anti-GAPDH, mouse IgG1 (clone 6C5, Abcam, 1 ng/ml); α-tubulin, mouse IgG1 (clone B-7, Santa Cruz Biotechnology, 250 ng/ml) and anti-nuclear matrix protein p84, mouse IgG2b (clone 5E10, Abcam, 1 µg/ml). Horse Radish Peroxidase (HRP)-conjugated affiniPure F(ab)2 fragment goat anti-mouse (GAM) IgG, Fc specific and HRP-conjugated affiniPure F(ab)2 fragment goat anti-rabbit (GAR) IgG, Fc specific antibodies, were purchased from Jackson Immunoresearch and HRP conjugated donkey anti-goat (DAG) antibody IgG, was purchased from Santa Cruz Biotechnology.
Flow cytometry
Flow cytometry was performed as previously described [85]. Briefly, trypsin-harvested cells, 2×105 cells/tube, were incubated with 25 µl of appropriate mAbs in wash buffer (0.2% CDFCS, 0.02% NaN3 in PBS) for 30 minutes at 4°C. Antibody binding was detected with phycoerythrin (PE) labeled goat anti-mouse (GAM) conjugate (Jackson Immunoresearch), followed by fixation in 1.0% paraformaldehyde (PFA) and analyzed using a FACS Calibur flow cytometer (Becton-Dickinson). For intracellular staining, the cells were fixed in 2% PFA and permeabilized with 0.2% Tween 20 in PBS (Sigma) prior to adding primary antibodies, diluted in wash buffer containing 0.2% Tween 20 and 0.5% BSA.
Western Blotting
Nuclear and cytoplasmic extracts were prepared using Nuclear Extract Kit (ActiveMotif) according to the manufacture's protocol. Whole cell lysates (WCL) were prepared in either Triton X-100 buffer (PBS pH 7.4,Triton X-100 1%, 0.5 M ethylene-diaminetetraaccetic acid) or RIPA buffer (PBS, pH7.4, 1% NP40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate) containing protease inhibitors aprotinin (1 µg/ml), leupeptin (1 µg/ml), pepstatin A (1 µg/ml) and phenylmethylsulfonyl fluoride (10 µg/ml). Proteins, quantified using a BCA protein assay kit (Thermo-Fisher Scientific), were reduced with 2-mercaptoethanol and electrophoresed (10 µg/lane) using 8–10% SDS PAGE, followed by western blotting. Membranes, treated with blocking buffer (5% milk powder in TBS-Tween (0.15 M NaCl, 0.05 M Tris pH 7.4, 0.05% Tween 20) for 1 hour, were incubated overnight with primary antibodies at 4°C. Antibody binding was detected with appropriate HRP-conjugated secondary antibodies and Immobilon Western Chemiluminescent HRP substrate (Millipore). Immunoreactivity was visualized and quantified by scanning densitometry using ImageQuant LAS 4000 and ImageQuant TL8.1 software, respectively (GE Healthcare).
Real-time RT-PCR
Total RNA, extracted using TRIzol Reagent (Invitrogen) and treated with Ambion® TURBO™ DNase to remove contaminating DNA, was quantified using NanoDrop (Thermo Scientific). The High Capacity cDNA Reverse Transcription kit (Applied Biosystems) was used for cDNA synthesis according to the manufacturer's protocol. Real time PCR was performed using TaqMan® Probe-Based Gene Expression Analysis kit for CIITA (Hs00172106_m1) and GAPDH (Hs99999905_m1) following the manufacturer's recommendations. Quantification was performed by the comparative threshold cycle (ΔΔCT) method and normalized to GAPDH using StepOnePlus™ (Applied Biosystems). A control sample without RNA and a reference sample (RAJI, B cell line) were included in each experiment.
siRNA Transfection
Cells, plated in a 6-well plate at 3×105 cells/well for 24 hours, were transfected with either 25 nM ON-TARGET plus SMART pool siRNA for ESR1 or non-targeting siRNA (Dharmacon, USA) using 4 µl DharmaFECT4 transfection reagent (Dharmacon, USA) per well according to the manufacturer's protocol. Forty-eight hours later, the cells were treated with E2 10−9 M or vehicle control (ethanol) and stimulated with IFN-γ, 100 units/ml, for 4 or 24 hours for mRNA and protein expression, respectively.
