

Abstract
Triterpenoids comprise a very diverse family of polycyclic molecules that is well-known to possess a myriad of medicinal properties. Therefore, triterpenoids constitute an attractive target for medicinal chemistry and diversity-oriented synthesis. Photochemical transformations provide a promising tool for the rapid, green and inexpensive generation of skeletal diversity in the construction of natural product-like libraries. With this in mind, we have developed a diversity-oriented strategy, whereby the parent triterpenoids bryonolic acid and lanosterol are converted to the pseudo-symmetrical polyketones by sequential allylic oxidation and oxidative cleavage of the bridging double bond at the B/C-ring fusion. The resultant polyketones were hypothesized to undergo divergent Norrish-Yang cyclization to produce unique 6/4/8-fused triterpenoid analogs. The subtle differences between parent triterpenoids led to dramatically different spatial arrangements of reactive functionalities. This finding was rationalized through conformational analysis to explain unanticipated photoinduced pinacolization, as well as the regio- and stereochemical outcome of the desired Norrish-Yang cyclization.
Introduction
Triterpenoids are a broad and structurally diverse class of natural products primarily derived from the plant kingdom.1 The triterpenoid family consists of nearly thirty thousand members with over two hundred unique carbocyclic skeletons.2–3 Consequently, triterpenoids are known to have a wide array of biological activities, including anti-fungal,4 anti-inflammatory,5 anti-cancer,6–7 as well as anti-viral8 and anti-bacterial properties.9 This remarkable molecular and medicinal diversity among triterpenoids is achieved through the variety of squalene and oxidosqualene cyclases that catalyze these reactions,10 as well as through further modification of the carbon skeletons by minor rearrangements including homologation, cleavage, and degradation (Figure 1).
Figure 1.

Selected members of triterpenoid family.
Considerable efforts in drug discovery have focused on the isolation and structural elucidation of novel triterpenoid molecules from the plant sources.11–13 Moreover, semi-synthetic triterpenoids, created by further manipulation of the exterior functional groups, have been shown to enhance the potency of their natural precursors.14–16 In this regard, the increase in structural complexity of triterpenoid-like molecules through alteration of their carbocyclic core skeleton can be viewed as a promising tool to study the chemical biology and medicinal chemistry of this natural product family.17–18
The use of photochemical transformations for the target-oriented synthesis of complex natural products has proven an effective approach.19–20 The advantages of these reactions are the green and low-cost nature of the reagent, as well as the fact that such transformations allow for the rapid construction of strained systems that otherwise would be very difficult to synthesize. However, the use of light as a reagent in diversity oriented synthesis is limited.21–22 In relation to natural products, a single report by the group of de la Torre23 describes an elegant application of a [2+2] cycloaddition and Paterno-Buchi reaction to the synthesis of diverse polycyclic terpene-like structures from readily available sesquiterpene Sclareolide.
We set out to apply Norrish-Yang photocyclization24–25 (Figure 2a) to the synthesis of triterpenoid analogs with unique 6/4/8-fused ring systems. The Norrish-Yang reaction was chosen because it allows for: (1) selective C-C bond formation from unactivated sp3-hybridized C–H bonds of saturated hydrocarbons; and (2) formation of particular 4/8 fused carbocycles from cyclodecanone substrates26–27 (Figure 2b). Additionally, the 1,2-diketone moiety is known to be an excitable chromophore that can undergo Yang cyclization as opposed to the Norrish type II fragmentation.28–33
Figure 2.

a. General mechanism of Norrish-Yang type II photocyclization. b. Formation of 4/8 fused carbocycle from 1,2-cyclodecanone. c. Structures and carbon numbering of bryonolic acid and lanosterol.
The application of Yang cyclization to the construction of additional four-membered rings in steroids has been reviewed by Kamernitskii et al.34 The diversity-oriented strategy35–36 which we have envisioned is different from earlier approaches in that the reactive keto groups are strategically positioned within a parent triterpenoid core structure by sequential oxidation reactions. This is followed by the generation of a new ring fusion via regioselective photocyclization.
Here, we report the successful application of this strategy to a pentacyclic triterpenoid, bryonolic acid, and a tetracyclic triterpenoid, lanosterol (Figure 2c). These molecules were chosen due to the unsaturation at the B/C-ring fusion, which allowed for the formation of the desired cyclodecane-1,2-diones by the allylic oxidation and subsequent oxidative cleavage reactions. The resultant ketones were systematically studied for structural elements dictating the regio- and stereoselectivity of the following Norrish-Yang reaction.
Results and Discussion
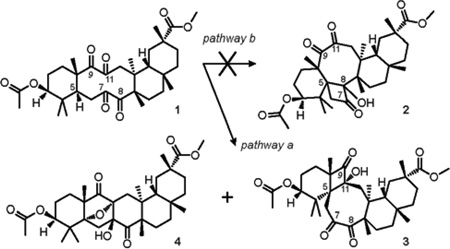
Tetraketone 1 was prepared according to our previously published method18 from doubly-protected bryonolic acid in two steps by oxidation of the Δ8,9 double bond with ruthenium tetroxide under Sharpless conditions. Early analysis of the bichromophoric structure of tetraketone 1 revealed that the ‘bowsprit’ hydrogen H-5 is in γ-position to the keto groups at C-8 and C-11. Thus, photoexcitation of the C-8 carbonyl bond could lead to the formation of cyclobutanol 2 via pathway b, and, concurrently, photoexcitation of C-11 carbonyl would result in the formation of cyclobutanol 3 via pathway a (Table 1). Taking into account this rationale, a solution of tetraketone 1 in dry deoxygenated C6D6 was irradiated in a quartz cuvette with 254 nm UV light until complete consumption of the starting material was detected by TLC (45 min, Table 1, entry 1). Further investigation of the reaction mixture disclosed showed the formation of two products, which were separable by column chromatography on silica.
TABLE 1.
Norrish-Yang Reactivity of Tetraketone 1.
 | ||||
|---|---|---|---|---|
| entry | λmax (nm) |
Time (min.) |
Isolated yield of product (%) | |
| 3 | 4 | |||
| 1 | 254 | 45 | 33 | 10 |
| 2 | 300 | 90 | 63 | 13 |
| 3 | 350 | 135 | 30 | 5 |
| 4 | 300a | 45 | 56 | <1 |
Reaction was carried out in the presence of 1.0 equiv of benzophenone
The study of the NMR spectra of the major reaction product revealed an AX system of two doublets at 3.40 and 2.75 ppm (J = 12 Hz) attached to isolated methylene carbon at 41.2 ppm. These protons correlate, as observed by HMBC, with two carbonyl carbons (C-7, δC 210.0; C-8, δC 211.5), as well as with four quaternary carbons (C-4, δC 43.8; C-5, δC 56.9; C-10, δC 62.3; C-11, δC 92.2). These correlations allowed the assignment of the AX system to position 6 of the cyclobutanol 3 and ruled out structure 2, in which H-6 could only be correlated to one carbonyl carbon by HMBC spectroscopy. Further investigation of the structure of cyclobutanol 3 disclosed an AX system of two doublets at 2.25 and 1.73 ppm (J = 18 Hz), attached to another isolated methylene carbon C-12 (δC 42.4). In the HMBC spectrum, H-12 showed cross-peaks with one carbonyl carbon (C-9, δC 218.9), as well as one methine carbon (C-18, δC 47.0), one methyl carbon (C-27, δC 19.0) and four quaternary carbons (C-5, C-11, C-13, δC 40.6; C-14, δC 55.4). Consequently, the NMR, as well as the HRMS data were consistent with the proposed structural framework of cyclobutanol 3 (Figure 3).
Figure 3.

Key HMBC (1H to 13C) and COSY correlations of cyclobutanol 3.
The routine NMR spectra of the minor reaction product were inconsistent with a structure of a product of Yang cyclization of tetraketone 1. Specifically, examination of 13C NMR spectrum revealed the signals of two carbonyl carbons (C-8, δC 212.6; C-9, δC 212.4), as well as three quaternary carbons (C-5, δC 90.6; C-7, δC 90.3; C-11, δC 77.3) in the region downfield from 50 ppm. Extensive 2D-NMR experiments allowed us to propose structure 4 for the minor product of the reaction. Especially useful were the cross-peaks shown by two doublets at 2.69 and 1.88 ppm (H-6, J = 14 Hz), attached to an isolated methylene carbon C-6 (δC 39.8). In the HMBC spectrum, H-6 correlated to C-5, C-7 , C-8, as well as C-10 (δC 48.7) and C-11. Additionally, the correlations of two doublets at 2.20 and 1.97 ppm (H-12) in the HMBC spectrum supported the elucidation of the carbon framework of structure 4 by showing the cross-peaks with C-7, C-9, C-11, as well as C-13 (δC 40.4), C-14 (δC 52.5), C-1 8 (δC 44.3) and C-27 (δC 22.3) (Figure 4). The structural assignment was also supported by HRMS data.
Figure 4.

