

Abstract
The effects of controlled release on immune response to an immunomodulating peptide were evaluated in a murine experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis (MS). The peptide, Ac-PLP-BPI-NH2-2 (Ac-HSLGKWLGHPDKF-(AcpGAcpGAcp)2-ITDGEATDSG-NH2; Ac = acetyl, Acp = aminocaproic acid) was designed to suppress T-cell activation in response to PLP139–151, an antigenic peptide in MS. Poly-lactide-co-glycolide (PLGA) microparticles containing Ac-PLP-BPI-NH2-2 (8±4 μm, 1.4±0.2% (w/w)) were prepared by a powder-in oil-in water emulsion-solvent evaporation method, sterilized and administered subcutaneously (s.c.) to SJL/J (H-2s) mice in which EAE had been induced by immunization with PLP139–151. Treatment groups received Ac-PLP-BPI-NH2-2: (i) in solution by repeated i.v. or s.c. injection, (ii) in solution co-administered with blank PLGA microparticles, (iii) in solution co-administered with Ac-PLP-BPI-NH2-2 loaded microparticles, and (iv) as Ac-PLP-BPI-NH2-2 loaded microparticles. Administration of Ac-PLP-BPI-NH2-2 as an s.c. solution produced clinical scores and maintenance of body weight comparable to i.v. solution, but with reduced overall survival, presumably due to anaphylaxis. Administration as s.c. microparticles provided a somewhat less effective reduction in symptoms but with no toxicity during treatment. Thus, the results suggest that s.c. administration of Ac-PLP-BPI-NH2-2 microparticles can provide pharmacological efficacy and reduction in dosing frequency without increased toxicity.
Keywords: PLP-BPI peptides; Poly(D,L-lactide); Microparticles; EAE
1. Introduction
Autoimmune diseases such as Type 1 diabetes (T1D), multiple sclerosis (MS) and rheumatoid arthritis (RA) are the result of an inappropriate immune response to endogenous proteins or protein fragments, which serve as autoantigens. A critical component of this response is the activation of T-cells that recognize a particular autoantigen. T-cell activation involves uptake of the antigenic protein by antigen presenting cells (APCs), which digest the protein into peptide fragments. The fragments are then “presented” on the APC cell surface bound to major histocompatibility complex (MHC) molecules. Binding of the T-cell receptor to these MHC-bound peptides constitutes the primary signal (“Signal 1”) for T-cell activation. A second, co-stimulatory signal (“Signal 2”) is also required, and involves higher affinity binding of adhesion molecules that bridge the T-cell and APC. The clustering of these two types of binding pairs and the subsequent translocation of the resulting “immunological synapse” provide the signal for clonal expansion of the T-cell line, ultimately leading to the autoimmune response [1-3].
Interfering with T-cell activation is a rational strategy for preventing and/or treating autoimmune disease. One of us (TJS) has developed a class of novel bifunctional peptide inhibitors (BPI) of T-cell activation, in which a peptide antigen involved in Signal-1 interactions is covalently linked to a second peptide that binds a Signal-2 adhesion molecule. For example, a BPI for preventing T-cell activation in experimental autoimmune encephalomyelitis (EAE), a murine model for MS, has the amino acid sequence HSLGKWLGHPDKF-AcpGAcpGAcp-ITDGEATDSG. The peptide HSLGKWLGHPDKF is an epitope of proteolipid protein (PLP139–151), an antigenic peptide in MS that is involved in Signal-1 interactions. The peptide ITDGEATDSG binds intercellular adhesion molecule 1 (ICAM-1), blocking its binding with lymphocyte function-associated antigen 1 (LFA-1), a Signal-2 interaction. The flexible linker AcpGAcpGAcp (Acp = aminocaproic acid) connects the two peptides and provides appropriate spacing between them. The resulting composite peptide, termed PLP-BPI, thus is designed to prevent T-cell activation by interfering with both Signal-1 and Signal-2, and by preventing translocation of the immunological synapse. Previous studies in an EAE mouse model have demonstrated that PLP-BPI reduces disease incidence and symptom severity when given prophylactically [4], and provides amelioration of symptoms when given therapeutically [5]. Specifically, following immunization with PLP on Day 0 to induce EAE, administration of 100 nmol of PLP-BPI on days 4, 7, 10 and 14 provided prophylactic suppression of EAE symptoms and incidence [4]. Similarly, i.v. administration of 100 nmol of PLP-BPI on three consecutive days following onset of symptoms showed therapeutic suppression of disease [5]. Promising results have also been obtained with a BPI peptide in an NOD mouse model of T1D [6]. Thus, BPI peptides show promise as vaccines for preventing autoimmune disease. However, unlike conventional vaccines which stimulate immune response to an administered antigen, BPI peptides act by suppressing the immune response and promoting immunotolerance.
The need for repeated i.v. administration of PLP-BPI suggests that implantable or injectable controlled release formulations may be warranted. Controlled release has long been used to provide more convenient dosing for drugs that must be administered frequently in other forms. A growing literature also suggests that controlled release of antigens from polymeric materials may be particularly effective in promoting tolerance. The maintenance of immunotolerance depends on antigen persistence in vivo, and is reversible if the antigen source is removed [7], a finding that suggests that sustained release of antigen may be effective in maintaining tolerance. Antigen release from polymeric materials has also been shown to alter the Th1/Th2 balance, in some cases promoting the tolerogenic Th2-like profile [8-10].
On the other hand, changes in the route of administration and the use of solid or particulate dosage forms may stimulate the immune response. For example, the immune response to a hepatitis B vaccine is influenced by the route of administration [11], and greater immune responses are often observed following s.c. or i.m. administration than by the i.v. route [12,13]. Micro- or nanoparticulate dosage forms may be selectively targeted to APCs [14-16], so that the peptide drug is digested and presented by the MHC complex rather than released intact. Poly-lactide-co-glycolide (PLGA) and other polymers used in controlled release devices may have adjuvant effects, recruiting APCs to the injection site and stimulating their maturation [7,10]. Thus, controlled release formulations of PLP-BPI may alter not only the frequency and route of administration, but also the extent and type of therapeutic response, so that their performance is not easily predicted a priori.
This report describes the development and evaluation of a PLGA microparticle formulation of Ac-PLP-BPI-NH2-2, a variant of PLP-BPI with the sequence Ac-HSLGKWLGHPDKF-(AcpGAcpGAcp)2-ITDGEATDSGNH2 (Ac = acetyl, Acp = aminocaproic acid). Microparticles were prepared by a suspension/solvent evaporation method and characterized with regard to particle size and morphology, peptide loading, residual solvent content and in vitro release rate. The Ac-PLP-BPI-NH2-2 loaded microparticles were then administered by s.c. injection in an EAE mouse model, and compared to a variety of controls for their ability to provide prophylactic suppression of disease symptoms. The results demonstrate that a single s.c. microparticle dose provides disease suppression comparable to an identical dose administered as four divided i.v. injections. The results also indicate an effect of route of administration on the therapeutic response and toxicity of Ac-PLP-BPI-NH2-2.
