

Abstract
The remodelling of a natural product core framework by means of diversity-oriented synthesis (DOS) is a valuable approach to access diverse/biologically relevant chemical space and to overcome the limitations of combinatorial-type compounds. Here we provide proof of principle and a thorough conformational analysis for a general strategy whereby the inherent complexity of a starting material is used to define the regio- and stereochemical outcomes of reactions in chemical library construction. This is in contrast to the traditional DOS logic employing reaction development and catalysis to drive library diversity
Introduction
The construction of combinatorial libraries coupled with high throughput screening has been a primary engine of the drug discovery process over the past three decades.1-2 A central tenet of this scientific model is that such libraries confer a massive number of diverse chemical entities and drug leads.3 Due to this reliance on library-derived molecules, natural products, despite their long history of success in pharmaceutical research4-5, have seen a decline in drug discovery efforts.6-7 The subsequent realization that combinatorial-type molecules are substantially less diverse in their chemical structures as compared to molecules from natural sources8-9 has led to the syntheses around “privileged scaffolds”10-13, chemical alteration of crude natural product extracts14-17, and diversity oriented synthesis (DOS), which was introduced to construct complex and diverse molecular skeletons from simple and similar starting materials.18-21
The synthetic approaches for the aforementioned library-types have generally followed the traditional logic of organic synthesis: generating libraries of complex molecules from simple substrates using new reaction development and/or catalysis. The approach we illustrate herein deviates from this traditional model in that we set out to identify chemical functionality that can be exploited to rearrange the carbocyclic skeleton of an abundant natural product. The central hypothesis behind our approach was that instead of relying on reaction development and catalysis to impart stereochemical and regiochemical selectivity, we postulated that the inherent complexity of the natural product-derived substrates can drive stereoselective and regioselective reactions. Herein we provide proof of principle for this concept. While a number of studies have produced novel scaffolds using the rearrangement of functionalities embedded within a natural product core structure22-27, this is the first report to carefully test and rationalize through conformational analysis the hypothesis that subtle perturbations to the structure of the parent natural product will have dramatic effects on the resultant products. Consequently, once a diversification strategy is devised, it can be applied to many members of a natural product family to afford significantly different resultant products. Coupled with a reliance on readily available natural products, we would argue that this general concept will prove to be a powerful and transferrable method for the development of diverse molecular libraries.
In this report, we describe a synthetic strategy for remodelling of a triterpenoid skeleton based on the reactivity patterns of lanosterol (Fig. 1), and application of the devised strategy to a pentacyclic triterpenoid, bryonolic acid. Lanosterol was initially chosen because of the unsaturated B/C ring fusion, which was shown by the groups of Snatzke28, Fox29, Sicinski30-31, and Marsaioli32-33 to undergo iterative allylic oxidation/oxidative cleavage to produce transannular polyketones, followed by the aldol addition reactions to form distinct molecular skeletons. A literature search revealed that bryonolic acid is the major documented pentacyclic triterpenoid with the unsaturated B/C ring fusion.34 We hypothesized that based on this structural similarity, bryonolic acid will react in a complementary fashion producing novel pentacyclic triterpenoids, each distinctively different from the others and the prototype structure. In addition, bryonolic acid can be isolated in gram-quantities from the sprouts of Cucurbita pepo L. (common zucchini)35 and is known to have anti-inflammatory properties.36
Figure 1.
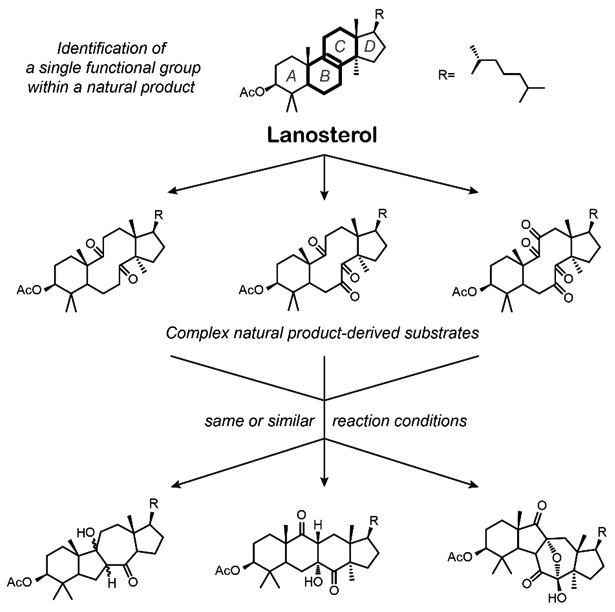
Diversity oriented synthesis strategy based on the reactivity patterns of lanosterol.