Key HMBC (1H to 13C) and COSY correlations of structure 4.
After elucidation of the structures of the reaction products, the yields of cyclobutanol 3 and the minor product of the photolysis 4 were found to be 33% and 10%, respectively (Table 1, entry 1). The changing of the light sources did not lead to substantial difference in product distribution; however, the highest combined yield of 3 and 4 was achieved with a 300 nm UV lamp (76%, Table 1, entry 2). It is noteworthy that no products of the type II photofragmentation were observed in any reactions performed.
To gain insight into the regio- and, more importantly, stereochemical outcome of the Yang cyclization of tetraketone 1, we next examined a plausible mechanism for the process. It has been determined by the Scheffer Group37–39 that in order for a ketone to undergo successful Norrish type II hydrogen abstraction reaction, the distance between the oxygen of the carbonyl and the γ-H has to be less than the sum of Van der Waals radii for the H and O (2.72 Å). Using B3LYP/6–311G(d,p) calculated geometry of tetraketone 1, we determined that only C-11 keto group met this requirement (d1 = 2.29 Å, Figure 5), while the carbonyl at C-8 exceeded the suggested distance (d2 = 2.95 Å). Consequently, irradiation of tetraketone 1 led exclusively to the formation of 1(C-5),4(C-11)-hydroxy biradical, which is geometrically predisposed to cyclize into cis-cyclobutanol 3. The intermediate hydroxy biradical can concurrently undergo photopinacolization40–43 with the C-7 carbonyl leading to C-5,O-7-centered biradical, which presumably can experience intermolecular H-exchange, followed by a ring-closure to from bridged structure 4 (Figure 5).
Figure 5.

Mechanistic considerations for the formation of 3 and 4.
The mechanistic rationale of the reaction is in complete agreement with the observed stereochemistry of the products 3 and 4, as determined by NOE correlation spectroscopy (Figure 6), demonstrating memory of chirality effect, whereby the chiral information of the tetraketone 1 is retained by the conformation of the biradical intermediate.44–47
Figure 6.

Key NOESY correlations of compounds 3 and 4.
In the NOESY spectrum of cyclobutanol 3, Hβ-6 correlates with H-25 and H-24, while Hα-6 correlates with H-23 and H-27, and the NOE interactions of Hβ-12 with H-25 and H-26 determine α-facial orientation of the hydroxyl group at C-11. In the case of the bridged structure 4, Hα-6 correlates with H-24 and H-27, while Hβ-6 correlates with H-25, as well as with the C7-OH hydroxyl group, which, in turn, shows an interaction with H-26. This provided evidence for the β-facial orientation of this hydroxyl group.
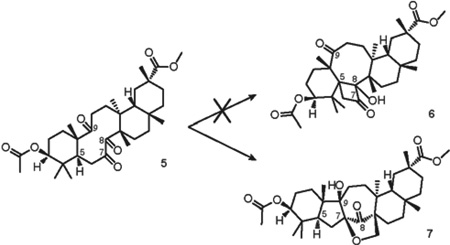
In an effort to force Yang cyclization into the alternative direction to form cyclobutanol 6, we took triketone 5 as a substrate for the photoreaction. The synthesis of triketone 5 by direct oxidation of doubly-protected bryonolic acid with ruthenium tetroxide under Sharpless conditions was previously reported by our group.18 The structure of triketone 5 preserves the main structural features of tetraketone 1, but lacks the keto group at C-11 (Table 2).
TABLE 2.
Norrish-Yang Reactivity of Triketone 5.
 | ||||
|---|---|---|---|---|
| entry | λmax (nm) |
Time (h) |
Yielda of 7 (% brsm) |
Conversiona of 5 (% ) |
| 1 | 254 | 72 | 12 | 57 |
| 2 | 300 | 72 | 15 | 69 |
| 3 | 350 | 72 | n/r | 0 |
| 4 | 300b | 72 | 39c | >99 |
| 5 | 254b | 72 | 18 | 72 |
Dermined by 1H NMR
Reaction was carried out in the presence of 1.0 equiv of benzophenone
Isolated yield
The computed distance between H-5 and the oxygen of the C-8 carbonyl of triketone 5 is 2.56 Å, suggesting the high possibility of the desired transformation. In addition, the 1(C-7),2(C-8)-dione system becomes the major chromophore of the polyketone. Accordingly, irradiation of triketone 5 with 254 nm UV light for 72 hours yielded a single reaction product (6% isolated yield, Table 2, entry 1). However, the structure of the product of the photolysis of triketone 5 was formulated as 7 on the basis of a thorough NMR investigation (Figure 7).
Figure 7.

Key HMBC (1H to 13C) and COSY correlations of structure 7.
The 13C NMR spectrum of 7 showed the presence of one carbonyl carbon C-8 (δC 212.9) as well as newly formed quaternary carbons C-7 (δC 87.3) and C-9 (δC 78.9), and a methylene carbon C-26 (δC 72.6) in the fingerprint region. This observation alone excludes structure 6, which would be expected to have two signals of the ketone groups in the 13C spectrum. The 1H NMR of 7 exhibited a pair of doublets at 4.29 and 4.04 ppm (J = 10 Hz), corresponding to a methylene carbon C-26 (δC 72.6). In the HMBC spectrum, H-26 showed cross-peaks with C-7, C-8, C-13 (δC 39.7), C-14 (δC 53.9) and a methylene carbon C-15 (δC 25.6). Structural assignment was completed by further analysis of cross-peaks shown by H-5, H-11 and H-25 (Figure 7), as well as HRMS data.
The formation of structure 7 was not anticipated. Presumably, the initial photoexcitation of the C-7 keto group is followed by a new carbon-carbon bond formation via pinacol-type coupling reaction with the C-9 carbonyl. The resulting oxygen-centered biradical intermediate, formation of which is commonly associated with the photochemical cleavage of cyclic peroxides,48–50 undergoes abstraction of ε-hydrogen H-26 by the oxygen of the C-9 carbonyl (d2 = 2.31 Å), followed by a radical cyclization process to form a furan ring (Figure 8).
Figure 8.

Mechanistic considerations for the formation of 7.
The changing of the light source did not significantly improve the efficiency of this reaction; however, almost complete conversion of the starting material was achieved by the use of benzophenone in combination with a 300 nm UV light (Table 2, entry 4). Low efficiency and long reaction times of the photolyses of triketone 5 are caused, perhaps, by the known reversibility of light-induced pinacolization and hydrogen-transfer processes.
The relative stereochemistry of 7 was confirmed by NOESY spectroscopy, which showed enhancements between the hydroxyl group at C-9 with Hβ-26, H-25 and Hβ-12. Hβ-12 in turn showed correlations with H-18 and Hβ-26, while Hα-26 showed a cross-peak with Hβ-16 (Figure 9). These NOESY data have determined the β-facial environment of the newly formed rings B and C.
Figure 9.

Key NOESY correlations of compound 7.
Having established the reactivity patterns of the polyketones derived from bryonolic acid, we then turned our attention to the tetracyclic triterpenoid lanosterol. We have previously found that the subtle structural differences between bryonolic acid and lanosterol led to dramatic differences in the aldol reactivity of the polyketones derived from these molecules.18 In this regard, we set out to predict using conformational analysis, and further confirm, the possibility of the formation of the alternative product of Yang cyclization via pathway b from lanosterol-derived polyketones 14 and 17.
The oxidation chemistry leading to tetraketone 14 was next investigated. After initial protection of the hydroxyl group at C-3 of lanosterol 8 to generate acetate 9, the Δ24,25 double bond of lanosterol acetate 9 was selectively oxidized according to the method developed by Reshetova et al.51 The use of KMnO4 and NaIO4 in aqueous tert-BuOH at 60 °C to rt for 15 h, followed by treatment of the resultant carboxylic acids with freshly prepared diazomethane in dry THF for 30 min, gave the desired methyl ester 10 in 65% yield, accompanied by α,β-unsaturated ketones 11 and 12; and enedione 13 in 9%, 7% and 4% yield, respectively. It is noteworthy that the formation of α,β-unsaturated ketone 12 was not observed by Reshetova et al. The ester 10 was further oxidized with KMnO4 and 18-crown-6 in aqueous DCM at rt to furnish enedione 13 in 78% yield after 24 h. Finally, tetraketone 14 was prepared from enedione 13 by catalytic oxidation with ruthenium tetroxide under Sharpless conditions.52 The use of RuCl3 with NaIO4 as a stoichiometric reoxidant in the mixture of CCl4, acetonitrile and water gave the desired product 14 in 87% yield after 15 h (Scheme 1).
SCHEME 1.

Sequential Oxidation of Δ24,25 and Δ8,9 of Lanosterol Acetate 9.
A literature search revealed that Marsaioli and co-workers have previously studied the aldol reactivity of a similar tetraketone derived from 24,25-dihydrolanosterol.53–54 Based on the analysis of 1H and 13C NMR spectra the authors have suggested that the tetraketone existed as a mixture of two conformational isomers. Rigorous verification of this suggestion was critical to our analysis and prediction of photochemical behavior of the tetraketone 14. Therefore, we conducted a variable temperature NMR study of tetraketone 14, which indeed was isolated as a mixture of two products (ca. 2.5:1 at 303.15 K), inseparable by various chromatographic means (Figure 10).
Figure 10.