2. Materials and methods
2.1. Materials
Poly(lactic- co-glycolic) acid was purchased from Durect Corporation (PLGA, 50:50, inherent viscosity of 0.15–0.25 dL/g, MW ~10,000; Cupertino, CA), and polyvinyl alcohol (PVA, MW 13,000–23,000, 98% hydrolyzed) was purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals or solvents are of analytical grade or better, and were used as received.
2.2. Peptide synthesis
PLP and Ac-PLP-BPI-NH2-2 peptides were synthesized using 9-fluorenylmethyloxycarbonyl- (FMOC) protected amino acid chemistry on appropriate polyethylene glycol-polystyrene resins (Applied Biosystems, Foster City, CA) using an automated peptide synthesis system (Pioneer; Perceptive Biosystems, Framingham, MA), as described previously [4,5]. All amino acids and aminocaproic acid were supplied by Peptide International (Kentucky, US). Cleavage of the peptides from the resin and removal of the protecting groups from the side chain were carried out using trifluoroacetic acid with scavengers. The crude peptides were purified by reversed-phase HPLC (Rainin, USA) using a C18 column with a gradient of solvent A [95%/5%, H2O (0.1% trifluoroacetic acid)/acetonitrile] and solvent B (100% acetonitrile). The purity of the peptide was analyzed by analytical HPLC (Rainin, USA) using a C18 column. The mass of the synthesized peptide was confirmed by matrix-assisted laser desorption ionization/time of flight (MALDI/TOF) mass spectrometry; the theoretical molecular weight of the C-terminally acetylated peptide is (i.e., Ac-PLP-BPI-NH2-2) is 3416.950.
2.3. Preparation of Ac-PLP-BPI-NH2-2 peptide-loaded microparticles
Ac-PLP-BPI-NH2-2 peptide-loaded microparticles were prepared by a powder-in oil-in water emulsion-solvent evaporation method, using a method similar to that reported by Kim and Burgess [17]. Briefly, 1.2 g of PLGA was added to 8 ml of methylene chloride (CH2Cl2) and the polymer was allowed to dissolve completely. 200 mg of Ac-PLP-BPI-NH2-2 peptide powder was then dispersed into the polymer solution and mixed using a homogenizer (Power Gen 500, Fisher Scientific) for 2 min at 7000 rpm, followed by the addition of 50 ml of a 1% PVA solution and homogenization for another 2 min at the same speed. The final emulsion was poured into 150 ml of 1% PVA solution and stirred for 3 h to evaporate the solvent. The microparticles were then harvested by centrifugation and washed three times with distilled water. The final dry microparticle product was obtained by lyophilization (Virtis, Gardiner, NY, USA). After pre-freezing at −70 °C overnight, the microparticles were lyophilized using the following protocol: −30 °C for 900 min, −15 °C for 480 min, −5 °C for 240 min, 5 °C for 240 min, 15 °C for 120 min, and 25 °C for an additional 120 min to complete dehydration. The products were then stored in sealed containers at −20 °C until further use. This protocol was also used to prepare empty microparticles, except that the polymer solution was directly emulsified with the 1% PVA solution without the addition of the Ac-PLP-BPI-NH2-2 peptide.
2.4. Characterization of microparticles
Peptide-loaded and blank microparticles were characterized to determine surface morphology, peptide content and residual methylene chloride content. In vitro peptide release was also quantified to establish that the microparticle formulation provides controlled release. Methods are described below.
2.4.1. Microparticle surface morphology and size distribution
The surface morphology of microparticles was evaluated by scanning electron microscopy (SEM, D. Leo Field Emission Scanning Electron Microscope, Hitachi S-4500, Tokyo, Japan) after sputter-coating the samples with a 5 nm layer of Au/Pd. The microsphere size distribution was determined from at least 500 particles per batch from SEM images using the public domain image processing software, ImageJ [18].
2.4.2. Peptide content
A ~25 mg sample of microparticles was dissolved in 2 ml of CH2Cl2. The peptide was extracted by addition of 3 ml of pH 13 solution (KCl/NaOH) three times. The extracts were combined and assayed by HPLC using a C18 column with a gradient of solvent A [95%/5%, H2O (0.1% trifluoroacetic acid)/acetonitrile] and solvent B (100% acetonitrile) at a flow rate of 1 ml/min with UV detection at 215 nm. Under these conditions, the retention time of the Ac-PLP-BPI-NH2-2 peptide is ~12.5 min and a calibration prepared using standards of known concentration is linear in the concentration range 10–100 μg/ml (r2≥0.995).
2.4.3. Residual CH2Cl2 content
Residual CH2Cl2 in the dry microparticles was determined by gas chromatography/mass spectrometry (GC/MS). Data were collected on an Agilent 6890N Gas Chromatrograph interfaced with a quadrupole mass analyzer (Quattro Micro GC, Waters Corp., Milford, MA). A 5% phenyl, methyl silicone stationary phase (HP-5MS) in a 15 m column with a 0.25″ ID was used. The carrier gas was He using constant flow control set to 1.5 ml/min. To prepare samples for analysis, ~25 mg of microparticles were dissolved in 0.5 ml of tetrahydrofuran (THF). 1.0 μl aliquots were then injected into the injector port (240 °C) using a 25:1 split ratio. The GC thermal gradient was set at an initial 35 °C with a 2 min hold, after which the temperature was increased 50 °C/min to a final temperature of 180 °C and held for 2 min. Ionization was by electron impact at 70 eV. The analyzers were tuned to 0.6 full-width at the half-height (i.e., 0.6 FWHH) and data were collected in the centroid mode. Signals were recorded as selected ion chromatographs for m/z=71, 72 (THF) and m/z=84, 86 (CH2Cl2) using a target dwell time of 60 ms for m/z=84 and 10 ms for m/z=71, 72. Standard solutions of CH2Cl2 were prepared by adding known amounts of CH2Cl2 to THF. Under these conditions, the calibration is linear in the concentration range 300–1500 ppm (r2≥0.985).
2.4.4. In vitro peptide release
Approximately 30 mg of Ac-PLP-BPI-NH2-2 loaded microparticles was weighed and placed in 1.5 ml of buffered medium (pH 7.4 PBS+0.05% Tween-20) in capped vials. The vials were then placed in a 37±0.1 °C incubator and the rotation speed was set at 50 rpm. A sample (0.5 ml) was taken after centrifugation at the various times up to 42 days. The volume removed was replaced with fresh medium, and the amount of peptide released was determined by the HPLC method described above.