Results and Discussion
Substrate 3 was prepared via methylation of C-29 carboxylic acid of bryonolic acid 1 with diazomethane to initially form ester 2, followed by the protection of the hydroxyl group at C3 to generate acetate 3 (Scheme 1).
Scheme 1. Oxidation of the double bond at the B/C ring fusion of the substrate 3.
(a) CH2N2, THF, rt for 5 min (2, 88% yield); (b) (CH3CO)2O, pyridine, 50 °C for 24 h (3, 84% yield)
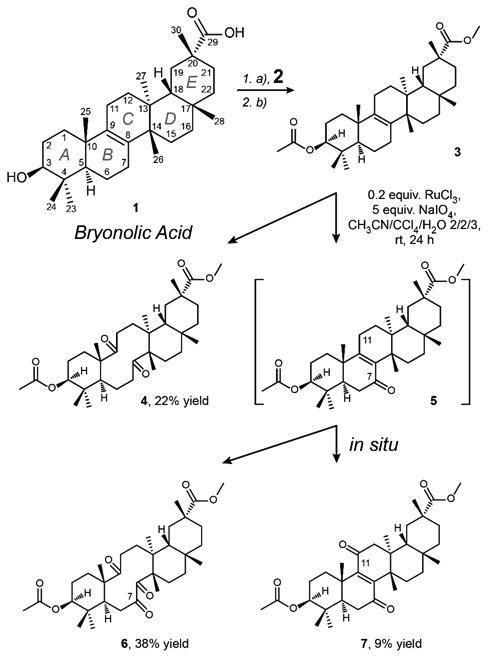
In order to gain access to ketones 4 and 6, protected bryonolic acid 3 was subjected to catalytic oxidation with ruthenium tetroxide under Sharpless conditions.37 Under these conditions, substrate 3 underwent competing oxidative cleavage of Δ8,9 double bond to form the desired diketone 4 and regioselective allylic oxidation at C7 to give α,β-unsaturated ketone 5 (Scheme 1). This transformation was fast and robust providing the complete conversion of starting material in 20 min as detected by TLC. However, with extended reaction time (24 h), intermediate 5 was further oxidized in situ by ruthenium tetroxide to give the desired triketone 6 and the product of competing CH activation 7.
Transannular aldol addition of diketone 4 was next investigated38-41 (Table 1). Examination of the structure of diketone 4 reveals two possible enolization sites, potentially leading to four different regioisomeric aldol adducts (pathways a and b). Since aldol addition is one of the most well-known reactions in organic synthesis42-43, our initial efforts were focused on the standard aldol protocols. The use of catalytic TFA at room temperature led to the formation of the mixture of syn product of pathway a (8) and syn product of pathway b (10) in good overall yield (85%, Table 1, entry 1). The use of sodium hydride as well as pyrrolidine as an organocatalyst for enamine-mediated aldol reaction gave a similar distribution of products 8 and 10, although reaction with sodium hydride led to some degradation (Table 1, entries 2 and 3). Predominant formation of aldol adduct 8 suggested that it was the more thermodynamically stable product of the reaction. It is noteworthy that no products of aldol condensation were observed.
Table 1. Aldol addition of diketone 4 via pathways a and b.
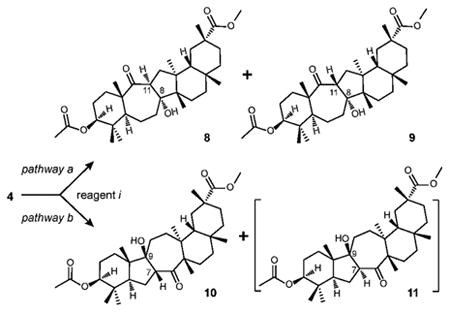
| |||||
|---|---|---|---|---|---|
| entry | reagent ia | isolated yield (%) | |||
| 8 | 9 | 10 | 11 | ||
| 1 | TFA | 45 | 0 | 40 | 0 |
| 2 | NaH | 56 | 0 | 6 | 0 |
| 3 | Pyrrolidine | 80 | 0 | 8 | 0 |
| 4 | LDA | 71 | 0 | 22 | 0 |
| 5 | LiHMDS | 65 | 0 | 25 | 0 |
| 6 | LiTMP | 48 | 0 | 43 | 0 |
| 7 | Ph3CLi | 79 | 0 | 5 | 0 |
| 8 | Al2O3 | 81 | 7 | 0 | 0 |
| 9 | TiCl4 | 50 | 0 | 32 | 0 |
See Experimental Section for detailed experimental procedures
Our next step was to force the transannular aldol reaction of diketone 4 into pathway b under the kinetic conditions. Upon reaction of diketone 4 with LDA (1.1 equiv., THF, -78 °C to rt), aldol adducts 8 and 10 were formed in 71% and 22% yield, respectively (Table 1, entry 4). The increase in the steric bulk of the amide base led to the increased formation of aldol adduct 10. Thus, the use of LiHMDS produced 10 in 25% yield and LiTMP proved to be the reagent of choice, giving the highest yield of aldol adduct 10 (43%, Table 1, entry 6).