Variable temperature 1H NMR study of tetraketone 14.
1H signals were assigned by the analysis of COSY, HMQC and HMBC spectra of tetraketone 14. 1H NMR experiments carried out in toluene-d8 at 0 °C revealed that H-3 signal exists as a doublet of doublets at 4.82 ppm (J1 = 11.4 Hz, J2 = 4.2 Hz) and a doublet of doublets at 4.60 ppm (J1 = 11.4 Hz, J2 = 3.6 Hz), H-12 splits into two doublets at 3.54 ppm (J = 13.2 Hz) and 3.48 ppm (J = 13.8 Hz), H-26 shows two singlets at 3.42 ppm and 3.40 ppm, while H-5 appears as a doublet of doublets at 3.21 ppm (J1 = J2 = 3.6 Hz) and a doublet of doublets at 3.03 ppm (J1 = 15.6 Hz, J2 = 9.6 Hz). With increasing temperature, all peaks broaden and at temperatures at and above 80 °C the doubled peaks merge and only time-average resonances are observed. We argue that the coalescence of the doubled peaks at higher temperatures confirms that tetraketone 14 exists as an equilibrium mixture of two conformational isomers. Lowering the temperature to −30 °C does not lead to any difference in the line shapes, indicating that the conformational equilibrium has reached the slow exchange rate.
Based on the empirical calculations using the torsional angles in the ten-membered ring Marsaioli and co-workers suggested the structures of the four possible conformers of the lanosterol-derived cyclodecatetraone (Figure 11). More accurate energy minimization calculations59 of the tetraketone 14 at the B3LYP/6–311G(d,p) level of theory disclosed that the lowest energy conformers CCC (‘chair-chair-chair’) and TB(‘twist-boat’)-1 are almost isoenergetic. Therefore, the energy difference of 0.44 kcal/mol between CCC and TB-1 would correspond well to the equilibrium mixture of two conformers in ca. 2.1:1 ratio at 303.15 K. The assignment of the structures of conformers CCC and TB-1 was further supported by comparison of the experimental J-values for H-5 of both conformers with the J-values predicted using the dihedral angles of −114° (TB-1, H5H6α), 128° (TB-1, H5H6β) and −73° (CCC, H5H6α) and 168° (CCC, H5H6β), which were determined with the GaussView 3.09 program. Unfortunately, NOESY experiments did not provide definitive evidence as to the structures of the conformers.
Figure 11.

Structures of the possible conformers of tetraketone 14.
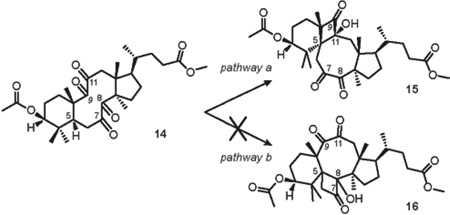
Next, we examined the photochemical reactivity of the bichromophoric structure 14. Analysis of the less populated conformational isomer CCC (Figure 11) revealed that the ‘bowsprit’ γ-hydrogen H-5 is anti-periplanar with C-8 and C-11 keto groups. Disfavorable orientation of reactive functionalities led to a conclusion that CCC must be unreactive in the desired Norrish-Yang photocyclization. On the contrary, examination of the more populated conformer TB-1 revealed that H-5 is anti-periplanar with the pi-bond of C-8 carbonyl, but synperiplanar with the C-11 keto group (Figure 12). Thus, favorable spatial position of H-5 and the C-11 carbonyl allowed for the prediction of regiospecific formation of the 1(C-5),4(C-11)-hydroxy biradical, which would be structurally biased to cyclize into cis-cyclobutanol 15 via pathway a. In addition, the distance between H-5 and the oxygen of the C-8 carbonyl of TB-1 was determined to be 2.27 Å, increasing the likelihood of the desired reaction. Concomitantly, disfavorable spatial arrangement of H-5 and C8=O should exclude the formation of cyclobutanol 16 via pathway b (Table 3).
Figure 12.

Mechanistic considerations for the formation of cyclobutanol 15.
TABLE 3.
Norrish-Yang reactivity of tetraketone 14.
 | ||||
|---|---|---|---|---|
| entry | λmax (nm) |
Time (h) |
Yield of 15 (%) |
Conversion of 14 (%) |
| 1 | 254 | 20 | 48 | 100 |
| 2 | 300 | 12 | 27 | 100 |
| 3 | 254a | 6 | 72b | 60 |
| 4 | 300a | 3 | 66 | 100 |
Reaction was carried out in the presence of 1.0 equiv of benzophenone
Yield of 15 is based on the recovered 14
In complete agreement with this rationale, irradiation of the solution of tetraketone 14 in dry deoxygenated C6D6 in a quartz cuvette with 254 nm UV light led to the formation of the sole product 15 in 48% yield after complete consumption of the starting material in 20 h.The change of the light source to 300 nm UV lamp led to a shorter reaction time, however cyclobutanol 15 was isolated in only 27% yield. The addition of 1.0 equiv benzophenone significantly improved the efficiency of the reaction, affording the highest yield of cyclobutanol 15 (66%, Table 3, entry 4). The structure of 15 was elucidated unambiguously after comprehensive analysis of 2-D NMR experiments and HRMS data (See Supporting Information). It is noteworthy that no other products were observed during the photolysis.
Our attention was then turned toward the oxidation reaction leading to triketone 17 (Scheme 2). In due course, when treated with our modified ruthenium conditions, α,β-unsaturated ketone 11 underwent competing cleavage of the Δ8,9 double bond to give the desired triketone 17 and regioselective allylic oxidation at C-11 to furnish enedione 13, which was further oxidized in the course of the reaction to yield tetraketone 14 as a minor reaction product. It is noteworthy that the dominating formation of aldol adduct 18 proved to be unavoidable during column chromatography of the crude reaction mixture on silica.
SCHEME 2.

Oxidation of Δ8,9 of α,β-Unsaturated Ketone 11
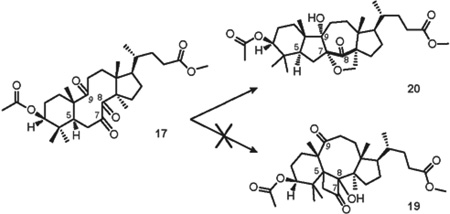
Conformational analysis of triketone 17 revealed that despite the fact that the keto group at C-8 is in γ-position to H-5, the C-8 carbonyl is anti-periplanar with the C5-H bond (Figure 13). This observation led to a conclusion that analogously with the CCC conformer of tetraketone 14, a prediction can be made that triketone 17 must be unreactive in the desired Norrish-Yang photocyclization and cyclobutanol 19 cannot be formed. However, the possibility of transannular photopinacolization remains plausible for triketone 17. By direct comparison with triketone 5, the excitation of the carbonyl at C-7 can be followed by the formation of oxygen-centered biradical. Based on the argument of closer spatial proximity, ε-hydrogen H-30 may then be abstracted by the oxygen of the C-9 carbonyl (Figure 13, d2 = 2.42 Å), followed by the formation of a furan ring with the oxygen of C-7 carbonyl.
Figure 13.