2.5. In vivo microparticle performance in an EAE mouse model
2.5.1. Mice
Female SJL/J (H-2s) mice were purchased from Charles River (Wilmington, MA), and housed under specific pathogen-free conditions at an AAALAC-approved facility at the University of Kansas. All protocols involving live mice were approved by the University’s Institutional Animal Care and Use Committee and are in compliance with the committee’s recommendations.
2.5.2. Induction of EAE and prophylactic suppression by Ac-PLP-BPI-NH2-2
Five- to 7-week-old SJL/J female mice were randomly divided into six groups, with ten mice in each group. All mice were immunized with PLP to induce EAE, as reported previously [4,5]. Immunization involved s.c. administration of 200 μg of PLP139–151 in a 0.2 ml emulsion composed of equal volumes of phosphate-buffered saline (PBS) and complete Freund’s adjuvant (CFA) containing killed Mycobacterium tuberculosis strain H37RA (final H37RA concentration 4 mg/ml; Difco, Detroit, MI). The PLP139–151/CFA solution was administered to four separate regions above the shoulder and on the flanks, with 50 μl injected at each site. In addition, 200 ng of pertussis toxin (PT; List Biological Laboratories Inc., Campbell, CA) was injected i.p. on the day of immunization (Day 0) and 2 days after immunization (Day 2). PT facilitates the development of EAE in this model, an effect that has been attributed to PT-induced activation of APCs [19].
The ability of various formulations of Ac-PLP-BPI-NH2-2 to suppress the development and progression of EAE was investigated using six treatment groups (Table 1, Phase I). Following initial immunization on Day 0 and pertussis toxin boosting on Day 2, treatments were administered on Days 4, 7, 10 and 14 (Table 1). Mice in Group 1 received four equal doses of 100 μL of PBS by i.v. injection, and served as a negative control. Mice in Group 2 received four i.v. injections of 100 nmol Ac-PLP-BPI-NH2-2 peptide in PBS, with a dose and route of administration used in previous studies of this peptide [4,5]. Mice in Group 3 received four s.c. injections of 100 nmol Ac-PLP-BPI-NH2-2 peptide. Comparison of Group 3 results with those of Group 2 is expected to provide information on route of administration effects. Mice in Group 4 received four s.c. injections of 100 nmol of Ac-PLP-BPI-NH2-2 peptide, with concomitant administration of 25 mg of blank PLGA microparticles. Comparison of results for Groups 3 and 4 is expected to identify possible adjuvant effects of blank microparticles. Mice in Groups 5 and 6 received microparticles loaded with the Ac-PLP-BPI-NH2-2 peptide. Those in Group 5 received an initial 100 nmol solution dose on Day 4, followed by a 300 nmol microparticle dose (~75 mg microparticles containing 300 nm Ac-PLP-BPI-NH2-2) on Day 7. Mice in Group 6 received a 400 nmol dose of peptide in microparticle form (~100 mg microparticles) on Day 4. Comparison of results for Groups 5 and 6 with solution treatments (Groups 2–4) addresses the ability of controlled peptide release to achieve prophylactic effects comparable to repeated solution injections. All six of these treatment groups received a total of 400 nmol Ac-PLP-BPI-NH2-2 peptide/mouse, and all microparticle formulations (i.e., Groups 4–6) were sterilized by overnight exposure to u.v. radiation in a clean bench prior to administration. Follow-up studies were conducted at a lower dose of 300 nmol Ac-PLP-BPI-NH2-2 peptide/mouse (Groups 7–9, Table 1).
Table 1.
Treatment schedule for SLJ mice receiving Ac-PLP-BPI-NH2-2 peptide in solution and microparticle formulations.
| Treatment schedule1
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Immunization phase | Treatment phase | Ana. phase2 | |||||||||
|
| |||||||||||
| Group | n | Treatment | Route | Dose3 | Day 0 | Day 2 | Day 4 | Day 7 | Day 10 | Day 14 | Day 45 |
| Phase I | |||||||||||
| 1 | 10 | PBS | i.v. | 400 μL | ■▲ | ▲ | S | S | S | S | P |
| 2 | 10 | PLP-BPI solution | i.v. | 400 nmol | ■▲ | ▲ | B | B | B | B | B |
| 3 | 10 | PLP-BPI solution | s.c. | 400 nmol | ■▲ | ▲ | B | B | B | B | B |
| 4 | 10 | PLP-BPI solution + blank microparticles | s.c. | 400 nmol | ■▲ | ▲ | B○ | B○ | B○ | B○ | B |
| 5 | 10 | PLP-BPI solution + PLP-BPI loaded microparticles | s.c. | 400 nmol | ■▲ | ▲ | B | ● | – | – | B |
| 6 | 10 | PLP-BPI loaded microparticles | s.c. | 400 nmol | ■▲ | ▲ | ● | – | – | – | B |
| Phase II | |||||||||||
| 7 | 10 | PBS | i.v. | 300 μL | ■▲ | ▲ | S | S | S | – | P |
| 8 | 10 | PLP-BPI solution | i.v. | 300 nmol | ■▲ | ▲ | B | B | B | – | B |
| 9 | 10 | PLP-BPI solution | s.c. | 300 nmol | ■▲ | ▲ | B | B | B | – | B |
Abbreviations: PLP-BPI = Ac-PLP-BPI-NH2-2, n = number of mice, i.v. = intravenous, s.c. = subcutaneous, ■ = immunization with PLP/CFA, ▲ = boosting with pertussis toxin (i.p.), S = solution dose of saline (100 μL), B = solution dose of Ac-PLP-BPI-NH2-2 (100 nmol), P = solution dose of PLP (100 nmol), ○ = dose of blank microparticles (25 mg), ● = dose of Ac-PLP-BPI-NH2-2 loaded microparticles (100 nmol Ac-PLP-BPI-NH2-2), and – = no dose administered.
Induction of anaphylaxis.
Total dose per mouse during the treatment phase; excludes anaphylactic dose.
Disease progression was evaluated blindly by the same observer, as reported previously [4,5], according to the following clinical scoring scale: 0 — no clinical signs of disease; 1 — tail weakness or limp tail; 2 — paraparesis (weakness or incomplete paralysis of one or two hind limbs); 3 — paraplegia (complete paralysis of two hind limbs); 4 — paraplegia with forelimb weakness or paralysis; and 5 — moribund. Mice were euthanized once they were found to be moribund. Body weight was measured daily.
2.5.3. Induction and monitoring of anaphylaxis
In addition to their beneficial therapeutic effects, antigen-derived peptides such as Ac-PLP-BPI-NH2-2 have the potential to induce fatal anaphylaxis, particularly on repeated injection. To compare the anaphylactic response to Ac-PLP-BPI-NH2-2 administered in different forms (solution vs. microparticle), by different routes (i.v. and s.c.) and with different dosing frequency, an “anaphylaxis dose” of 100 nmol was administered at the end of the treatment period to challenge the immune system by repeated exposure to a previously administered antigen. At Day 45, animals in Groups 2–6 (Table 1) received 100 nmol Ac-PLP-BPI-NH2-2 peptide/mouse, while those in Group 1 received 100 nmol PLP peptide/mouse. For each group, the route of administration of the anaphylactic dose was the same as that used for earlier treatment. The incidence of anaphylactic response is reported as the percentage of mice in each group that died within 30 min after the anaphylactic dose. Characteristic signs of immediate hypersensitivity (i.e., piloerection; prostration; erythema of the tail, ears, and footpads; shallow breathing; and lethargy) observed within a few minutes after the anaphylactic dose were also recorded.