However, upon treatment of diketone 4 with Al2O3 (basic) in DCM, aldol adduct 8 was isolated in 81% yield but 10 was not observed. Instead, the anti product of pathway a (9) was formed in 7% yield (Table 1, entry 8). By the virtue of substrate bias, the lowest energy conformation of the cyclodecane ring of diketone 4 “boat (with bow at C-5 and stern between C-8 and C-11) -chair-chair” which determines the α-face position of the carbonyl group at C-8 and β-face position of the carbonyl at C-9 (Figure 2). In both pathways a and b the pseudo-axial α-hydrogen is antiperiplanar with the carbonyl and the pseudo-equatorial α-hydrogen is orthogonal to the C=O bonds, thus producing the trans (Z)-enolate of diketone 4 as the key intermediate after deprotonation by the loss of the pseudo-equatorial hydrogen. The intermediate enolate is geometrically predisposed for the formation of transannular C-C bond with syn configuration at the ring junction. To rationalize the formation of anti product 9, we hypothesized that when treated with a Lewis acid such as Al2O3, the key intermediate engages in the Zimmerman-Traxler transition state44 (Figure 2) which determines the β-face position of H-11 and α-face position of the keto group at C-9. When purified aldol adduct 8 was resubjected to the same reaction conditions (100 equiv. Al2O3, DCM, rt, 16 h), no interconversion of these products was observed. The absence of product interconversion under the same set of conditions led to a conclusion that the relative stereochemistry of anti product 9 is the result of Lewis acid-controlled transition state of the aldol addition reaction of the diketone 4.
Figure 2.
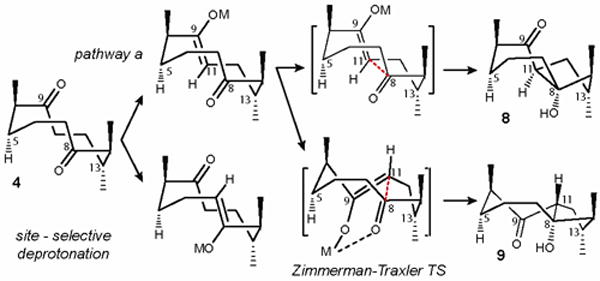
Working model demonstrates the role of the enolate geometry and the conformation of cyclodecane ring of diketone 4 towards stereoselectivity.
It is worth noting that the lithium cation, which is generally known to follow the Zimmerman-Traxler model, did not give any anti products in reactions with amide bases (Table 1, entries 4-6). The reaction of diketone 4 with TiCl4 in the presence of tertiary amine (1.1 equiv., -78 °C to rt) led to the formation of aldol adducts 8 and 10 in 1.6:1 ratio and no anti products were observed (Table 1, entry 9). In due course, treatment of diketone 4 with BF3·Et2O (1.1 equiv., -78 °C to rt) unexpectedly gave aliphatic ketones 12 and 13 and αβ-unsaturated ketone 14 (Scheme 2, pathway c).
Scheme 2.
Aldol addition/condensation of diketone 4 via pathways c and d.
(a) BF3·Et2O (1.1 equiv.), DCM, -78 °C to rt, 4 days; (b) BF3·Et2O (10 equiv.), DCM, -78 °C to rt, 18 h.