Mechanistic considerations for the formation of product 20.
As a result, irradiation of the solution of triketone 17 in dry deoxygenated C6D6 in a quartz cuvette with 254 nm UV light for 24 h led to the exclusive formation of furan 20 in 18% isolated yield (41% conversion, Table 4, entry 1), and no other products of the reaction were observed. The change of the λmax alone did not enhance the efficiency of the reaction, however the use of 1.0 equiv of benzophenone with 300 nm UV light yielded photoproduct 20 in 25% isolated yield (82% conversion, Table 4, entry 3). The structure of furan 20 was confirmed after extensive 2-D NMR study and by HRMS data (See Supporting Information).
TABLE 4.
Photoreactivity of Triketone 17.
 | ||||
|---|---|---|---|---|
| entry | λmax (nm) |
Time (h) |
Yield of 20 (% brsm) | Conversion of 17 (% ) |
| 1 | 254 | 24 | 44 | 41 |
| 2 | 300 | 24 | 19 | 69 |
| 3 | 300a | 24 | 31 | 82 |
| 4 | 54a | 24 | 39 | 55 |
Reaction was carried out in the presence of 1.0 equiv of benzophenone
Conclusion
Here we have devised and successfully executed a DOS strategy, whereby the substrates for divergent Norrish-Yang photocyclization were prepared from parent triterpenoids bryonolic acid and lanosterol by sequential allylic oxidation and oxidative cleavage reactions. Irradiation of bryonolic acid-derived tetraketone 1 brought a new 6/6/6-fused structural type 4 to the resultant chemical library of triterpenoid analogs by the virtue of an unanticipated photopinacolization. The unexpected structure 4 was accompanied by the expected formation of 6/4/8-fused product of Yang cyclization 3 via pathway a. The use of bryonolic acid-derived triketone 5 to force Yang cyclization into pathway b led to predominant pinacol-type coupling reaction to give 6 /5/7-fused structure 7, despite the desirable arrangement of the ‘bowsprit’ H-5 and the keto group at C-8. The subtle differences between the parent triterpenoids resulted in completely different conformational preferences of the polyketones and concomitantly, completely different spatial orientations of the reactive functionalities. Consequently, Norrish-Yang photocyclization of lanosterol-derived polyketones 14 and 17 via pathway b was proven to be unfeasible, but the pinacol-type coupling reaction of triketone 17 was proven to be predictable. Thus, under our irradiation conditions, tetraketone 14 formed a single photocycloadduct 15 via pathway a, while triketone 17 yielded the anticipated product of photopinacolization 20.
We would argue that these findings constitute a general approach, whereby the identification of substrate-dictated conformational preferences of similar substrates will allow for a reliable prediction of the outcome of Norrish-Yang type II photocyclization, as well as the feasibility of transannular photoinduced pinacol-type coupling reaction. Moreover, we hope that this study will inspire the development of the divergent strategies for the synthesis of polycyclic compounds from other natural product families to screen against different biological targets.
Experimental Section
(2R,4aS,6aS,9aS,11S,13aS,16aS,16bR)-methyl-11-acetoxy-2,4a,6a,10,10,13a,16a–heptamethyl-7,8,14,15-tetraoxodocosahydrobenzo[6,7]cyclodeca[1,2-a]naphthalene-2-carboxylate (1)
Tetraketone 1 was prepared from bryonolic acid in four steps by a previously published method18 (74 mg, 14% overall yield). Identity of 1 was confirmed by 1H and 13C NMR. Purity of 1 was determined by 1H NMR, TLC and mp. Physical and spectroscopic data were found to match lit.18 data.
General Procedure for Photolyses of Polyketones 1, 5, 14, and 17
A solution of a specified polyketone (20 mg) in dry C6D6 (0.6 mL) in a 4 mL rectangular quartz cuvette (0.1 cm× 0.1 cm×0.4 cm) was deoxygenated by purging with dry argon gas for 15 min while stirring. When specified, benzophenone (1.0 equiv) was added to a reaction mixture. A cuvette was then sealed with a Teflon stopper and placed in the middle of a chamber photoreactor (10 cm distance from the light source), equipped with six UV-lamps (8 W each, specified λmax) and a cooling fan. Solutions were irradiated at rt for a specified amount of time, until specified polyketone was consumed or decomposition was observed, as determined by either TLC or 1H NMR. After removal of the solvent in vacuo, photoproducts 3, 4, 7, 15, and 20 were isolated by column chromatography on silica with specified ethyl acetate-hexanes mixtures.
Preparation of 3 and 4
Table 1 (Entry 1)
Following the general procedure for photolyses, the use of tetraketone 1 (0.035 mmol) and 254 nm UV light for 45 min gave 3 (7 mg, 33%) and 4 (2 mg, 10%).
Table 1 (Entry 2)
Following the general procedure for photolyses, the use of tetraketone 1 (0.035 mmol) and 300 nm UV light for 90 min gave 3 (12.5 mg, 63%) and 4 (2.6 mg, 13%).
Table 1 (Entry 3)
Following the general procedure for photolyses, the use of tetraketone 1 (0.035 mmol) and 350 nm UV light for 135 min gave 3 (6 mg, 30%) and 4 (1 mg, 5%).
Table 1 (Entry 4)
Following the general procedure for photolyses, the use of tetraketone 1 (0.035 mmol), benzophenone (6.4 mg, 0.035 mmol) and 300 nm UV light for 45 min gave 3 (11.2 mg, 56%) and 4 (0.1 mg, <1%).
(2aS,5R,6aR,6bS,7aR,8aS,11S,12aR,15aS)-methyl-11-acetoxy-7a-hydroxy-2a,5,6b,8a,12,12,15a-heptamethyl-8,14,15-trioxoicosahydro-1H-benzo[1’,4’] cyclobuta[1’,2’:6,7]cycloocta[1,2-a]naphthalene-5-carboxylate (3)
Transparent oil. Rf = 0.37 (EA/hex = 3/7). 1H NMR (600 MHz, CDCl3, δ): 5.70 (dd, J1 = J2 = 9 Hz, 1H), 3.60 (s, 3H), 3.40 (d, J = 12.6 Hz, 1H), 3.10 (br. s, 1H), 2.75 (d, J = 12.6 Hz, 1H), 2.35 (d, J = 16.2 Hz, 1H), 2.25 (d, J = 18 Hz, 1H), 2.17 (ddd, J1 = J2 = 14.4 Hz, J3 = 4.2 Hz, 1H), 2.02 (s, 3H), 1.73 (d, J = 18 Hz, 1H), 1.35 (s, 3H), 1.32 (s, 3H), 1.23 (s, 3H), 1.16 (s, 3H), 1.06 (s, 3H), 1.04 (s, 3H), 1.02 (s, 3H), 0.95 (m, 1H). 13C NMR (150 MHz, CDCl3, δ): 218.9 (CO), 211.5 (CO), 210.0 (CO), 178.7 (CO), 170.4 (CO), 92.2 (COH), 72.4 (CH), 62.3 (C), 56.9 (C), 55.4 (C), 52.4 (CH3), 47.0 (CH), 43.8 (C), 42.4 (CH2), 41.2 (CH2), 40.7 (C), 40.6 (C), 35.4 (CH2), 33.4 (CH2), 32.8 (CH3), 32.7 (C), 31.22 (CH3), 31.20 (CH2), 29.8 (CH2), 29.1 (CH2), 24.5 (CH2), 23.2 (CH2), 22.4 (CH3), 21.8 (CH3), 21.6 (CH3), 21.3 (CH3), 19.0 (CH3), 17.2 (CH3). HRMS (ESI) m/z calcd for C33H48O8Na+ [M+Na]+ 595.3247, found 595.3236.
(2R,4aS,6aS,7aS,8aR,10S,12aS,13aR,14aS,14bR)-methyl-10-acetoxy-7a-hydroxy-2,4a,6a,9,9,12a,14a-heptamethyl-7,13-dioxoicosahydro-8a,13a-epoxybenzo[a]tetracene-2-carboxylate (4)
Transparent oil. Rf = 0.32 (EA/hex = 3/7). 1H NMR (600 MHz, CDCl3, δ): 4.81 (dd, J1 =10.8 Hz, J2 = 4.8 Hz, 1H), 3.61 (s, 3H), 2.69 (d, J = 14.4 Hz, 1H), 2.36 (d, 15 Hz, 1H), 2.05 (s, 3H), 1.31 (s, 3H), 1.29 (s, 3H), 1.19 (s, 3H), 1.07 (s, 3H), 1.00 (s, 3H), 0.92 (s, 3H), 0.77 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 212.6 (CO), 212.4 (CO), 178.6 (CO), 170.6 (CO), 90.6 (C), 90.3 (C), 77.3 (C), 76.5 (CH), 52.7 (C), 51.6 (CH3), 48.7 (C), 44.3 (CH), 40.4 (C), 40.4 (C), 39.8 (CH2), 38.5 (C), 35.4 (CH2), 33.9 (CH2), 32.5 (CH3), 31.5 (CH3), 31.3 (C), 31.1 (CH2), 30.7 (CH2), 29.91 (CH2), 29.87 (CH2), 24.2 (CH2), 23.1 (CH2), 22.3 (CH3), 21.4 (CH3), 20.8 (CH3), 19.4 (CH3), 18.5 (CH3), 17.7 (CH3). HRMS (ESI) m/z calcd for C33H48O8Na+ [M+Na]+ 595.32414, found 595.32423.