3. Results
3.1. Microparticle properties and in vitro release
PLGA microparticles containing Ac-PLP-BPI-NH2-2 were successfully prepared by the powder-in oil-in water solvent evaporation method. This method produced Ac-PLP-BPI-NH2-2 loaded microparticles with a mean size of 8±4 μm, as shown by SEM micrographs and corresponding size distributions (Fig. 1A). Blank PLGA microparticles were roughly twice this size, with a mean size of 16±5 μm (Fig. 1B). Both peptide-loaded and blank microparticles were spherical with a smooth surface, though occasional pores in the surface of the peptide-loaded microparticles may be the result of peptide leaching during fabrication (Fig. 1A). Peptide leaching, perhaps followed by particle contraction, may also contribute to the smaller size of the peptide-loaded vs. blank microparticles. Both types of microparticles are less than 30 μm, and thus are expected to pass freely through a 20 gauge needle and are suitable for s.c. injection. The peptide content of the Ac-PLP-BPI-NH2-2 loaded microparticles was 1.4±0.2% (w/w) and the peptide encapsulation efficiency was 8.2±1.2% (n=3,±S.D.). This low encapsulation efficiency and loading suggest poor retention of the hydrophilic Ac-PLP-BPI-NH2-2 peptide on exposure to aqueous washing solutions during microparticle fabrication. Residual CH2Cl2 in the microparticle formulation, as determined by GC/MS, was 580±30 ppm (n=3,±S.D.), below the recommended ICH limit of 600 ppm [20].
Fig. 1.
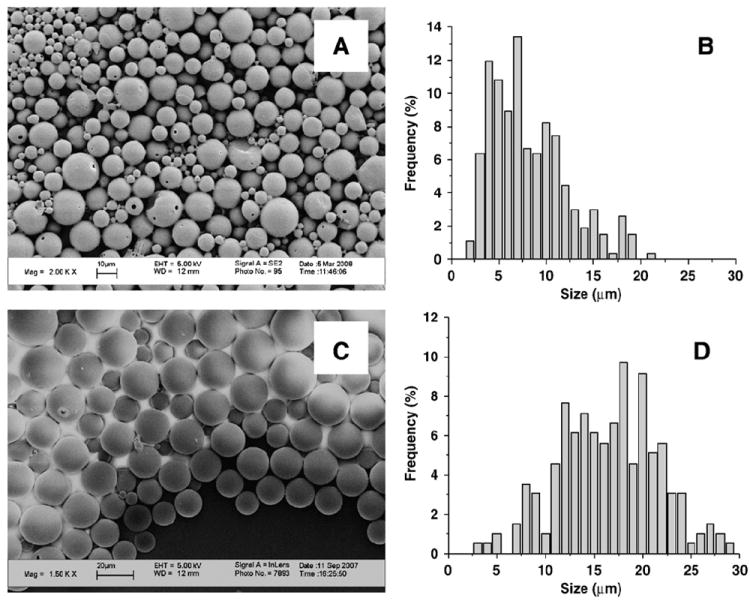
SEM micrographs of microparticles (A,C) and corresponding particle size distributions (B,D). (A) SEM micrograph of Ac-PLP-BPI-NH2-2 loaded microparticles; scale bar is 10 μm. (B) Particle size distribution of Ac-PLP-BPI-NH2-2 loaded microparticles. Mean size = 8 ± 4 μm; median size = 9 μm. (C) SEM micrograph of blank microparticles, scale bar is 20 μm. (D) Particle size distribution of blank microparticles. Mean size = 16 ± 5 μm; median size = 18 μm.
The in vitro release of Ac-PLP-BPI-NH2-2 from peptide-loaded microparticles shows an initial burst of ~15%, sustained release of ~1% per day through Day 21, followed by more rapid release during Days 21 to 44 (Fig. 2). This triphasic release profile is typical for PLGA microspheres [21,22]. The initial burst is widely believed to be caused by rapid release of drug from the particle surface [23], while the subsequent period of slow release is thought to reflect release by diffusion through the polymer matrix. The final, more rapid release phase is typically attributed to extensive degradation of the polymer matrix and release by erosion. For our purposes, the in vitro release profile demonstrates that the microparticle formulation is effective in providing sustained release over a period of more than forty days in buffered solution. It is therefore reasonable to expect that this formulation will also provide sustained release when injected s.c., and will generate a release profile distinct from s.c. injection of the peptide in solution.
Fig. 2.
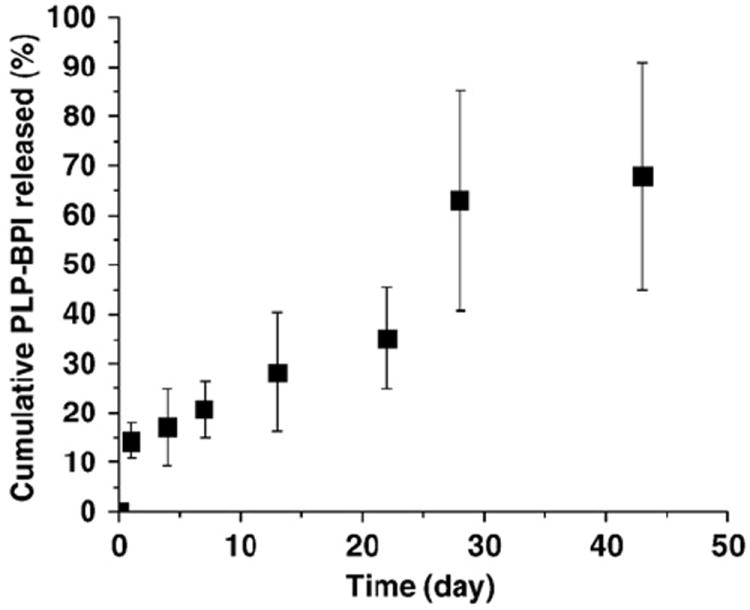
In vitro release of Ac-PLP-BPI-NH2-2 from PLGA microparticles (pH 7.4 PBS + 0.05% Tween-20, 37 °C, 50 rpm stirring). n = 3 ± S.D.