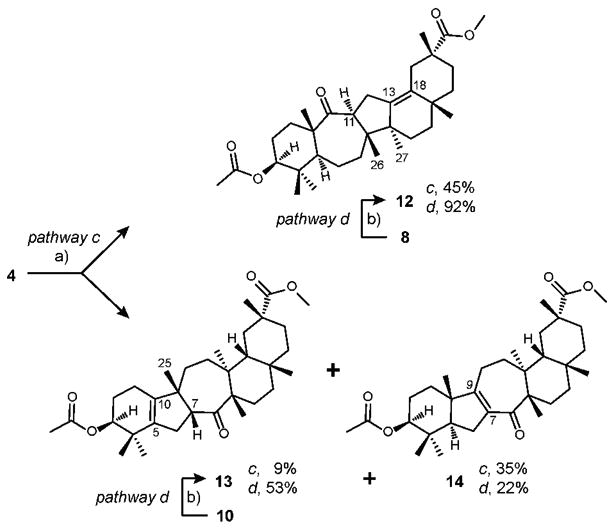
The absolute stereochemistry of H-11 suggested that ketone 12 was the result of aldol addition via pathway a to form aldol adduct 8 followed by a cascade of methide shifts and an elimination of H-18 to form internal double bond at Δ13, 18 (Scheme 3). The control experiment (Scheme 2, pathway d) confirmed our prediction when isolated aldol adduct 8 was treated with BF3·Et2O to form aliphatic ketone 12 in 92% yield. Thermodynamic stability of 8 and trans arrangement of the C-8 hydroxyl group and the methyl at C-26 add to the robustness of this transformation. A similar rationale was then proposed for the formation of ketones 13 and 14 via pathway c in 1:4 ratio. β-Facial position of H-7 in ketone 13 predisposed for the formation of aldol adduct 10 via pathway b to be followed by either a C-25 methide shift or an elimination of H-7 through a common carbocationic intermediate and E1-type transition state to yield ketones 13 or 14, respectively.
Scheme 3. Mechanistic considerations for the formation of ketones 12, 13 and 14 from corresponding aldol adducts 8, 10 and 11.
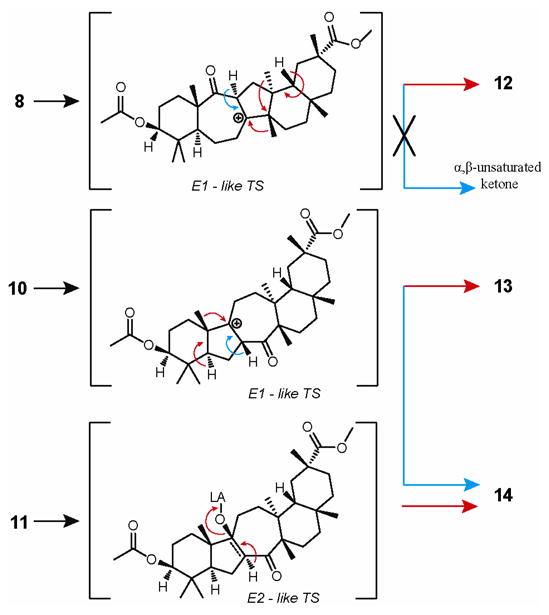
Both of these possibilities seem equally plausible due to the fact that both the C-25 methyl group and the H-7 are in cis arrangement with the hydroxyl group at C-9. The control experiment (Scheme 2, pathway d) with isolated aldol adduct 10 yielded compounds 13 and 14 in 2.4:1 ratio. The reversal of selectivity observed for the production of ketone 13 over 14 via pathway d led to the conclusion that the reaction through pathway c gives intermediately aldol adduct 10 and an anti product of pathway b (11), which has the antiperiplanar arrangement of H-7 and the hydroxyl group at C-9 (Scheme 3), thus raising the possibility of the increased production of ketone 14, if the overall elimination of water is to occur through E2-type transition state.
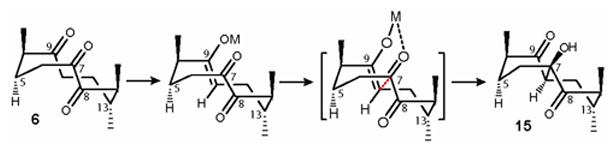
In the case of triketone 6 (Scheme 4), we also located the lowest energy conformation, the cyclodecane part of which closely resembles that of diketone 4 (Figure 3). Abstraction of pseudo-equatorial α-hydrogen at C-11 upon treatment of triketone 6 with Al2O3 in DCM at room temperature produces trans (Z)-enolate, which is structurally biased to adopt the Zimmerman-Traxler transition state with C-7 carbonyl thus forming anti product 15.
Scheme 4. Aldol addition reaction of triketone 6.