Reporter gene assays
The CIITA promoter IV firefly luciferase construct [79] and the 8 X GAS firefly luciferase construct [86] were kind gifts from Dr. Jenny Ting and Dr. Eleanor N. Fish, respectively. Transfection conditions were optimized using Fugene HD (Roche) transfection reagent according to the manufacturer's protocol: briefly a master mix was prepared by diluting the appropriate plasmid with Opti-MEM (Gibco) to a concentration of 0.02 µg/µl; Fugene HD was added to the same mixture in the ratio of 7∶2 (Fugene HD in µl:Plasmid DNA in µg) and left for 20 minutes at ambient temperature. Cells, plated in a 96-well plate at 2×104 cells/well for 24 hours, at 37°C were transfected with 5 µl of this mixture and incubated for an additional 24 hours. The medium was then replaced with medium containing the appropriate treatments and incubated for 12 hours for CIITA pIV or 6 hours for 8 X GAS constructs. Transfection efficiency was estimated by co-transfecting the cells with SV-40 Renilla luciferase or green fluorescent protein (GFP). Luciferase activity was measured using the dual luciferase assay system (Promega) and a 96-well luminometer (Fluoroskan Ascent Fl, Labsystems).
Generation of CIITA pIV deletion constructs
Different sets of deletion mutants of P-346/+50 CIITA pIV were generated by site-directed mutagenesis using QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions. Mutagen primers (Table 1) were designed with Agilent's web-based QuikChange Primer Design Program Sequences. Sequences, deleted from the original template, are bolded and underlined. All deletions were confirmed by sequencing.
Table 1. Mutagen primers used to generate the CIITA PIV deletion constructs.
| Site 1 | Original template | ctcaacctctctttgtctctgggtgggtccccacccctg |
| Primers | Del −328/−324 Fw | 5′-ttggagagaaacagcacccaggggtggg-3′ |
| Del −328/−324 Rv | 5′-cccacccctgggtgctgtttctctccaa-3′ | |
| Site 2 | Original template | gacgttgagtcctgaacgtctagtgaacgggttcaccgaggga |
| Primers | Del −280/−276 Fw | 5′-caactcaggacttgcacttgcccaagtggctc-3′ |
| Del −280/−276 Rv | 5′-gagccacttgggcaagtgcaagtcctgagttg-3′ | |
| Site 3 | Original template | agaggggcttcaccccgaccggtgacactccttggctgacctccgtccctg |
| Primers | Del −209/−191 Fw | 5′-ccccgaagtgggggactggaggcagg-3′ |
| Del −209/−191 Rv | 5′-cctgcctccagtcccccacttcgggg-3′ | |
| Site 4 | Original template | cttgacgcccctccgcccctccatcctactggtcg cctgctcgacggtgt |
| Primers | Del −33/−14 Fw | 5′-ctgcggggaggcggacgagctgcc-3′ |
| Del −33/−14 Rv | 5′-ggcagctcgtccgcctccccgcag-3′ |
Statistics
Statistical analysis was performed using Microsoft excel 2010 software. One-way analysis of variance (ANOVA) and Tukey post hoc tests were used for comparisons within a group. The student t-test was used for comparing two different treatments for one cell. All tests were two-sided and p<0.05 was considered significant.