(2R,4aS,6aS,9aS,11S,13aS,16aS,16bR)-methyl-11-acetoxy-2,4a,6a,10,10,13a,16a-heptamethyl-7,8,14-trioxodocosahydrobenzo[6,7]cyclodeca[1,2-a]naphthalene-2-carboxylate (5)
Triketone 5 was prepared from bryonolic acid in three steps by a previously published method18 (184 mg, 23% overall yield). Identity of 5 was confirmed by 1H and 13C NMR and HRMS. Purity of 5 was determined by 1H NMR, TLC and mp. Physical and spectroscopic data were found to match lit.18 data.
Preparation of 7
Table 2 (Entry 1)
Following the general procedure for photolyses, the use of triketone 5 (0.036 mmol) and 254 nm UV light for 72 h yielded an inseparable mixture (10 mg) of 7 (1.4 mg, 12% brsm, determined by 1H NMR) and unreacted 5 (8.6 mg, 57% conversion, determined by 1H NMR). Rf = 0.4 (EA/hex = 3/7).
Table 2 (Entry 2)
Following the general procedure for photolyses, the use of triketone 5 (0.036 mmol) and 300 nm UV light for 72 h yielded an inseparable mixture (8.3 mg) of 7 (2.1 mg, 15% brsm, determined by 1H NMR) and unreacted 5 (6.2 mg, 69% conversion, determined by 1H NMR). Rf = 0.4 (EA/hex = 3/7).
Table 2 (Entry 3)
Following the general procedure for photolyses, the use of triketone 5 (0.036 mmol) and 350 nm UV light for 72 h yielded unreacted 5 (20 mg, 0% conversion). Rf = 0.4 (EA/hex = 3/7).
Table 2 (Entry 4)
Following the general procedure for photolyses, the use of triketone 5 (0.036 mmol), benzophenone (6.5 mg, 0.036 mmol) and 300 nm UV light for 72 h yielded 7 (7.8 mg, 39%). Rf = 0.4 (EA/hex = 3/7).
Table 2 (Entry 5)
Following the general procedure for photolyses, the use of triketone 5 (0.036 mmol), benzophenone (6.5 mg, 0.036 mmol) and 254 nm UV light for 72 h yielded an inseparable mixture (8.2 mg) of 7 (2.5 mg, 18% brsm, determined by 1H NMR) and unreacted 5 (5.7 mg, 72% conversion, determined by 1H NMR). Rf = 0.4 (EA/hex = 3/7).
(2R,4aS,6aS,8aS,9aS,11S,13aS,13bR,15aS,15bR)-methyl-11-acetoxy-13b-hydroxy-2,4a,10,10,13a,15a-hexamethyl-16-oxoicosahydro-6a,8a-methanoindeno[2,1-b]naphtho[1,2-f]oxocine-2-carboxylate (7)
Analytically pure 7 was obtained by the following procedure. A round-bottom flask open to atmosphere was charged with basic alumina (365 mg, 3.58 mmol), followed by the addition of DCM (0.5 mL). A mixture (20 mg), containing 5 (17.2 mg, 0.031 mmol) and 7 (2.8 mg, 5 µmol) as determined by 1H NMR, was dissolved in DCM (0.5 mL) and subsequently added to the resulting suspension. The flask was sealed with a glass stopper and vigorously stirred at rt overnight, at which time the suspension was filtered over a fine sinter funnel and washed successively with ethyl acetate. After removal of the solvent in vacuo, the column chromatography on silica yielded 7 as a transparent oil (2 mg, 71% recovery). Rf = 0.38 (EA/hex = 25/75). 1H NMR (600 MHz, CDCl3, δ): 4.51 (dd, J1 = 11.4 Hz, J2 = 4.2 Hz, 1H), 4.29 (d, J = 9.6 Hz, 1H), 4.04 (d, J = 9.6 Hz, 1H), 3.64 (s, 3H), 3.07 (br.s, 1H), 2.05 (s, 3H), 1.19 (s, 3H), 1.00 (s, 3H), 0.97 (s, 3H), 0.95 (s, 3H), 0.93 (s, 3H), 0.85 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 212.9 (CO), 179.3 (CO), 171.1 (CO), 87.3 (C), 80.5 (CH), 78.9 (C), 72.6 (CH2), 53.9 (C), 52.0 (CH3), 48.3 (CH), 47.4 (C), 46.7 (CH), 40.7 (C), 39.7 (C), 37.3 (C), 36.8 (CH2), 34.0 (CH2), 33.5 (CH2), 32.2 (CH2), 31.8 (C), 31.3 (CH3), 30.96 (CH2), 30.90 (CH3), 29.3 (CH2), 29.2 (CH2), 29.1 (CH3), 29.0 (CH2), 25.6 (CH2), 25.3 (CH2), 21.4 (CH3), 17.5 (CH3), 17.2 (CH3), 16.6 (CH3). HRMS (ESI) m/z calcd for C33H50O7Na+ [M+Na]+ 581.34488, found 581.34503. The starting material 5 reacted with basic alumina to give the corresponding product of aldol addition. This product of aldol addition retained on silica during column chromatography due to the presence of the hydroxyl group and was not isolated. The synthesis, purification and structural analysis of the product of aldol addition of 5 were previously studied by our group18.
(3S,5R,10S,13R,14R,17R)-4,4,10,13,14-pentamethyl-17-((R)-6-methylhept-5-en-2-yl)-2,3,4,5,6,7,10,11,12,13,14,15,16,17-tetradecahydro-1H–cyclopenta[a]phenanthren-3-yl acetate (9)
In a round-bottom flask, lanosterol 8 (10g, 23.4 mmol; 60% in the mixture with 24,25-dihydrolanosterol, purchased from various commercial sources) and DMAP (143 mg, 1.17 mmol) were dissolved in 47 mL of dry pyridine. Acetic anhydride (7.18 g, 70.3 mmol, 6.6 mL) was added to the resulting solution by a quick syringe transfer. The reaction mixture was stirred at 50 °C overnight, after which time pyridine was removed at reduced pressure and the crude mixture was taken up in DCM. The organic layer was washed successively with dilute HCl, saturated solution of sodium bicarbonate and brine. It was subsequently dried over Na2SO4 and the solvent removed in vacuo. The crude product was purified by column chromatography to give 9 (60% in the mixture with 24,25-dihydrolanosterol acetate) as a white solid (9.7 g, 88%). Rf = 0.55 (EA/hex = 10/90). Identity and purity of 9 were confirmed by 1H and 13C NMR. Spectroscopic data were found to match lit.55 data.
Preparation of 10–13
Using modified literature protocol,51 lanosterol acetate 9 (10 g, 60% in the mixture with 24,25-dihydrolanosterol acetate, 12.8 mmol) was dissolved in 1 L of tert-BuOH at 60 °C, followed by the addition of K2CO3 (10g, 72.4 mmol). Meanwhile, a solution of KMnO4 (1.42 g, 9 mmol) and NaIO4 (24.1 g, 0.113 mol) in 800 mL of H2O was heated to 60 °C and subsequently added to the resulting suspension in one portion. The flask was removed from heat, and the reaction mixture was allowed to cool to rt and stirred at rt overnight, at which time the reaction was interrupted by the addition of saturated solution of sodium thiosulfate until complete discoloration was observed (about 250 mL). After the organic layer was separated and the solvent removed in vacuo, the crude residue was acidified with dilute HCl and extracted with DCM. The organic layer was washed successively with water and brine. It was subsequently dried over Na2SO4 and the solvent removed in vacuo. The crude mixture of products was separated by gradient elution column chromatography on silica with ethyl acetate-hexanes mixtures. The use of hexanes as an eluting solvent yielded pure 24,25-dihydrolanosterol acetate as a white solid (3.36 g, 84%). Rf = 0.75 (EA/hex = 20/80). mp: 118–120 °C ( lit.56 mp 120–121 °C). Elution with progressively more polar combinations of eluting solvent, followed by the use of ethyl acetate as an eluting solvent, gave a crude mixture (6.5 g) of carboxylic acids. In a round-bottom flask open to atmosphere, the mixture of carboxylic acids (6.5 g) was dissolved in 50 mL of freshly distilled THF. Freshly prepared diazomethane in diethyl ether (15 mmol) was added dropwise to the resulting solution by a pipette, and the reaction mixture was stirred at rt until full conversion was detected by TLC (approximately 30 min). The reaction was quenched by dropwise addition of glacial acetic acid until the yellow color of the solution disappeared. The solvent was removed in vacuo and the crude mixture of products was separated by careful column chromatography on silica to yield 10 as a white solid (3.9 g, 65%), 11 as a white solid (610 mg, 9%), 12 as a white solid (420 mg, 7%), and 13 as a yellow solid (270 mg, 4%).