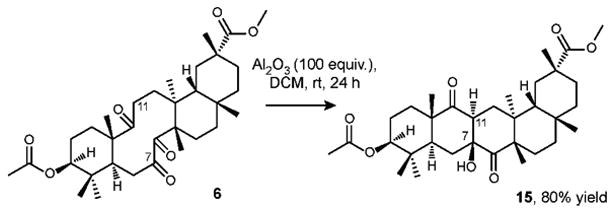
Figure 3. Working model demonstrates the role of the enolate geometry and the conformation of cyclodecane ring of triketone 6 towards stereoselectivity.
In an attempt to obtain the last transannular polyketone in the desired series, we performed the oxidative cleavage of the enone 7 (Scheme 5). As in the case of protected bryonolic acid 3, ruthenium tetroxide under Sharpless conditions proved to be the reagent of choice for this otherwise hardly achievable transformation. However, larger amounts of ruthenium catalyst and reoxidant and a longer reaction time were required to afford, after careful column chromatography, tetraketone 16 together with the product of oxidative destruction 17 in 18% and 21% yield respectively.
Scheme 5. Oxidative cleavage of 7 with ruthenium tetroxide under Sharpless conditions.
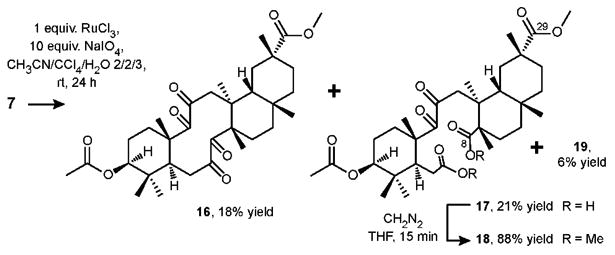
The structure of the carboxylic acid 17 was tentatively suggested after a thorough NMR study but appeared to be difficult to establish due to slow conformational equilibrium and identical chemical shifts of C-8 and C-29 carbonyls at 179.5 ppm. The structure was established unambiguously after carboxylic acid 17 was treated with diazomethane to form the ester 18, which showed three singlets centered around 3.6 ppm in 1H spectrum as well as four ester carbonyls and two keto groups in the 13C spectrum.
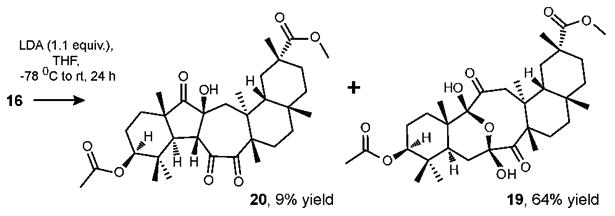
In order to obtain the final aldol adduct in the proposed molecular library, tetraketone 16 was treated with LDA (1.1 equiv., THF, -78 °C to rt) to give the aldol adduct 20 and the product of transannular hemiketalization 19 in 9% and 64% yield, respectively (Scheme 6). As expected, tetraketone 16 was not deprotonated at C-12 since it is considerably more sterically hindered as compared to C-6.
Scheme 6. Aldol addition reaction of tetraketone 16.
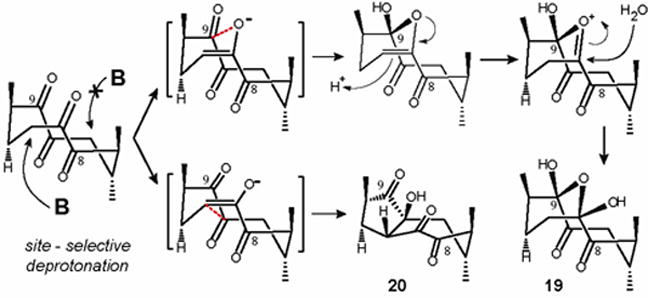
In the ground-state conformation46 of tetraketone 16, pseudo-axial H-6 is coplanar with the carbonyl at C-7, giving rise to the cis (E)-enolate as the key intermediate upon the abstraction of pseudo-equatorial H-6 by the base (Figure 4). The higher yield of 19 over the aldol adduct 20 can be explained in view of the fact that the transition state leading to the aldol adduct 20 seems to be markedly higher in energy in contrast with the transition state leading to the product of transannular hemiketalization 19. The formation of 19 suggests a mechanism which presumably proceeds through an intermediate with the charge localized on the oxygen of the pyranyl ring45, thereby bringing a novel triterpenoid structure into the library by virtue of an unanticipated reaction. This proposed mechanism was confirmed when the reaction was quenched with D2O, resulting in a partial deuterium incorporation at C-6.