Funding Statement
This work was supported by Canadian Institute of Health Research (www.cihr-irsc.gc.ca) grant number ROP 82352 and Canadian Breast Cancer Foundation/Atlantic Chapter (http://www.cbcf.org/atlantic/Pages/default.aspx) grant numbers R08-D11 and R09-F20. AAM was supported by a Health Professionals fellowship from Canadian Institute of Health Research Regional Partnership Program (www.cihr-irsc.gc.ca). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Armstrong TD, Clements VK, Ostrand-Rosenberg S (1998) MHC class II-transfected tumor cells directly present antigen to tumor-specific CD4+ T lymphocytes. J Immunol 160: 661–666. [PubMed] [Google Scholar]
- 2. Meazza R, Comes A, Orengo AM, Ferrini S, Accolla RS (2003) Tumor rejection by gene transfer of the MHC class II transactivator in murine mammary adenocarcinoma cells. Eur J Immunol 33: 1183–1192. [DOI] [PubMed] [Google Scholar]
- 3. Accolla RS, Frangione V, De Lerma Barbaro A, Mortara L (2010) New strategies of mammary cancer vaccination. Breast Journal 16: S42–S44. [DOI] [PubMed] [Google Scholar]
- 4. Accolla RS, Tosi G (2012) Optimal MHC-II-restricted tumor antigen presentation to CD4+ T helper cells: The key issue for development of anti-tumor vaccines. J Transl Med 10: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Collins T, Korman AJ, Wake CT, Boss JM, Kappes DJ, et al. (1984) Immune interferon activates multiple class II major histocompatibility complex genes and the associated invariant chain gene in human endothelial cells and dermal fibroblasts. Proc Natl Acad Sci U S A 81: 4917–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jabrane-Ferrat N, Faille A, Loiseau P, Poirier O, Charron D, et al. (1990) Effect of gamma interferon on HLA class-I and -II transcription and protein expression in human breast adenocarcinoma cell lines. Int J Cancer 45: 1169–1176. [DOI] [PubMed] [Google Scholar]
- 7. Martins I, Deshayes F, Baton F, Forget A, Ciechomska I, et al. (2007) Pathologic expression of MHC class II is driven by mitogen-activated protein kinases. Eur J Immunol 37: 788–797. [DOI] [PubMed] [Google Scholar]
- 8. Warabi M, Kitagawa M, Hirokawa K (2000) Loss of MHC class II expression is associated with a decrease of tumor-infiltrating T cells and an increase of metastatic potential of colorectal cancer: Immunohistological and histopathological analyses as compared with normal colonic mucosa and adenomas. Pathol Res Pract 196: 807–815. [DOI] [PubMed] [Google Scholar]
- 9. Bustin SA, Li SR, Phillips S, Dorudi S (2001) Expression of HLA class II in colorectal cancer: Evidence for enhanced immunogenicity of microsatellite-instability-positive tumours. Tumour Biol 22: 294–298. [DOI] [PubMed] [Google Scholar]
- 10. Concha A, Ruiz-Cabello F, Cabrera T, Nogales F, Collado A, et al. (1995) Different patterns of HLA-DR antigen expression in normal epithelium, hyperplastic and neoplastic malignant lesions of the breast. Eur J Immunogenet 22: 299–310. [DOI] [PubMed] [Google Scholar]
- 11. Oldford SA, Robb JD, Watson PH, Drover S (2004) HLA-DRB alleles are differentially expressed by tumor cells in breast carcinoma. Int J Cancer 112: 399–406. [DOI] [PubMed] [Google Scholar]
- 12. Oldford SA, Robb JD, Codner D, Gadag V, Watson PH, et al. (2006) Tumor cell expression of HLA-DM associates with a Th1 profile and predicts improved survival in breast carcinoma patients. Int Immunol 18: 1591–1602. [DOI] [PubMed] [Google Scholar]
- 13. Cabrera T, Angustias FM, Sierra A, Garrido A, Herruzo A, et al. (1996) High frequency of altered HLA class I phenotypes in invasive breast carcinomas. Hum Immunol 50: 127–134. [DOI] [PubMed] [Google Scholar]
- 14. Calabro A, Beissbarth T, Kuner R, Stojanov M, Benner A, et al. (2009) Effects of infiltrating lymphocytes and estrogen receptor on gene expression and prognosis in breast cancer. Breast Cancer Res Treat 116: 69–77. [DOI] [PubMed] [Google Scholar]
- 15. Sedlak J, Speiser P, Zeillinger R, Krugluger W, Wiltschke C, et al. (1992) Cytokine (IFN-alpha, IFN-gamma, IL-1-alpha, TNF-alpha)-induced modulation of HLA cell surface expression in human breast cancer cell lines. Neoplasma 39: 269–272. [PubMed] [Google Scholar]