3.2. In vivo microparticle performance in an EAE mouse model
Mice were immunized on Day 0 to induce disease, then treated with various Ac-PLP-BPI-NH2-2 or control formulations over a two-week period (Table 1; Phase I). Figs. 3 and 4 show body weight and clinical scores for mice treated prophylactically for suppression of EAE symptoms; a statistical comparison of the treatment groups at each time point is available online as Supplementary Material. Deaths were observed both during treatment and on administration of the anaphylactic dose; Table 2 presents percent survival values for each group at the end of the treatment period (Day 14) and following the anaphylactic dose (Day 45). Group 1 received repeated i.v. injections of PBS (Table 1), and serves as an untreated control. Mice in Group 1 showed a nearly 20% decrease in weight during Days 10–12 (Fig. 3), with corresponding worsening of neurological symptoms as indicated by increasing clinical scores (Fig. 4). Both body weight and clinical scores show improvements following Day 15, an observation consistent with the typical relapse/remission pattern of the EAE model. There were no fatalities in Group 1 at the end of the treatment period (Day 14, Table 2).
Fig. 3.
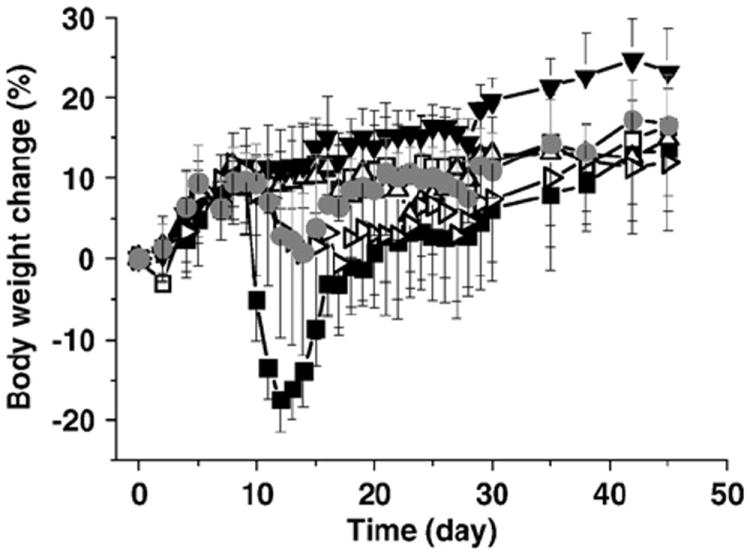
Body weight change as percentage of initial weight in EAE mice receiving Ac-PLP-BPI-NH2-2 microparticle formulations or controls at 400 nmol total dose. Treatment groups correspond to Groups 1–6 in Table 1: Group 1 (■); Group 2 (□); Group 3 (Δ); Group 4 (▼); Group 5 (▷); and Group 6 (●). n = 10 ± S.D. See Supplementary Material, Table S1, for statistical comparison of treatment and control groups on each day.
Fig. 4.

Clinical scores for EAE mice receiving Ac-PLP-BPI-NH2-2 microparticle formulations or controls at 400 nmol total dose. Treatment groups correspond to Groups 1–6 (Phase I) in Table 1: Group 1 (■); Group 2 (□); Group 3 (Δ); Group 4 (▼); Group 5 (▷); and Group 6 (●). n = 10 ± S.D. See Supplementary Material, Table S2, for statistical comparison of treatment and control groups on each day.
Table 2.
Percent survival of mice in treatment groups 1–9 (see Table 1).
| Group | Treatment Day
|
||
|---|---|---|---|
| Day 0 | Day 14 | Day 45 | |
| Phase I | |||
| 1 | 100 | 100 | 20 |
| 2 | 100 | 90 | 30 |
| 3 | 100 | 60 | 0 |
| 4 | 100 | 60 | 0 |
| 5 | 100 | 100 | 10 |
| 6 | 100 | 100 | 10 |
| Phase II | |||
| 7 | 100 | 90 | 10 |
| 8 | 100 | 100 | 20 |
| 9 | 100 | 100 | 0 |
Only 20% of mice in Group 1 survived the anaphylactic dose, however (Day 45, Table 2), suggesting a high potential for anaphylactic toxicity on repeated i.v. administration of the PLP peptide. Mice in Group 2 received repeated i.v. injections of Ac-PLP-BPI-NH2-2 in aqueous solution (Table 1). As in previous studies of PLP-BPI [4,5], these mice showed no appreciable weight loss (Fig. 3) and had no clinical signs of disease (score = 0; Fig. 4), confirming that repeated i.v. administration of Ac-PLP-BPI-NH2-2 provides effective suppression of EAE symptoms. 90% of mice in Group 2 survived at the end of the treatment period (Day 14, Table 2) and 30% survived the analphylactic dose (Day 45, Table 2), suggesting that i.v. Ac-PLP-BPI-NH2-2 has a potential to induce anaphylaxis comparable to PLP. Mice in Group 3 received repeated s.c. injections of Ac-PLP-BPI-NH2-2 in aqueous solution (Table 1), a treatment protocol that differs from that for Group 2 only in the route of administration. Body weights and clinical scores in Groups 2 and 3 were similar, suggesting similar therapeutic effects by the two routes (Figs. 3 and 4). However, survival was markedly reduced in Group 3, suggesting increased toxicity and/or a reduction in the effective dose range when Ac-PLP-BPI-NH2-2 is administered s.c. rather than i.v. (Group 3 vs. Group 2; Table 2). Mice in Group 4 received repeated s.c. injections of Ac-PLP-BPI-NH2-2 in solution, together with injections of blank PLGA microparticles (Table 1). Body weights and clinical scores for Group 4 were equivalent or superior to those for Groups 2 and 3, indicating that blank PLGA microparticles have no appreciable effects on treatment. Survival of mice in Group 4 was comparable to that for Group 3, again suggesting route of administration effects on Ac-PLP-BPI-NH2-2 toxicity and/or dose range (Groups 3 and 4 vs. Group 2; Table 2) and the absence of microparticle effects. Mice in Group 5 received an s.c. injection of Ac-PLP-BPI-NH2-2 in solution on Day 4, followed by an s.c. injection of microparticles on Day 7 (Table 1). Body weights (Fig. 3) and clinical scores (Fig. 4) were somewhat lower for mice in Group 5 than for those in groups receiving Ac-PLP-BPI-NH2-2 in solution only (Groups 2–4, Table 1), though clearly better than the untreated control (Group 1, Table 1). Interestingly, 100% of mice in Group 5 survived the treatment period (Day 14, Table 2), suggesting that the toxicities associated with repeated s.c. injection of Ac-PLP-BPI-NH2-2 in solution (Groups 3,4; Table 2) are reduced by slower and/or incomplete release from the microparticle dosage form. Sterlization by u.v. irradiation may contribute to incomplete release, an effect observed previously for TGFß3-loaded PLGA microspheres [24]. Only 10% of Group 5 mice survived the anaphylactic dose, however (Table 2), suggesting that the potential for anaphylactic toxicity remains. Mice in Group 6 received a single dose of Ac-PLP-BPI-NH2-2 loaded microparticles (Table 1). Body weights for mice in Group 6 were comparable to those of Group 5 (Fig. 3) and clinical scores somewhat superior, particularly late in the treatment period (Fig. 4). Survival percentages for Group 6 were identical to those of Group 5 (Table 2), again suggesting that Ac-PLP-BPI-NH2-2 toxicity is reduced by slow release and/or lower dose from the microparticle dosage form.