(R)-methyl-4-((3S,5R,10S,13R,14R,17R)-3-acetoxy-4,4,10,13,14-pentamethyl-2,3,4,5,6,7,10,11,12,13,14,15,16,17-tetradecahydro-1H–cyclopenta[a]phenanthren-17-yl)pentanoate (10)
mp: 174–177 °C (lit.51 mp 226–227 °C, lit.57 mp 174–176 °C ). Rf = 0.58 (EA/hex = 20/80). Identity and purity of 10 were confirmed by 1H and 13C NMR, HRMS and TLC. Spectroscopic data did not duplicate lit.51 data. 1H NMR (400 MHz, CDCl3, δ): 4.50 (dd, J1 = 11.6 Hz, J2 = 4.4 Hz, 1H), 3.66 (s, 3H), 2.37 (m, 1H), 2.22 (m, 1H), 2.05 (s, 3H), 1.00 (s, 3H), 0.89 (d, J = 6 Hz, 3H), 0.88 (s, 3H), 0.88 (s, 3H), 0.87 (s, 3H), 0.68 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 174.9, 171.1, 134.6, 134.4, 81.1, 51.6, 50.6, 50.4, 50.0, 44.7, 38.0, 37.0, 36.2, 35.4, 31.41, 31.39, 31.1, 30.9, 28.2, 28.1, 26.5, 24.4, 24.3, 21.5, 21.1, 19.3, 18.4, 18.3, 16.7, 15.9. HRMS (ESI) m/z calcd for C30H48O4Na+ [M+Na]+ 495.34448, found 495.34456.
(R)-methyl-4-((3S,5R,10S,13R,14R,17R)-3-acetoxy-4,4,10,13,14-pentamethyl-7-oxo-2,3,4,5,6,7,10,11,12,13,14,15,16,17-tetradecahydro-1H–cyclopenta[a]phenanthren-17-yl)pentanoate (11)
mp: 200–203 °C (lit.51 mp 255–258 °C). Rf = 0.28 (EA/hex = 20/80). Rf = 0.39 (EA/hex = 25/75). Identity of 11 was confirmed by 1H and 13C NMR and HRMS. Purity of 11 was determined by 1H NMR and TLC. 1H NMR data did not duplicate lit.51 data. 13C NMR data were not reported in chemical literature. 1H NMR (600 MHz, CDCl3, δ): 4.52 (dd, J1 = 11.6 Hz, J2 = 4.4 Hz, 1H), 3.66 (s, 3H), 2.06 (s, 3H), 1.18 (s, 3H), 0.95 (s, 3H), 0.91 (m, 3H), 0.91 (s, 3H), 0.88 (s, 3H), 0.65 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 198.8 (CO), 174.8 (CO), 171.0 (CO), 164.7 (C), 139.1 (C), 79.7 (CH), 51.6 (CH3), 50.0 (CH), 48.9 (CH), 47.9 (C), 45.1 (C), 39.8 (C), 37.9 (C), 36.6 (CH2), 36.1 (CH), 34.6 (CH2), 32.1 (CH2), 31.4 (CH2), 31.4 (CH2), 30.2 (CH2), 28.8 (CH2), 27.5 (CH3), 25.1 (CH3), 24.0 (CH2), 23.8 (CH2), 21.4 (CH3), 18.6 (CH3), 18.5 (CH3), 16.5 (CH3), 15.9 (CH3). HRMS (ESI) m/z calcd for C30H47O5+ [M+H]+ 487.34180, found 487.34188.
(R)-methyl-4-((3S,5R,10S,13R,14R,17R)-3-acetoxy-4,4,10,13,14-pentamethyl-11-oxo-2,3,4,5,6,7,10,11,12,13,14,15,16,17-tetradecahydro-1H–cyclopenta[a]phenanthren-17-yl)pentanoate (12)
mp: 144–147 °C (lit.57 mp 145–149 °C ). Rf = 0.33 (EA/hex = 20/80). Rf = 0.42 (EA/hex = 25/75). Identity of 12 was confirmed by 1H and 13C NMR and HRMS. Purity of 12 was determined by 1H NMR, TLC and mp. Spectroscopic data for 12 were not previously reported. 1H NMR (600 MHz, CDCl3, δ): 4.49 (dd, J1 = 10.2 Hz, J2 = 6 Hz, 1H), 3.65 (s, 3H), 2.99 (ddd, J1 = 13.8 Hz, J2 = J3 = 3.6 Hz, 1H), 2.64 (d, J = 16.2 Hz, 1H), 2.44 (d, J = 16.2 Hz, 1H), 2.03 (s, 3H), 1.13 (s, 3H), 1.10 (s, 3H), 0.88 (s, 3H), 0.88 (s, 3H), 0.85 ( d, J = 6.6 Hz, 3H), 0.80 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 199.1 (CO), 174.6 (CO), 171.1 (CO), 164.2 (C), 139.5 (C), 80.7 (CH), 52.0 (CH), 51.9 (CH2), 51.7 (CH3), 51.6 (C), 50.1 (CH), 47.4 (C), 38.0 (C), 37.6 (C), 35.8 (CH), 34.1 (CH2), 31.2 (CH2), 31.0 (CH2), 31.0 (CH2), 29.9 (CH2), 28.4 (CH3), 27.0 (CH2), 25.9 (CH3), 24.3 (CH2), 21.4 (CH3), 19.1 (CH3), 18.1 (CH3), 17.3 (CH2), 16.9 (CH3), 16.8 (CH3). HRMS (ESI) m/z calcd for C30H47O5+ [M+H]+ 487.3423, found 487.3426.
(R)-methyl-4-((3S,5R,10S,13R,14R,17R)-3-acetoxy-4,4,10,13,14-pentamethyl-7,11-dioxo-2,3,4,5,6,7,10,11,12,13,14,15,16,17-tetradecahydro-1H–cyclopenta[a]phenanthren-17-yl)pentanoate (13)
A 100 mL round-bottom flask open to atmosphere was charged with KMnO4 (2.67 g, 16.92 mmol) and 18-crown-6 (4.47 g, 16.92 mmol), followed by the addition of 33.9 mL of water. A solution of 10 (1.6 g, 3.385 mmol) in 33.9 mL of DCM was then added to the resulting suspension by a quick syringe transfer. The flask was sealed with a glass stopper, and the reaction mixture was vigorously stirred at rt for 24 h, at which time the mixture was filtered over a fine sinter funnel and washed with DCM. Layers were separated, and the aqueous layer was extracted with DCM. The organic layer was dried over Na2SO4 and the solvent removed in vacuo. The crude product was purified by column chromatography on silica to give 13 as a yellow solid (1.32 g, 78%). mp: 142–145 °C (lit.51 mp 178–179 °C, lit.58 mp 141–143 °C). Rf = 0.25 (EA/hex = 20/80). Rf = 0.37 (EA/hex = 25/75). Identity of 13 was confirmed by 1H and 13C NMR and HRMS. Purity of 13 was determined by 1H NMR, TLC and mp. 13C NMR data was found to match lit.51 data. 1H NMR data did not duplicate lit.51,58 data. 1H NMR (400 MHz, CDCl3, δ): 4.51 (dd, J1 = 11.2 Hz, J2 = 5.2 Hz, 1H), 3.65 (s, 3H), 2.88 (ddd, J1 = 13.6 Hz, J2 = J3 = 3.6 Hz, 1H), 2.75 (d, J = 16 Hz, 1H), 2.58 (d, J = 16 Hz, 1H), 2.04 (s, 3H), 1.31 (s, 3H), 1.15 (s, 3H), 0.94 (s, 3H), 0.88 (s, 3H), 0.87 (d, J = 5.2 Hz, 3H), 0.78 (s, 3H). HRMS (ESI) m/z calcd for C30H45O6+ [M+H]+ 501.32107, found 501.32111.
(R)-methyl-4-((1R,3aR,6aS,8S,10aS,13aR)-8-acetoxy-3a,7,7,10a,13a-pentamethyl-4,5,11,12-tetraoxohexadecahydro-1H-benzo[a]cyclopenta[f][10]annulen-1-yl)pentanoate (14)
Using modified literature protocol,54 RuCl3 (62 mg, 0.299 mmol) was added in one portion to the solution of NaIO4 (539 mg, 2.52 mmol) in 18 mL of water, and the resulting suspension was stirred open to atmosphere for 15 min, followed by the addition of 12 mL of acetonitrile. The solution of 13 (300 mg, 0.599 mmol) in 12 mL of CCl4 was then added to the reaction mixture by a quick syringe transfer. The flask was sealed with a glass stopper, and the resulting biphasic mixture was vigorously stirred at rt for 15 h, at which time the reaction was interrupted by the addition of 5 mL of ethanol. The layers were separated and the aqueous layer was extracted with DCM. The organic layer was dried over Na2SO4 and the solvent removed in vacuo. The crude product was purified by column chromatography on silica to give 14 as a yellow solid (278 mg, 87%). mp: 140–143 °C. Rf = 0.39 (EA/hex = 25/75). 1H NMR (600 MHz, toluene-d8, 273.15 K) major conformer, δ: 4.82 (dd, J1 = 11.4 Hz, J2 = 4.2 Hz, 1H), 3.54 (d, J = 13.2 Hz, 1H), 3.42 (s, 3H), 3.21 (dd, J1 = J2 = 3.6 Hz, 1H), 2.69 (ddd, J1 = J2 = 12.6 Hz, J3 = 6.6 Hz, 1H), 1.67 (s, 3H), 1.34 (s, 3H), 1.01 (s, 3H), 0.98 (s, 3H), 0.75 (s, 3H), 0.71 (d, J = 6.6 Hz, 3H), 0.64 (s, 3H); minor conformer, δ: 4.60 (dd, J1 = 11.4 Hz, J2 = 3.6 Hz, 1H), 3.48 (d, J = 13.8 Hz, 1H), 3.40 (s, 3H), 3.03 (dd, J1 = 15.6 Hz, J2 = 9.6 Hz, 1H), 1.65 (s, 3H), 1.42 (s, 3H), 1.30 (s, 3H), 0.87 (s, 3H), 0.77 (d, J = 6.6 Hz, 3H), 0.63 (s, 3H). 13C NMR (150 MHz, toluene-d8, 273.15 K) major conformer, δ: 208.7, 206.1, 205.1, 203.2, 173.7, 169.8, 79.6, 62.3, 55.1, 53.1, 51.5, 49.7, 48.2, 44.3, 38.9, 37.6, 36.3, 33.8, 31.7, 31.61, 31.59, 30.9, 28.6, 24.3, 23.9, 20.3, 20.2, 19.6, 17.9, 16.5; minor conformer, δ: 207.3, 207.1, 202.6, 201.3, 173.3, 169.2, 78.7, 60.6, 52.3, 50.98, 50.0, 49.4, 46.9, 41.4, 38.7, 35.6, 35.2, 34.8, 34.5, 30.7, 27.3, 24.4, 22.7, 19.64, 18.9, 17.8, 17.0, 16.2. HRMS (ESI) m/z calcd for C30H44O8Na+ [M+Na]+ 555.29284, found 555.29296.