- 16. Rohn W, Tang LP, Dong Y, Benveniste EN (1999) IL-1 beta inhibits IFN-gamma-induced class II MHC expression by suppressing transcription of the class II transactivator gene. J Immunol 162: 886–896. [PubMed] [Google Scholar]
- 17. Tzortzakaki E, Spilianakis C, Zika E, Kretsovali A, Papamatheakis J (2003) Steroid receptor coactivator 1 links the steroid and interferon gamma response pathways. Mol Endocrinol 17: 2509–2518. [DOI] [PubMed] [Google Scholar]
- 18. Bernard DJ, Maurizis JC, Chassagne J, Chollet P, Plagne R (1986) Effect of prolactin on class II HLA antigen expression by MCF7 cell line. Anticancer Res 6: 79–83. [PubMed] [Google Scholar]
- 19. Tabibzadeh SS, Sivarajah A, Carpenter D, Ohlsson-Wilhelm BM, Satyaswaroop PG (1990) Modulation of HLA-DR expression in epithelial cells by interleukin 1 and estradiol-17 beta. J Clin Endocrinol Metab 71: 740–747. [DOI] [PubMed] [Google Scholar]
- 20. Maudsley DJ (1991) Role of oncogenes in the regulation of MHC antigen expression. Biochem Soc Trans 19: 291–296. [DOI] [PubMed] [Google Scholar]
- 21. Muhlethaler-Mottet A, Otten LA, Steimle V, Mach B (1997) Expression of MHC class II molecules in different cellular and functional compartments is controlled by differential usage of multiple promoters of the transactivator CIITA. EMBO J 16: 2851–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Setterblad N, Peterlin BM, Andersson G (1997) Role of the X2 box in activated transcription from the DRA promoter in B cells. Immunogenetics 46: 318–325. [DOI] [PubMed] [Google Scholar]
- 23. Moreno CS, Beresford GW, Louis-Plence P, Morris AC, Boss JM (1999) CREB regulates MHC class II expression in a CIITA-dependent manner. Immunity 10: 143–151. [DOI] [PubMed] [Google Scholar]
- 24. Muhlethaler-Mottet A, Krawczyk M, Masternak K, Spilianakis C, Kretsovali A, et al. (2004) The S box of major histocompatibility complex class II promoters is a key determinant for recruitment of the transcriptional co-activator CIITA. J Biol Chem 279: 40529–40535. [DOI] [PubMed] [Google Scholar]
- 25. Ting JP, Trowsdale J (2002) Genetic control of MHC class II expression. Cell 109 Suppl: S21–S33 [DOI] [PubMed] [Google Scholar]
- 26. van der Stoep N, Quinten E, van den Elsen PJ (2002) Transcriptional regulation of the MHC class II trans-activator (CIITA) promoter III: Identification of a novel regulatory region in the 5′-untranslated region and an important role for cAMP-responsive element binding protein 1 and activating transcription factor-1 in CIITA-promoter III transcriptional activation in B lymphocytes. J Immunol 169: 5061–5071. [DOI] [PubMed] [Google Scholar]
- 27. Otten LA, Steimle V, Bontron S, Mach B (1998) Quantitative control of MHC class II expression by the transactivator CIITA. Eur J Immunol 28: 473–478. [DOI] [PubMed] [Google Scholar]
- 28. Masternak K, Barras E, Zufferey M, Conrad B, Corthals G, et al. (1998) A gene encoding a novel RFX-associated transactivator is mutated in the majority of MHC class II deficiency patients. Nat Genet 20: 273–277. [DOI] [PubMed] [Google Scholar]
- 29. Zhu XS, Linhoff MW, Li G, Chin KC, Maity SN, et al. (2000) Transcriptional scaffold: CIITA interacts with NF-Y, RFX, and CREB to cause stereospecific regulation of the class II major histocompatibility complex promoter. Mol Cell Biol 20: 6051–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hake SB, Tobin HM, Steimle V, Denzin LK (2003) Comparison of the transcriptional regulation of classical and non-classical MHC class II genes. Eur J Immunol 33: 2361–2371. [DOI] [PubMed] [Google Scholar]
- 31. Steimle V, Otten LA, Zufferey M, Mach B (1993) Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell 75: 135–146. [PubMed] [Google Scholar]
- 32. Reith W, Mach B (2001) The bare lymphocyte syndrome and the regulation of MHC expression. Annu Rev Immunol 19: 331–373 10.1146/annurev.immunol.19.1.331. [DOI] [PubMed] [Google Scholar]
- 33. Muhlethaler-Mottet A, Di BW, Otten LA, Mach B (1998) Activation of the MHC class II transactivator CIITA by interferon-gamma requires cooperative interaction between Stat1 and USF-1. Immunity 8: 157–166. [DOI] [PubMed] [Google Scholar]