To probe the effects of dose, a second phase of animal studies was conducted in which a reduced dose of Ac-PLP-BPI-NH2-2 (300 nm) was administered in solution by either the i.v. or s.c. route (Phase II, Table 1). Effective suppression of EAE disease progression, as indicated by stable body weight (Fig. 5A) and low clinical score (Fig. 5B), was achieved with both i.v. and s.c. administration of three 100 nm doses of Ac-PLP-BPI-NH2-2, rather than the four 100 nm doses used in Phase I. In addition, there was no evidence of fatal toxicity during the treatment period, with 100% survival of mice receiving either the 300 nm i.v. dose (Group 8; Tables 1, 2) or the 300 nm s.c. dose (Group 9; Tables 1, 2). Reponses to the anaphylactic dose (Day 45, Table 2) showed somewhat greater fatalities in mice receiving the s.c. dose (Group 9, Table 2) than the i.v. dose (Group 8, Table 2), consistent with the results at higher dose in Phase I.
Fig. 5.

Body weight change (A) and clinical scores (B) for EAE mice receiving Ac-PLP-BPI-NH2-2 solutions or controls at 300 nmol total dose. Treatment groups correspond to Groups 7–9 (Phase II) in Table 1: Group 7 (■); Group 8 (□); and Group 9 (Δ). n = 10±S.D. See Supplementary Material, Tables S3 and S4, for statistical comparison of treatment and control groups on each day.
4. Discussion
MS involves the activation of CD4+ T-cells leading to inflammation and the destruction of myelin in the central nervous system. Current drug treatments for MS include corticosteroids to control inflammation; interferons (e.g., Betaseron) as general immunodulators; Tysabri® (natalizumab), a monoclonal antibody to α4 integrin thought to prevent the entry of activated T-cells into the CNS [25,26]; and Copaxone® (glatiramer acetate), a peptide copolymer related to the myelin basic protein (MBP) that induces Th2 regulatory cells [27,28]. These drugs offer considerable benefits to MS patients, but carry the risk of interference with normal immune function. For example, Tysabri® was temporarily withdrawn from the market after patients in clinical trials were infected with the JC virus. This viral infection causes progressive multifocal leukoencephalopathy (PML), a brain-wasting disease that can produce dementia, paralysis and even death [26,29,30], and that occurs most often in immunocompromised patients. The PLP-BPI peptide and its derivatives (e.g., Ac-PLP-BPI-NH2-2) being developed by co-author Siahaan and his group [4,5] and related inhibitors of T-cell activation reported by other groups [31-33] offer the promise of specific modulation of the pathological autoimmune response while preserving the immune system’s ability to respond to foreign pathogens. PLP-BPI peptides are hypothesized to induce immunotolerance in an antigen-specific manner by preventing the translocation of Signal-1 and Signal-2, which form the immunological synapse at the interface of the T-cell and the APC. The goal of treatment with PLP-BPI peptides thus is the induction of tolerance to PLP139–151, an MS associated antigen, by preventing the activation of T-cells that respond to it.
The method of delivery of PLP-BPI peptides may be important for balancing their efficacy and toxicity in vivo. In the studies reported here, we have compared the effects of three routes of administration (i.v. solution, s.c. solution, and s.c. microparticle) on the EAE suppressive activity of Ac-PLP-BPI-NH2-2 by measuring body weight, clinical score and survival. Interestingly, the results demonstrate that the in vivo activity of Ac-PLP-BPI-NH2-2 is influenced by route of administration. Specifically, s.c. solution administration of Ac-PLP-BPI-NH2-2 provides symptom amelioration comparable to i.v. solution administration (Fig. 3), but with greater incidence of toxicity (Table 2). In contrast, s.c. administration of Ac-PLP-BPI-NH2-2 in microparticle form provides slightly lower activity in reducing disease symptoms (Fig. 3), but does not show any toxicity during treatment (Table 2). All routes of administration show low survival rates on repeat challenge with the antigen as the anaphylactic dose (Day 45, Table 2). The results thus suggest that s.c. administration of PLP-BPI peptides in microparticle form can provide pharmacological efficacy and reduction in dosing frequency without increased toxicity.
It should be noted that the microparticles used in these studies were sterilized by exposure to u.v. radiation, a method less commonly used than exposure to ethylene oxide or γ-irradiation. The u.v. method was the only method readily available to us and is known to be effective for inactivating bacteria and viruses on surfaces. However, lack of complete sterility and u.v.-induced changes in the PLGA and/or PLP-BPI cannot be ruled out. Moioli et al. observed incomplete release from TGFß3-loaded PLGA microspheres following u.v. sterilization [24]. If the PLP-BPI peptide is similarly affected, this could contribute to the reduced toxicity of the microparticle forms.
Controlled and/or sustained release of antigens from polymeric materials have been investigated for altering the immune response and inducing immunotolerance by modulating Th1/Th2 balance, in some cases promoting the tolerogenic Th2-like profile [8-10]. It has been suggested that the maintenance of immunotolerance depends on antigen persistence in vivo, and can be reversed if the antigen source is removed [7]. Particulate delivery systems have been used for selective targeting to APCs, so that incorporated antigens are more effectively presented by MHC molecules and therefore more likely to be immunogenic. For example, in cancer-immunotherapy, a ten-fold lower dose was required when tetanus-toxoid (TT) was delivered in PLGA microspheres compared to solution delivery, suggesting antigen targeted APC delivery by microspheres [34]. Although there is growing evidence that PLGA and other polymeric materials have inherent adjuvant effects for stimulating immune response [7,10], the effect of particle size, morphology, surface chemistry and site of administration influence have not been fully elucidated [8-10]. Previous reports have documented uptake of PLGA microparticles in the 0.5 to 10 μm size range by dendritic cells [14,34,35] and macrophages [36]. This provides opportunities to study the effects of particle characteristics and delivery sites during antigen delivery on the immune response and its modulation.