Preparation of 15
Table 3 (Entry 1)
Following the general procedure for photolyses, the use of tetraketone 14 (0.0375 mmol) and 254 nm UV light for 20 h yielded 15 (9.6 mg, 48%).
Table 3 (Entry 2)
Following the general procedure for photolyses, the use of tetraketone 14 (0.0375 mmol) and 300 nm UV light for 12 h yielded 15 (5.3 mg, 27%).
Table 3 (Entry 3)
Following the general procedure for photolyses, the use of tetraketone 14 (0.0375 mmol), benzophenone (6.8 mg, 0.0375 mmol) and 254 nm UV light for 6 h yielded unreacted 14 (8.1 mg, 60% conversion) and 15 (8.6 mg, 72% brsm).
Table 3 (Entry 4)
Following the general procedure for photolyses, the use of tetraketone 14 (0.0375 mmol), benzophenone (6.8 mg, 0.0375 mmol) and 300 nm UV light for 3 h yielded 15 (13.1 mg, 66%).
(R)-methyl-4-((1aS,4S,5aR,8aR,11R,11aR,12aR)-4-acetoxy-12a-hydroxy-1a,5,5,8a,11a-pentamethyl-1,7,8-trioxohexadecahydrobenzo[1,4]cyclobuta[1,2-a]cyclopenta[d][8]annulen-11-yl)pentanoate (15)
Transparent oil. Rf = 0.25 (EA/hex = 25/75). 1H NMR (400 MHz, CDCl3, δ): 5.65 (m, 1H), 3.69 (s, 3H), 2.90 (d, J = 13.9 Hz, 1H), 2.83 (br s, 1H), 2.60 (d, J = 13.9 Hz, 1H), 2.07 (s, 3H), 1.37 (s, 3H), 1.28 (s, 3H), 1.16 (s, 3H), 1.16 (s, 3H), 1.10 (d, J = 6.7 Hz, 3H), 0.93 (s, 3H). 13C NMR (150 MHz, CDC13, δ): 213.3 (CO), 210.0 (CO), 209.5 (CO), 174.3 (CO), 171.3 (CO), 94.9 (C), 78.7 (CH), 63.4 (C), 59.5 (C), 56.2 (C), 53.9 (CH), 51.8 (CH3), 47.8 (C), 43.1 (CH2), 40.9 (CH2), 40.0 (C), 35.6 (CH2), 34.2 (CH), 31.9 (CH2), 30.9 (CH2), 29.3 (CH2), 23.0 (CH2), 22.7 (CH2), 21.6 (CH3), 21.4 (CH3), 20.5 (CH3), 20.3 (CH3), 19.8 (CH3), 19.5 (CH3), 17.7 (CH3). HRMS (ESI) m/z calcd for C30H44O8Na+ [M+Na]+ 555.2934, found 555.2933.
Preparation of 17 and 18
In a 100 mL single-neck round-bottom flask, RuCl3 (30 mg, 0.144 mmol) was added in one portion to a solution of NaIO4 (646 mg, 3.02 mmol) in 22 ml of H2O, and the resulting suspension was stirred open to atmosphere for 15 min, followed by the addition of 14.5 mL of acetonitrile. The solution of 11 (350 mg, 0.719 mmol) in 14.5 mL of CCl4 was then added dropwise to the reaction mixture by a syringe-pump. The flask was sealed with a glass stopper and the resulting biphasic mixture was vigorously stirred for 6 hours, at which time 3 mL of ethanol was added to the solution. The layers were separated and the aqueous layer was extracted with DCM. The organic layer was dried over Na2SO4, concentrated under vacuum and the crude mixture of products was further separated by column chromatography on silica to yield tetraketone 14 as a yellow solid (24 mg, 6%), Rf = 0.39 (EA/hex = 25/75), triketone 17 as a yellow oil (103 mg, 28%), and aldol adduct 18 as a white solid (238 mg, 64%).
(R)-methyl 4-((1R,3aR,6aS,8S,10aS,13aR)-8-acetoxy-3a,7,7,10a,13a-pentamethyl-4,5,11-trioxohexadecahydro-1H-benzo[a]cyclopenta[f][10]annulen-1-yl)pentanoate (17)
Rf = 0.26 (EA/hex = 25/75). 1H NMR (400 MHz, CDCl3, δ): 4.66 (dd, J1 = 11.6 Hz, J2 = 4.4 Hz, 1H), 3.67 (s, 3H), 2.81 (dd, J1 = 16.4 Hz, J2 = 7.6 Hz, 1H), 2.06 (s, 3H), 1.37 (s, 3H), 1.30 (s, 3H), 0.98 (s, 3H), 0.96 (d, J = 6.8 Hz, 3H), 0.90 (s, 3H), 0.79 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 214.0, 209.2, 203.2, 174.4, 170.7, 79.4, 59.0, 53.4, 53.1, 52.2, 51.7, 50.4, 39.2, 36.2, 35.5, 34.3, 33.8, 33.2, 31.7, 30.8, 28.0, 26.1, 25.1, 23.4, 21.3, 20.3, 19.4, 17.7, 17.3, 16.8. HRMS (ESI) m/z calcd for C30H47O7+ [M+H]+9 519.33163, found 519.33173.
(R)-methyl-4-((1R,3aR,4aR,5aS,7S,9aS,10aR,11aR)-7-acetoxy-4a-hydroxy-3a,6,6,9a,11a-pentamethyl-4,10-dioxohexadecahydro-1H-cyclopenta[b]anthracen-1-yl)pentanoate (18)
mp: 260–264 °C. Rf = 0.15 (EA/hex = 25/75). 1H NMR (600 MHz, CDCl3, δ): 4.48 (dd, J1 = 11.4 Hz, J2 = 4.6 Hz, 1H), 3.65 (s, 3H), 3.10 (dd, J1 = 12.4 Hz, J2 = 2.6 Hz, 1H), 2.13 (d, J = 13.3 Hz, 1H), 2.04 (s, 3H), 1.28 (s, 3H), 1.12 (s, 3H), 0.99 (s, 3H), 0.95 (d, J = 6.4 Hz, 3H), 0.88 (s, 3H), 0.68 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 211.8 (CO), 211.6 (CO), 174.6 (CO), 171.0 (CO), 80.0 (CH), 79.2 (C), 59.3 (C), 51.7 (CH3), 50.2 (CH), 49.5 (CH), 47.9 (C), 47.3 (C), 45.5 (CH), 38.5 (C), 35.3 (CH), 31.2 (CH2), 31.1 (CH2), 30.7 (CH2), 29.3 (CH2), 28.9 (CH2), 28.4 (CH2), 28.1 (CH3), 26.9 (CH2), 23.6 (CH2), 22.8 (CH3), 21.3 (CH3), 19.5 (CH3), 18.7 (CH3), 18.6 (CH3), 17.3 (CH3). HRMS (ESI) m/z calcd for C30H46O7Na+ [M+Na]+ 541.31357, found 541.31373.
Preparation of 20
Table 4 (Entry 1)
Following the general procedure for photolyses, the use of triketone 17 (0.0386 mmol) and 254 nm UV light for 24 h yielded unreacted 17 (11.8 mg, 41% conversion) and 20 (3.6 mg, 44% brsm).
Table 4 (Entry 2)
Following the general procedure for photolyses, the use of triketone 17 (0.0386 mmol) and 300 nm UV light for 24 h yielded unreacted 17 (6.3 mg, 69% conversion) and 20 (2.6 mg, 19% brsm).
Table 4 (Entry 2)
Following the general procedure for photolyses, the use of triketone 17 (0.0386 mmol), benzophenone (7 mg, 0.0386 mmol) and 300 nm UV light for 24 h yielded unreacted 17 (3.7 mg, 82% conversion) and 20 (5 mg, 31% brsm).
Table 4 (Entry 4)
Following the general procedure for photolyses, the use of triketone 17 (0.0386 mmol), benzophenone (7 mg, 0.0386 mmol) and 254 nm UV light for 24 h yielded unreacted 17 (9 mg, 55% conversion) and 20 (4.3 mg, 39% brsm).
(R)-methyl-4-((1R,3aR,5aR,6aS,8S,10aS,10bS,12aR)-8-acetoxy-10b-hydroxy-7,7,10a,12a-tetramethyl-13-oxotetradecahydro-1H-3a,5a-methanocyclopenta[f]indeno[2,1-b]oxocin-1-yl)pentanoate (20)
Transparent oil. Rf = 0.23 (EA/hex = 25/75). 1H NMR (600 MHz, CDCl3, δ): 4.56 (dd, J1 = 11.9 Hz, J2 = 4.7 Hz, 1H), 4.26 (d, J = 9.3 Hz, 1H), 3.80 (d, J =9.3 Hz, 1H), 3.66 (s, 3H), 3.63 (s, 1H), 2.05 (s, 3H), 0.96 (s, 3H), 0.92 (d, J = 6.4 Hz, 3H), 0.88 (s, 3H), 0.86 (s, 3H), 0.85 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 209.9 (CO), 174.5 (CO), 171.1 (CO), 89.5 (C), 80.89 (CH), 80.88 (C), 74.1 (CH2), 61.4 (C), 53.5 (CH), 51.7 (CH3), 49.9 (C), 49.2 (CH), 49.1 (C), 37.0 (C), 34.8 (CH), 31.3 (CH2), 31.2 (CH2), 30.4 (CH2), 29.8 (CH2), 29.7 (CH2), 29.6 (CH2), 29.4 (CH3), 27.4 (CH2), 26.2 (CH2), 24.4 (CH2), 21.4 (CH3), 19.0 (CH3), 16.8 (CH3), 15.4 (CH3), 15.3 (CH3). HRMS (ESI) m/z calcd for C30H46O7Na+ [M+Na]+ 541.3141, found 541.3142.
Supplementary Material
Acknowledgements
This work was supported by grants from the National Cancer Institute CA157735 & CA132168, NSF grant MCB-084480, Reuter Foundation, and Landon Foundation INNOVATOR award from AACR to G.P.T.
Footnotes
Supporting Information
General experimental details, complete ref 59, conformational analyses of 14 and 17, 13C VT NMR of 14, atomic coordinates for the optimized geometry for 1, 5, 14, 17; copies of 1H and 13C NMR spectra of all new compounds, as well as all known compounds that were prepared by new or modified synthetic protocols; copies of HMQC, HMBC, COSY, and NOESY NMR spectra, as well as Tables of 1H and 13C peak assignments and HMQC correlations of 3, 4, 7, 11, 12, 15, 18, 20; key COSY, HMBC, and NOESY correlations of compounds 11, 12, 15, 18, 20. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Connolly JD, Hill RA. Nat. Prod. Rep. 2010;27:79. doi: 10.1039/b808530g. [DOI] [PubMed] [Google Scholar]