Figure 4. Working model demonstrates the role of the enolate geometry and the conformation of cyclodecane ring of tetraketone 16 towards regio- and stereoselectivity.
Conclusion
Here we have illustrated what we consider to be a new principle in the construction of molecular libraries. We have shown a strategy for the remodelling the triterpenoid skeleton devised on the basis of the reactivity of the steroidal triterpenoid lanosterol. We hypothesized that the subtle differences between the steroid and triterpeoid core structures would impart differential reactivity. This hypothesis was confirmed by illustrating that despite the common unsaturation at the B/C-ring fusion, the differences between the parent molecules led to dramatic differences in reactivity and concomitantly, the composition of the resultant chemical library. Specifically, the aldol addition of diketone 4 was shown to follow three different pathways. Moreover, the aldol reactivity of the tetraketone 16 appeared to be completely different from that derived from lanosterol, giving rise to the novel bridged structure 19 as a major reaction product. We would argue that the strategy outlined here is a general approach that could be applied to a wide range of natural product families, allowing access to broad categories of novel and potentially biologically relevant molecules that would be otherwise very difficult to attain.
Experimental Section
(2R,4aS,6aS,8aR,10S,12aS,14aS,14bR)-10-hydroxy-2,4a,6a,9,9,12a,14a-heptamethyl-1,2,3,4,4a,5,6,6a,7,8,8a,9,10,11,12,12a,13,14,14a,14b-icosahydropicene-2-carboxylic acid (1)
Bryonolic acid 1 was isolated in gram-quantities from the sprouts of “Spineless beauty” zucchini squash by previously published method35. mp 274-278 °C (lit35. 274-278 °C).
(2R,4aS,6aS,8aR,10S,12aS,14aS,14bR)-methyl 10-hydroxy-2,4a,6a,9,9,12a,14a-heptamethyl-1,2,3,4,4a,5,6,6a,7,8,8a,9,10,11,12,12a,13,14,14a,14b-icosahydropicene-2-carboxylate (2)
In a round-bottom flask open to atmosphere, bryonolic acid 1 (3 g, 6.57 mmol) was dissolved in 65 ml of freshly distilled THF. Freshly prepared diazomethane in diethyl ether (15 mmol) was added dropwise to the resulting solution by a pipette and the reaction mixture was stirred until full conversion detected by TLC (approximately 5 min). The reaction was quenched by dropwise addition of glacial acetic acid until the yellow color of the solution disappeared. The reaction mixture was concentrated under vacuum and the crude product was purified by column chromatography on silica to give 2 as a white solid (2.72 g, 88%). mp 142-144 °C. Rf = 0.5 (EA/hex, 2/3). 1H NMR (400 MHz, CDCl3, δ): 3.61 (s, 3H), 3.23 (dd, J1 = 12 Hz, J2 = 4 Hz, 1H), 2.37 (d, J = 12 Hz, 1H), 2.19 (m, 1H), 2.1 (s, 3H), 1.02 (s, 3H), 0.99 (s, 3H), 0.95 (s, 3H), 0.94 (s, 3H), 0.79 (s, 3H), 0.74 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 179.4, 134.1, 133.9, 79.0, 51.6, 50.5, 44.8, 41.9, 40.5, 38.9, 37.6, 37.2, 37.1, 35.1, 34.5, 33.0, 31.3, 31.0, 30.9, 30.4, 30.0, 28.1, 27.9, 27.7, 25.1, 22.1, 20.8, 20.0, 19.3, 17.2, 15.7. HRMS (EI) m/z calcd for C31H50O3+ [M]+ 470.37600, found: 470.37553.