- 34. Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW (2002) Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 285: 1–24. [DOI] [PubMed] [Google Scholar]
- 35. Ahmed CM, Johnson HM (2006) IFN-gamma and its receptor subunit IFNGR1 are recruited to the IFN-gamma-activated sequence element at the promoter site of IFN-gamma-activated genes: Evidence of transactivational activity in IFNGR1. J Immunol 177: 315–321. [DOI] [PubMed] [Google Scholar]
- 36. Sadzak I, Schiff M, Gattermeier I, Glinitzer R, Sauer I, et al. (2008) Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc Natl Acad Sci U S A 105: 8944–8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marino M, Galluzzo P, Ascenzi P (2006) Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics 7: 497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ramsauer K, Sadzak I, Porras A, Pilz A, Nebreda AR, et al. (2002) p38 MAPK enhances STAT1-dependent transcription independently of ser-727 phosphorylation. Proc Natl Acad Sci U S A 99: 12859–12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Giroux M, Schmidt M, Descoteaux A (2003) IFN-gamma-induced MHC class II expression: Transactivation of class II transactivator promoter IV by IFN regulatory factor-1 is regulated by protein kinase C-alpha. J Immunol 171: 4187–4194. [DOI] [PubMed] [Google Scholar]
- 40. Hardy PO, Diallo TO, Matte C, Descoteaux A (2009) Roles of phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase in the regulation of protein kinase C-alpha activation in interferon-gamma-stimulated macrophages. Immunology 128: e652–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Simpson ER (2003) Sources of estrogen and their importance. J Steroid Biochem Mol Biol 86: 225–230. [DOI] [PubMed] [Google Scholar]
- 42. Andersen P, Pedersen MW, Woetmann A, Villingshoj M, Stockhausen MT, et al. (2008) EGFR induces expression of IRF-1 via STAT1 and STAT3 activation leading to growth arrest of human cancer cells. Int J Cancer 122: 342–349. [DOI] [PubMed] [Google Scholar]
- 43. Levin ER (2003) Bidirectional signaling between the estrogen receptor and the epidermal growth factor receptor. Mol Endocrinol 17: 309–317. [DOI] [PubMed] [Google Scholar]
- 44. Adamski J, Ma Z, Nozell S, Benveniste EN (2004) 17beta-Estradiol inhibits class II major histocompatibility complex (MHC) expression: influence on histone modifications and cbp recruitment to the class II MHC promoter. Mol Endocrinol 18: 1963–1974. [DOI] [PubMed] [Google Scholar]
- 45. Adamski J, Benveniste EN (2005) 17beta-estradiol activation of the c-jun N-terminal kinase pathway leads to down-regulation of class II major histocompatibility complex expression. Mol Endocrinol 19: 113–124. [DOI] [PubMed] [Google Scholar]
- 46. Pietras RJ, Marquez-Garban DC (2007) Membrane-associated estrogen receptor signaling pathways in human cancers. Clin Cancer Res 13: 4672–4676. [DOI] [PubMed] [Google Scholar]
- 47. Tonetti DA, Rubenstein R, DeLeon M, Zhao H, Pappas SG, et al. (2003) Stable transfection of an estrogen receptor beta cDNA isoform into MDA-MB-231 breast cancer cells. J Steroid Biochem Mol Biol 87: 47–55. [DOI] [PubMed] [Google Scholar]
- 48. Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ (1997) Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 45: 607–617. [DOI] [PubMed] [Google Scholar]
- 49. Filardo EJ (2002) Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: A novel signaling pathway with potential significance for breast cancer. J Steroid Biochem Mol Biol 80: 231–238. [DOI] [PubMed] [Google Scholar]
- 50. Morris AC, Beresford GW, Mooney MR, Boss JM (2002) Kinetics of a gamma interferon response: Expression and assembly of CIITA promoter IV and inhibition by methylation. Mol Cell Biol 22: 4781–4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Morrow AN, Schmeisser H, Tsuno T, Zoon KC (2011) A novel role for IFN-stimulated gene factor 3II in IFN-gamma signaling and induction of antiviral activity in human cells. J Immunol 186: 1685–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. O'Donnell PW, Haque A, Klemsz MJ, Kaplan MH, Blum JS (2004) Cutting edge: Induction of the antigen-processing enzyme IFN-gamma-inducible lysosomal thiol reductase in melanoma cells is STAT1-dependent but CIITA-independent. J Immunol 173: 731–735. [DOI] [PubMed] [Google Scholar]