In the studies reported here, Ac-PLP-BPI-NH2-2 was loaded into microparticles (<10 μm, Fig. 1) for delivery either via uptake by APCs or by controlled release of Ac-PLP-BPI-NH2-2 to extracellular receptor of the APCs. The therapeutic response to the microparticle dosage forms is consistent with sustained extracellular release of Ac-PLP-BPI-NH2-2 or, alternatively, to APC uptake without digestion of the Ac-PLP-BPI-NH2-2 peptide; the former is deemed more likely. Controlled release from these ~10 μm particles rather than APC uptake and digestion may be due to the large total mass of microparticles administered, which may agglomerate to form a depot in situ. Similarly, the data are not consistent with a marked adjuvant effect of the microparticles, since the therapeutic and toxic effects of s.c. injection of Ac-PLP-BPI-NH2-2 in solution were not affected by co-injection of microparticles (Table 1, Groups 3 and 4; Fig. 3, Table 2). That survival rates during treatment with the microparticle formulations are higher than for s.c. solutions (Groups 3,4 vs. Groups 5,6; Day 14, Table 2) suggests that sustained delivery of Ac-PLP-BPI-NH2-2 has promoted tolerance, while low survival following anaphylactic challenge on Day 45 is consistent with the overwhelming of tolerance to induce anaphylaxis. However, differences in total dose delivered by microparticle and solution forms cannot be ruled out. Additional studies to fully define the dose/response profile for Ac-PLP-BPI-NH2-2, including the effects of route of administration on the therapeutic window, are in progress.
Like MS, the EAE mouse model is an autoimmune inflammatory and demyelinating disease of the central nervous system. Local cytokine production varies significantly during EAE disease progression, and increases in discrete sets of cytokines are associated with the acute response and the recovery/chronic phase of the disease [37]. In EAE mice treated with PLP-BPI by i.v. injection, levels of CD4+CD25+TGF-β+, CD4+CD25+IL-4+ and CD4+CD25+IL-10+ T-cells were significantly greater than those in untreated controls in the lymphocyte population recovered from isolated spleens [4]. Elevated levels of IL-17 have been detected in the cerebrospinal fluid and in brain lesions of MS patients, and the IL-17 level is highly correlated with disease activity [38-40]. In mice receiving Ac-PLP-BPI-NH2-2 as an i.v. solution, IL-17 levels were significantly lower than those in PBS-treated controls [5]. In the current studies, the observed differences in therapeutic and anaphylactic responses following Ac-PLP-BPI-NH2-2 administration by different routes (s.c. vs. i.v. injection) and with different dosing frequency (microparticle vs. solution) suggest that cytokine profiles may be influenced by these dosing factors, although further investigation is needed.
5. Conclusions
The results presented here demonstrate that administration of Ac-PLP-BPI-NH2-2 by any of three different routes (i.e., i.v. solution, s.c. solution, and s.c. microparticle) produces significant suppression of disease onset in EAE mice relative to controls receiving PBS alone. Administration of Ac-PLP-BPI-NH2-2 as an s.c. solution produced clinical scores and maintenance of body weight comparable to the i.v. solution, but with reduced overall survival, while administration as s.c. microparticles provided a somewhat less effective reduction in symptoms but with no toxicity during treatment. The results thus suggest that s.c. administration of Ac-PLP-BPI-NH2-2 in microparticle form can provide pharmacological efficacy and reduction in dosing frequency without increased toxicity.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the assistance of the University of Kansas Mass Spectrometry Service Laboratory in GC/MS analysis of residual solvent levels. This work was supported by a grant from the Juvenile Diabetes Research Foundation (2006-JDRF00INNOV00008591; P.I. E.M. Topp).
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jconrel.2009.09.002.
References
- 1.Goldsby RA, Kindt TJ, Osborne BA, Kuby J. Immunology. W.H. Freeman and Company; New York: 2003. [Google Scholar]
- 2.Dunehoo AL, Anderson M, Majumdar S, Kobayashi N, Berkland C, Siahaan TJ. Cell adhesion molecules for targeted drug delivery. J Pharm Sci. 2006;95:1856–1872. doi: 10.1002/jps.20676. [DOI] [PubMed] [Google Scholar]
- 3.Seminario M-C, Bunnell SC. Signal initiation in T-cell receptor microclusters. Immunol Rev. 2008;221:90–106. doi: 10.1111/j.1600-065X.2008.00593.x. [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi N, Kobayashi H, Gu L, Malefyt T, Siahaan TJ. Antigen-specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. J Pharmacol Exp Ther. 2007;322:879–886. doi: 10.1124/jpet.107.123257. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi N, Kiptoo P, Kobayashi H, Ridwan R, Brocke S, Siahaan TJ. Prophylactic and therapeutic suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. Clin Immunol. 2008;129:69–79. doi: 10.1016/j.clim.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murray JS, Oney S, Page JE, Kratochvil-Stava A, Hu Y, Makagiansar IT, Brown JC, Kobayshi N, Siahaan TJ. Suppression of type 1 diabetes in NOD mice by bifunctional peptide inhibitor: modulation of the immunological synapse formation. Chem Biol Drug Des. 2007;70:227–236. doi: 10.1111/j.1747-0285.2007.00552.x. [DOI] [PubMed] [Google Scholar]
- 7.Lofthouse S. Immunological aspects of controlled antigen delivery. Adv Drug Del Rev. 2002;54:863–870. doi: 10.1016/s0169-409x(02)00073-x. [DOI] [PubMed] [Google Scholar]
- 8.Matzelle MA, Babensee JE. Humoral immune responses to model antigen co-delivered with biomaterials used in tissue engineering. Biomaterials. 2004;25:295–304. doi: 10.1016/s0142-9612(03)00531-3. [DOI] [PubMed] [Google Scholar]
- 9.Bennewitz NL, Babensee JE. The effect of the physical form of poly(lactic-co-glycolic acid) carriers on the humoral immune response to co-delivered antigen. Biomaterials. 2005;26:2991–2999. doi: 10.1016/j.biomaterials.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida M, Babensee JE. Poly(lactic-co-glycolic acid) enhances maturation of human monocyte-derived dendritic cells. J Biomed Mater Res A. 2004;71:45–54. doi: 10.1002/jbm.a.30131. [DOI] [PubMed] [Google Scholar]
- 11.de Lalla F, Rinaldi E, Santoro D, Pravettoni G. Immune response to hepatitis B vaccine given at different injection sites and by different routes: a controlled randomized study. Eur J Epidemiol. 1988;4:256–258. doi: 10.1007/BF00144763. [DOI] [PubMed] [Google Scholar]
- 12.Schellekens H. Factors influencing the immunogenicity of therapeutic proteins. Nephrol Dial Transplant. 2005;20:vi3–vi9. doi: 10.1093/ndt/gfh1092. [DOI] [PubMed] [Google Scholar]