(2R,4aS,6aS,8aR,10S,12aS,14aS,14bR)-methyl 10-acetoxy-2,4a,6a,9,9,12a,14a-heptamethyl-1,2,3,4,4a,5,6,6a,7,8,8a,9,10,11,12,12a,13,14,14a,14b-icosahydropicene-2-carboxylate (3)
In a round-bottom flask, bryonolic acid methyl ester 2 (2.35 g, 5 mmol) was dissolved in 10 ml of dry pyridine. Acetic anhydride (1.02 g, 10 mmol) was added to the resulting solution by a quick syringe transfer. The reaction mixture was stirred at 50 °C for 24 hours, after which time pyridine was removed at reduced pressure and the crude mixture was taken into ethyl acetate. The organic layer was washed successively with dilute HCl, water and saturated solution of sodium bicarbonate. It was subsequently dried with Na2SO4 and evaporated in vacuo. The crude product was purified by column chromatography to give 3 as a white solid (2.16 g, 84%). mp 164-167 °C. Rf = 0.62 (EA/hex = 3/7). 1H NMR (400 MHz, CDCl3, δ): 4.47 (dd, J1 = 12 Hz, J2 = 4 Hz, 1H), 3.62 (s, 3H), 2.37 (d, J = 16 Hz, 1H), 2.19 (d, J = 12 Hz, 1H), 2.05 (s, 3H), 1.17 (s, 3H), 1.02 (s, 3H), 0.96 (s, 3H), 0.94 (s, 3H), 0.87 (s, 3H), 0.86 (s, 3H), 0.73 (s, 3H). 13C NMR (100 MHz, CDCl3, δ): 179.3, 171.1, 134.1, 134.0, 81.0, 51.7, 50.7, 44.9, 41.9, 40.6, 37.8, 37.5, 37.3, 37.1, 34.8, 34.5, 33.0, 31.3, 31.0, 30.9, 30.4, 30.0, 28.1, 27.5, 25.1, 24.3, 22.2, 21.5, 20.8, 20.1, 19.2, 17.1, 16.8. HRMS (EI) m/z calcd for C33H52O4+ [M]+ 512.38656, found: 512.38532.
Preparation of 4, 6 and 7
In a 200 ml single-neck round-bottom flask, RuCl3 (81 mg, 0.39 mmol) was added in one portion to the solution of NaIO4 (2.09 g, 9.75 mmol) in 59 ml of H2O, and the resulting suspension was stirred open to atmosphere for 15 min, followed by the addition of 39 ml of acetonitrile. The solution of 3 (1 g, 1.95 mmol) in 39 ml of CCl4 was then added dropwise to the reaction mixture by a syringe-pump. The flask was sealed with a glass stopper and the resulting biphasic mixture was vigorously stirred for 24 hours, at which time 10 ml of ethanol was added to the solution, layers were separated and the aqueous layer was extracted with DCM. The organic layer was dried with Na2SO4, concentrated under vacuum and the products further separated by chromatography. Silica column chromatography yielded an inseparable mixture (351 mg) of 4 and 7, Rf = 0.45 (EA/hex = 25/75); and pure 6 as yellow solid (416 mg, 38%). mp 168-171 °C. Rf = 0.38 (EA/hex = 25/75). 4 and 7 were further separated by semi-prep HPLC (Agilent C18 column 21.2×250 mm, isocratic elution CH3CN/H2O = 9/1, flow rate 5 ml/min) to yield 7 as a yellow oil (96 mg, 9%), Rt = 52 min; and 4 as white foam (231 mg, 22%). Rt = 66 min.
(2R,4aS,6aS,9aS,11S,13aS,16aS,16bR)-methyl 11-acetoxy-2,4a,6a,10,10,13a,16a-heptamethyl-7,14-dioxodocosahydrobenzo[6,7]cyclodeca[1,2-a]naphthalene-2-carboxylate (4)
1H NMR (400 MHz, CDCl3, δ): 4.61 (m, 1H), 3.67 (s, 3H), 2.65-2.57 (2H), 2.39 (d, J = 16 Hz, 1H), 2.22 (d, J = 16 Hz, 1H), 2.08 (s, 3H), 1.174 (s, 3H), 1.170 (s, 3H), 1.14 (s, 3H), 1.12 (s, 3H), 1.04 (s, 3H), 0.94 (s, 3H), 0.87 (s, 3H). 13C NMR (100 MHz, CDCl3, δ): 218.1, 216.6, 179.0, 171.1, 80.2, 54.1, 52.2, 52.0, 45.52, 45.51, 44.2, 40.8, 38.7, 36.4, 36.3, 35.2, 34.7, 32.9, 31.7, 31.6, 31.5, 29.83, 29.79, 29.2, 28.5, 28.4, 28.1, 23.5, 21.4, 19.3, 17.4, 16.4, 16.0. HRMS (EI) m/z calcd for C33H52O6+ [M]+ 544.37639, found: 544.37685.