- 53. Aka JA, Lin SX (2012) Comparison of functional proteomic analyses of human breast cancer cell lines T47D and MCF7. PLoS One 7: e31532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Renoir JM (2012) Estradiol receptors in breast cancer cells: Associated co-factors as targets for new therapeutic approaches. Steroids 77: 1249–1261. [DOI] [PubMed] [Google Scholar]
- 55. Matthews J, Gustafsson JA (2006) Estrogen receptor and aryl hydrocarbon receptor signaling pathways. Nucl Recept Signal 4: e016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhao C, Matthews J, Tujague M, Wan J, Strom A, et al. (2007) Estrogen receptor beta2 negatively regulates the transactivation of estrogen receptor alpha in human breast cancer cells. Cancer Res 67: 3955–3962. [DOI] [PubMed] [Google Scholar]
- 57. Vladusic EA, Hornby AE, Guerra-Vladusic FK, Lakins J, Lupu R (2000) Expression and regulation of estrogen receptor beta in human breast tumors and cell lines. Oncol Rep 7: 157–167. [DOI] [PubMed] [Google Scholar]
- 58. Sastre-Serra J, Nadal-Serrano M, Pons DG, Valle A, Oliver J, et al. (2012) The effects of 17beta-estradiol on mitochondrial biogenesis and function in breast cancer cell lines are dependent on the ERalpha/ERbeta ratio. Cell Physiol Biochem 29: 261–268. [DOI] [PubMed] [Google Scholar]
- 59. Heldring N, Pike A, Andersson S, Matthews J, Cheng G, et al. (2007) Estrogen receptors: How do they signal and what are their targets. Physiol Rev 87: 905–931. [DOI] [PubMed] [Google Scholar]
- 60. Zika E, Fauquier L, Vandel L, Ting JP (2005) Interplay among coactivator-associated arginine methyltransferase 1, CBP, and CIITA in IFN-gamma-inducible MHC-II gene expression. Proc Natl Acad Sci U S A 102: 16321–16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wright KL, Ting JP (2006) Epigenetic regulation of MHC-II and CIITA genes. Trends Immunol 27: 405–412. [DOI] [PubMed] [Google Scholar]
- 62. Holling TM, van Eggermond MC, Jager MJ, van den Elsen PJ (2006) Epigenetic silencing of MHC2TA transcription in cancer. Biochem Pharmacol 72: 1570–1576. [DOI] [PubMed] [Google Scholar]
- 63. Kanaseki T, Ikeda H, Takamura Y, Toyota M, Hirohashi Y, et al. (2003) Histone deacetylation, but not hypermethylation, modifies class II transactivator and MHC class II gene expression in squamous cell carcinomas. J Immunol 170: 4980–4985. [DOI] [PubMed] [Google Scholar]
- 64. Londhe P, Zhu B, Abraham J, Keller C, Davie J (2012) CIITA is silenced by epigenetic mechanisms that prevent the recruitment of transactivating factors in rhabdomyosarcoma cells. Int J Cancer 131: e437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shi B, Vinyals A, Alia P, Broceno C, Chen F, et al. (2006) Differential expression of MHC class II molecules in highly metastatic breast cancer cells is mediated by the regulation of the CIITA transcription implication of CIITA in tumor and metastasis development. Int J Biochem Cell Biol 38: 544–562. [DOI] [PubMed] [Google Scholar]
- 66. Truax AD, Thakkar M, Greer SF (2012) Dysregulated recruitment of the histone methyltransferase EZH2 to the class II transactivator (CIITA) promoter IV in breast cancer cells. PLoS One 7: e36013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lacroix M (2008) Persistent use of “false” cell lines. Int J Cancer 122: 1–4. [DOI] [PubMed] [Google Scholar]
- 68. Rae JM, Creighton CJ, Meck JM, Haddad BR, Johnson MD (2007) MDA-MB-435 cells are derived from M14 melanoma cells—a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res Treat 104: 13–19. [DOI] [PubMed] [Google Scholar]
- 69. Zhou Q, Shaw PG, Davidson NE (2009) Epigenetics meets estrogen receptor: regulation of estrogen receptor by direct lysine methylation. Endocr Relat Cancer 16: 319–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jang ER, Lim SJ, Lee ES, Jeong G, Kim TY, et al. (2004) The histone deacetylase inhibitor trichostatin A sensitizes estrogen receptor alpha-negative breast cancer cells to tamoxifen. Oncogene 23: 1724–1736. [DOI] [PubMed] [Google Scholar]