- 13.Crommelin DJA, Sindelar RD, Meibohm B. Pharmaceutical Biotechnology: Fundamentals and Applications. 3. Informa Health Care; New York: 2007. [Google Scholar]
- 14.Little SR, Lynn DM, Ge Q, Anderson DG, Puram SV, Chen J, Eisen HN, Langer R. Poly-beta amino ester-containing microparticles enhance the activity of nonviral genetic vaccines. Proc Natl Acad Sci U S A. 2004;101:9534–9539. doi: 10.1073/pnas.0403549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang W, Gupta RK, Deshpande MC, Schwendeman SP. Biodegradable poly (lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Adv Drug Deliv Rev. 2005;57:391–410. doi: 10.1016/j.addr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Combadiere B, Mahe B. Particle-based vaccines for transcutaneous immunization. Comp Immunol Microbiol Infect Dis. 2008;31:293–315. doi: 10.1016/j.cimid.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 17.Kim H, Burgess DJ. Effect of drug stability on the analysis of release data from controlled release microspheres. J Microencapsul. 2002;19:631–640. doi: 10.1080/02652040210140698. [DOI] [PubMed] [Google Scholar]
- 18.Collins TJ. ImageJ for microscopy. BioTechniques. 2007;43:25–30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- 19.Hofstetter HH, Shive CL, Forsthuber TG. Pertussis toxin modulates the immune response to neuroantigens injected in incomplete Freund’s adjuvant: induction of Th1 cells and experimental autoimmune encephalomyelitis in the presence of high frequencies of Th2 cells. J Immunol. 2002;169:117–125. doi: 10.4049/jimmunol.169.1.117. [DOI] [PubMed] [Google Scholar]
- 20.ICH-Working-Group. Impurities: Guideline for Residual Solvents Q3C(R3) 1997 Jul;:20. [Google Scholar]
- 21.Zolnik BS, Burgess DJ. Evaluation of in vivo–in vitro release of dexamethasone from PLGA microspheres. J Control Release. 2008;127:127–145. doi: 10.1016/j.jconrel.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 22.Faisant N, Siepmann J, Benoit JP. PLGA-based microparticles: elucidation of mechanisms and a new, simple mathematical model quantifying drug release. Eur J Pharm Sci. 2002;15:355–366. doi: 10.1016/s0928-0987(02)00023-4. [DOI] [PubMed] [Google Scholar]
- 23.Woo BH, Kostanski JW, Gebrekidan S, Dani BA, Thanoo BC, DeLuca PP. Preparation, characterization and in vivo evaluation of 120-day poly(d, l-lactide) leuprolide microspheres. J Control Release. 2001;75:307–315. doi: 10.1016/s0168-3659(01)00403-5. [DOI] [PubMed] [Google Scholar]
- 24.Moioli EK, Hong L, Guardado J, Clark PA, Mao JJ. Sustained release of TGF-beta3 from PLGA microspheres and its effect on early osteogenic differentiation of human mesenchymal stem cells. Tissue Eng. 2006;12:537–546. doi: 10.1089/ten.2006.12.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodin DS, Cohen BA, O’Connor P, Kappos L, Stevens JC. Assessment: the use of natalizumab (Tysabri) for the treatment of multiple sclerosis (an evidence-based review) Neurology. 2008;71:766–773. doi: 10.1212/01.wnl.0000320512.21919.d2. [DOI] [PubMed] [Google Scholar]
- 26.Hoag H. Tysabri’s troubles return. Nat Biotechnol. 2008;26:1061. doi: 10.1038/nbt1008-1061. [DOI] [PubMed] [Google Scholar]
- 27.Aharoni R, Teitelbaum D, Leitner O, Meshorer A, Sela M, Arnon R. Specific Th2 cells accumulate in the central nervous system of mice protected against experimental autoimmune encephalomyelitis by copolymer 1. Proc Natl Acad Sci U S A. 2000;97:11472–11477. doi: 10.1073/pnas.97.21.11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blanchette F, Neuhaus O. Glatiramer acetate: evidence for a dual mechanism of action. J Neurol. 2008;255:26–36. doi: 10.1007/s00415-008-1005-5. [DOI] [PubMed] [Google Scholar]
- 29.Langer-Gould A, Steinman L. Progressive multifocal leukoencephalopathy and multiple sclerosis: lessons from natalizumab. Curr Neurol Neurosci Rep. 2006;6:253–258. doi: 10.1007/s11910-006-0013-z. [DOI] [PubMed] [Google Scholar]
- 30.Langer-Gould A, Steinman L. What went wrong in the natalizumab trials? Lancet. 2006;367:708–710. doi: 10.1016/S0140-6736(06)68279-2. [DOI] [PubMed] [Google Scholar]
- 31.Wang C, Gold BG, Kaler LJ, Yu X, Afentoulis ME, Burrows GG, Vandenbark AA, Bourdette DN, Offner H. Antigen-specific therapy promotes repair of myelin and axonal damage in established EAE. J Neurochem. 2006;98:1817–1827. doi: 10.1111/j.1471-4159.2006.04081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luca ME, Kel JM, van Rijs W, Drijfhout J, Koning F, Nagelkerken L. Mannosylated PLP(139–151) induces peptide-specific tolerance to experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;160:178–187. doi: 10.1016/j.jneuroim.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 33.Falk K, Rotzschke O, Santambrogio L, Dorf ME, Brosnan C, Strominger JL. Induction and suppression of an autoimmune disease by oligomerized T cell epitopes: enhanced in vivo potency of encephalitogenic peptides. J Exp Med. 2000;191:717–730. doi: 10.1084/jem.191.4.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waeckerle-Men Y, von Allmen EU, Gander B, Scandella E, Schlosser E, Schmidtke G, Merkle HP, Groettrup M. Encapsulation of proteins and peptides into biodegradable poly(d, l-lactide-co-glycolide) microspheres prolongs and enhances antigen presentation by human dendritic cells. Vaccine. 2006;24:1847–1857. doi: 10.1016/j.vaccine.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 35.Jhunjhunwala S, Raimondi G, Thomson AW, Little SR. Delivery of rapamicin to dendritic cells using degradable microparticles. J Control Release. 2009;133:191–197. doi: 10.1016/j.jconrel.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luzardo-Alvarez A, Blarer N, Peter K, Romero JF, Reymond C, Corradin G, Gander B. Biodegradable microspheres alone do not stimulate murine macrophages in vitro, but prolong antigen presentation by macrophages in vitro and stimulate a solid immune response in mice. J Control Release. 2005;109:62–76. doi: 10.1016/j.jconrel.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 37.Kennedy M, Torrance D, Picha K, Mohler K. Analysis of cytokine mRNA expression in the central nervous system of mice with experimental autoimmune encephalomyelitis reveals that IL-10 mRNA expression correlates with recovery. J Immunol. 1992;149:2496–2505. [PubMed] [Google Scholar]
- 38.Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–508. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 39.Hedegaard CJ, Krakauer M, Bendtzen K, Lund H, Sellebjerg F, Nielsen CH. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology. 2008;125:161–169. doi: 10.1111/j.1365-2567.2008.02837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gold R, Luehder F. Interleukin-17-extended features of a key player in multiple sclerosis. Am J Pathol. 2008;172:8–10. doi: 10.2353/ajpath.2008.070862. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.