- 2.Xu R, Fazio GC, Matsuda SPT. Phytochemistry. 2004;65:261. doi: 10.1016/j.phytochem.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 3.Degenhardt J, Köllner TG, Gershenzon J. Phytochemistry. 2009;70:1621. doi: 10.1016/j.phytochem.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 4.Onishi J, Meinz M, Thompson J, Curotto J, Dreikorn S, Rosenbach M, Douglas C, Abruzzo G, Flattery A, Kong L, Cabello A, Vicente F, Pelaez F, Diez MT, Martin I, Bills G, Giacobbe R, Dombrowski A, Schwartz R, Morris S, Harris G, Tsipouras A, Wilson K, Kurtz MB. Antimicrob. Agents Chemother. 2000;44:368. doi: 10.1128/aac.44.2.368-377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gatbonton-Schwager TN, Letterio JJ, Tochtrop GP. J. Nat. Prod. 2012;75:591. doi: 10.1021/np200823p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liby KT, Yore MM, Sporn MB. Nat. Rev. Cancer. 2007;7:357. doi: 10.1038/nrc2129. [DOI] [PubMed] [Google Scholar]
- 7.Petronelli A, Pannitteri G, Testa U. Anti-Cancer Drugs. 2009;20:880. doi: 10.1097/CAD.0b013e328330fd90. [DOI] [PubMed] [Google Scholar]
- 8.Zhou WB, Tao JY, Xu HM, Chen KL, Zeng GZ, Ji CJ, Zhang YM, Tan NHZ. Naturforsch., B: J. Chem. Sci. 2010;65:1393. [Google Scholar]
- 9.Wolska KI, Grudniak AM, Fiecek B, Kraczkiewicz-Dowjat A, Kurek A. Cent. Eur. J. Biol. 2010;5:543. [Google Scholar]
- 10.Thoma R, Schulz-Gasch T, D’Arcy B, Benz J, Aebi J, Dehmlow H, Hennig M, Stihle M, Ruf A. Nature. 2004;432:118. doi: 10.1038/nature02993. [DOI] [PubMed] [Google Scholar]
- 11.Wang C-Q, Wang L, Fan C-L, Zhang D-M, Huang X-J, Jiang R-W, Bai L-L, Shi J-M, Wang Y, Ye W-C. Org. Lett. 2012;14:4102. doi: 10.1021/ol301745b. [DOI] [PubMed] [Google Scholar]
- 12.Meng F-Y, Sun J-X, Li X, Yu H-Y, Li S-M, Ruan H-L. Org. Lett. 2011;13:1502. doi: 10.1021/ol200188n. [DOI] [PubMed] [Google Scholar]
- 13.Wang C-F, Liu J-Q, Yan Y-X, Chen J-C, Lu Y, Guo Y-H, Qiu M-H. Org. Lett. 2010;12:1656. doi: 10.1021/ol100062b. [DOI] [PubMed] [Google Scholar]
- 14.Sporn MB, Liby KT, Yore MM, Fu L, Lopchuk JM, Gribble GW. J. Nat. Prod. 2011;74:537. doi: 10.1021/np100826q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Honda T, Rounds BV, Gribble GW, Suh N, Wang Y, Sporn MB. Bioorg. Med. Chem. Lett. 1998;8:2711. doi: 10.1016/s0960-894x(98)00479-x. [DOI] [PubMed] [Google Scholar]
- 16.Honda T, Rounds BV, Bore L, Finlay HJ, Favaloro FG, Jr, Suh N, Wang Y, Sporn MB, Gribble GW. J. Med. Chem. 2000;43:4233. doi: 10.1021/jm0002230. [DOI] [PubMed] [Google Scholar]
- 17.Kumar N, Kiuchi M, Tallarico JA, Schreiber SL. Org. Lett. 2005;7:2535. doi: 10.1021/ol0504345. [DOI] [PubMed] [Google Scholar]
- 18.Ignatenko VA, Han Y, Tochtrop GP. J. Org. Chem. 2013;78:410. doi: 10.1021/jo302211f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffmann N. Chem. Rev. 2008;108:1052. doi: 10.1021/cr0680336. [DOI] [PubMed] [Google Scholar]
- 20.Bach T, Hehn JP. Angew. Chem., Int. Ed. 2011;50:1000. doi: 10.1002/anie.201002845. [DOI] [PubMed] [Google Scholar]
- 21.Hehn JP, Herdtweck E, Bach T. Org. Lett. 2011;13:1892. doi: 10.1021/ol2004462. [DOI] [PubMed] [Google Scholar]
- 22.Mukhina OA, Bhuvan KNN, Arisco TM, Valiulin RA, Metzel GA, Kutateladze AG. Angew. Chem., Int. Ed. 2011;50:9423. doi: 10.1002/anie.201103597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de la Torre MC, Garcia I, Sierra MA. J. Org. Chem. 2003;68:6611. doi: 10.1021/jo034177y. [DOI] [PubMed] [Google Scholar]
- 24.Norrish RGW. Trans. Faraday Soc. 1937;33:1521. [Google Scholar]
- 25.Yang NC, Yang D-DH. J. Am. Chem. Soc. 1958;80:2913. [Google Scholar]
- 26.Urry WH, Trecker DJ, Winey DA. Tetrahedron Lett. 1962;3:609. [Google Scholar]
- 27.Olovsson G, Scheffer JR, Trotter J, Wu C-H. Tetrahedron Lett. 1997;38:6549. [Google Scholar]
- 28.Urry WH, Trecker DJ. J. Am. Chem. Soc. 1962;84:118. [Google Scholar]
- 29.Hamer NK. Tetrahedron Lett. 1986;27:2167. [Google Scholar]
- 30.Obayashi M, Mizuta E, Noguchi S. Chem Pharm. Bull. 1979;27:1679. doi: 10.1248/cpb.27.1352. [DOI] [PubMed] [Google Scholar]
- 31.Herrera AJ, Rondon M, Suarez E. Synlett. 2007:1851. [Google Scholar]
- 32.Herrera AJ, Rondon M, Suarez E. J. Org. Chem. 2008;73:3384. doi: 10.1021/jo702663w. [DOI] [PubMed] [Google Scholar]
- 33.Kamijo S, Hoshikawa T, Inoue M. Tetrahedron Lett. 2010;51:872. [Google Scholar]
- 34.Kamernitskii AV, Ignatov VN, Levina IS. Usp. Khim. 1988;57:474. [Google Scholar]
- 35.Schreiber SL. Scienc. 2000;287:1964. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 36.Burke MD, Schreiber SL. Angew. Chem., Int. Ed. 2004;43:46. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- 37.Scheffer JR, Garcia-Garibay M, Nalamasu O. Org. Photochem. 1987;8:249. [Google Scholar]
- 38.Gudmundsdottir AD, Lewis TJ, Randall LH, Scheffer JR, Rettig SJ, Trotter J, Wu C-H. J. Am. Chem. Soc. 1996;118:6167. [Google Scholar]
- 39.Ihmels H, Scheffer JR. Tetrahedron. 1999;55:885. [Google Scholar]
- 40.Horspool W, Armesto D. Organic Photochemistry: A Comprehensive Treatment; Paramount Publ. 1993 [Google Scholar]
- 41.Mori T, Yang KH, Kimoto K, Nozaki H. Tetrahedron Lett. 1970:2419. [Google Scholar]
- 42.Rubin MB. Tetrahedron Lett. 1982;23:4615. [Google Scholar]
- 43.Netto-Ferreira JC, Lopes da Silva ESC, de Lucas N. J. Photochem. Photobiol., A. 2011;225:135. [Google Scholar]
- 44.Dalgard JE, Rychnovsky SD. Org. Lett. 2004;6:2713. doi: 10.1021/ol049038x. [DOI] [PubMed] [Google Scholar]
- 45.Giese B, Wettstein P, Stahelin C, Barbosa F, Neuburger M, Zehnder M, Wessig P. Angew. Chem., Int. Ed. 1999;38:2586. [PubMed] [Google Scholar]
- 46.Griesbeck AG, Mauder H, Stadtmueller S. Acc. Chem. Res. 1994;27:70. [Google Scholar]
- 47.Sinicropi A, Barbosa F, Basosi R, Giese B, Olivucci M. Angew. Chem., Int. Ed. 2005;44:2390. doi: 10.1002/anie.200461898. [DOI] [PubMed] [Google Scholar]
- 48.Turro NJ, Lechtken P. Tetrahedron Lett. 1973;14:565. [Google Scholar]
- 49.Srinivasan R, Brown KH, Ors JA, White LS, Adam W. J. Am. Chem. Soc. 1979;101:7424. [Google Scholar]
- 50.Maheshwari KK, De Mayo P, Wiegand D. Can. J. Chem. 1970;48:3265. [Google Scholar]
- 51.Reshetova IG, Tkhaper RK, Kamernitskii AV, Bogdanov VS. Izv. Akad. Nauk SSSR, Ser. Khim. 1990:687. [Google Scholar]
- 52.Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. J. Org. Chem. 1981;46:3936. [Google Scholar]
- 53.Castellano EE, Zukerman-Schpector J, Rehder VLG, Marsaioli AJ. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1989;C45:966. [Google Scholar]
- 54.Fujiwara FY, Rehder VG, Marsaioli AJ. Magn. Reson. Chem. 1992;30:500. [Google Scholar]
- 55.Emmons GT, Wilson WK, Schroepfer GJ., Jr Magn. Reson. Chem. 1989;27:1012. [Google Scholar]
- 56.Corey EJ, Yamamoto H. Tetrahedron Lett. 1970;11:2385. doi: 10.1016/s0040-4039(01)98235-8. [DOI] [PubMed] [Google Scholar]
- 57.Woodward RB, Patchett AA, Barton DHR, Ives DAJ, Kelly RB. J. Chem. Soc. 1957:1131. [Google Scholar]
- 58.Rodewald WJ, Jagodzinski JJ. Pol. J. Chem. 1979;53:1203. [Google Scholar]
- 59.All computational work described herein was carried out using Gaussian 09, Revision A.02 (see Supporting Information for full reference)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.