- 71. Zhou Q, Shaw PG, Davidson NE (2009) Inhibition of histone deacetylase suppresses EGF signaling pathways by destabilizing EGFR mRNA in ER-negative human breast cancer cells. Breast Cancer Res Treat 117: 443–451. [DOI] [PubMed] [Google Scholar]
- 72. Bayliss J, Hilger A, Vishnu P, Diehl K, El-Ashry D (2007) Reversal of the estrogen receptor negative phenotype in breast cancer and restoration of antiestrogen response. Clin Cancer Res 13: 7029–7036. [DOI] [PubMed] [Google Scholar]
- 73. Duong V, Licznar A, Margueron R, Boulle N, Busson M, et al. (2006) ERalpha and ERbeta expression and transcriptional activity are differentially regulated by HDAC inhibitors. Oncogene 25: 1799–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhu Y, Singh B, Hewitt S, Liu A, Gomez B, et al. (2006) Expression patterns among interferon regulatory factor-1, human X-box binding protein-1, nuclear factor kappa B, nucleophosmin, estrogen receptor-alpha and progesterone receptor proteins in breast cancer tissue microarrays. Int J Oncol 28: 67–76. [PubMed] [Google Scholar]
- 75. Bi X, Hameed M, Mirani N, Pimenta EM, Anari J, et al. (2011) Loss of interferon regulatory factor 5 (IRF5) expression in human ductal carcinoma correlates with disease stage and contributes to metastasis. Breast Cancer Res 13: R111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schwartz JL, Shajahan AN, Clarke R (2011) The role of interferon regulatory factor-1 (IRF1) in overcoming antiestrogen resistance in the treatment of breast cancer. Int J Breast Cancer 2011: 912102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chan SR, Vermi W, Luo J, Lucini L, Rickert C, et al. (2012) STAT1-deficient mice spontaneously develop estrogen receptor alpha-positive luminal mammary carcinomas. Breast Cancer Res 14: R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lee YJ, Benveniste EN (1996) Stat1 alpha expression is involved in IFN-gamma induction of the class II transactivator and class II MHC genes. J Immunol 157: 1559–1568. [PubMed] [Google Scholar]
- 79. Piskurich JF, Linhoff MW, Wang Y, Ting JP (1999) Two distinct gamma interferon-inducible promoters of the major histocompatibility complex class II transactivator gene are differentially regulated by STAT1, interferon regulatory factor 1, and transforming growth factor beta. Mol Cell Biol 19: 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kennedy AM, Shogren KL, Zhang M, Turner RT, Spelsberg TC, et al. (2005) 17beta-estradiol-dependent activation of signal transducer and activator of transcription-1 in human fetal osteoblasts is dependent on src kinase activity. Endocrinology 146: 201–207. [DOI] [PubMed] [Google Scholar]
- 81. Gough DJ, Sabapathy K, Ko EY, Arthur HA, Schreiber RD, et al. (2007) A novel c-jun-dependent signal transduction pathway necessary for the transcriptional activation of interferon gamma response genes. J Biol Chem 282: 938–946. [DOI] [PubMed] [Google Scholar]
- 82. Johnson HM, Noon-Song E, Ahmed CM (2011) Controlling nuclear jaks and stats for specific gene activation by ifn gamma and other cytokines: A possible steroid-like connection. J Clin Cell Immunol 2: 1000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lampson LA, Levy R (1980) Two populations of ia-like molecules on a human B cell line. J Immunol 125: 293–299. [PubMed] [Google Scholar]
- 84. Camacho-Carvajal MM, Klingler S, Schnappauf F, Hake SB, Steimle V (2004) Importance of class II transactivator leucine-rich repeats for dominant-negative function and nucleo-cytoplasmic transport. Int Immunol 16: 65–75. [DOI] [PubMed] [Google Scholar]
- 85. Spurrell DR, Oldford SA, Frost T, Larsen B, Codner D, et al. (2004) Discordant expression of HLA class II-associated co-chaperones and HLA-DRB alleles in cultured fibroblast-like synoviocytes. Hum Immunol 65: 1516–1529. [DOI] [PubMed] [Google Scholar]
- 86. Kaur S, Uddin S, Platanias LC (2005) The PI3' kinase pathway in interferon signaling. J Interferon Cytokine Res 25: 780–787. [DOI] [PubMed] [Google Scholar]