(2R,4aS,6aS,9aS,11S,13aS,16aS,16bR)-methyl 11-acetoxy-2,4a,6a,10,10,13a,16a-heptamethyl-7,8,14-trioxodocosahydrobenzo[6,7]cyclodeca[1,2-a]naphthalene-2-carboxylate (6)
1H NMR (400 MHz, CDCl3, δ): 4.67 (dd, J1 = 12 Hz, J2 = 4 Hz, 1H), 3.67 (s, 3H), 2.78 (dd, J1 = 20 Hz, J2 = 8 Hz, 1H), 2.71 (dd, J1 = 16 Hz, J2 = 4 Hz, 1H), 2.32 (d, J = 16 Hz, 1H), 2.27 (dd, J1 = 8 Hz, J2 = 4 Hz, 1H), 2.23 (d, J = 12 Hz, 1H), 2.06 (s, 3H), 1.79 (dd, J1 = 16 Hz, J2 = 4 Hz, 1H), 1.73 (dd, J1 = 16 Hz, J2 = 8 Hz, 1H), 1.54 (d, J = 8 Hz, 1H), 1.25 (s, 3H), 1.18 (s, 3H), 1.07 (s, 3H), 1.03 (s, 3H), 1.01 (s, 3H), 0.98 (s, 3H), 0.91 (s, 3H). 13C NMR (100 MHz, CDCl3, δ): 214.1, 212.6, 203.1, 179.2, 171.0, 79.6, 53.1, 52.0, 50.7, 46.3, 44.4, 43.5, 40.8, 39.3, 39.2, 35.4, 33.6, 33.0, 32.3, 31.9, 31.8, 31.7, 31.2, 30.9, 29.6, 28.5, 27.5, 23.4, 21.4, 17.6, 16.9, 16.6, 16.1. HRMS (ESI) m/z calcd for C33H50O7Na+ [M+Na]+ 581.3454, found: 581.3465.
(2R,4aS,6aS,8aR,10S,12aS,14aS,14bR)-methyl 10-acetoxy-2,4a,6a,9,9,12a,14a- heptamethyl-7,13-dioxo-1,2,3,4,4a,5,6,6a,7,8,8a,9,10,11,12,12a,13,14,14a,14b-icosahydropicene-2-carboxylate (7)
1H NMR (400 MHz, CDCl3, δ): 4.56 (m, 1H), 3.66 (s, 3H), 2.58 (ddd, J1 = 12 Hz, J2 = J3 = 4 Hz, 1H), 2.51-2.47 (3H), 2.07 (s, 3H), 1.93 (ddd, J1 = J2 = 12 Hz, J3 = 4 Hz, 1H), 1.28 (s, 3H), 1.24 (s, 3H), 1.20 (s, 3H), 1.06 (s, 3H), 0.989 (s, 3H), 0.987 (s, 3H), 0.96 (s, 3H), 0.89 (s, 3H). 13C NMR (100 MHz, CDCl3, δ): 200.0, 199.8, 179.1, 171.1, 154.9, 152.7, 79.6, 52.1, 52.0, 48.1, 44.2, 42.1, 40.5, 39.1, 38.1, 37.6, 36.8, 36.3, 34.1, 33.6, 32.5, 31.0, 30.9, 30.6, 29.8, 29.6, 27.5, 25.3, 23.9, 21.4, 21.0, 20.6, 17.9, 16.1. HRMS (ESI) m/z calcd for C33H48O6Na+ [M+Na]+ 563.3348, found: 563.3352.
Preparation of 8, 9 and 10
Table 1 (entry1)
In a round-bottom flask open to atmosphere, trifluoroacetic acid (1 μl, 35 mol%) was added to the solution of 4 (20 mg, 0.0367 mmol) in 1 ml of DCM. The flask was sealed with a glass stopper and the mixture was stirred vigorously for 72 hours, at which time the solvent was removed in vacuo and the mixture of products was separated by column chromatography on silica without additional work-up, to yield 8 (9 mg, 45%) and 10 (8 mg, 40%).
Table 1 (entry 2)
NaH (95%, 2 mg, 0.0792 mmol) was placed in a flame-dried (under vacuum) round-bottom flask, followed by the addition of 0.5 ml of dry THF. The resulting suspension was cooled to 0 °C and the solution of 4 (36 mg, 0.066 mmol) in 0.5 ml of dry THF was added dropwise at this temperature. The reaction mixture was stirred at 0 °C for 30 min, then warmed up to r.t and stirred at r.t for 19 hours, at which time the solvent was removed in vacuo and the crude mixture was treated with 5% acetic acid in H2O and the aqueous layer was extracted with DCM. The organic layer was dried with Na2SO4, concentrated under vacuum and the crude mixture of products was separated by column chromatography on silica to give 8 (20 mg, 56%) and 10 (2 mg, 6%).
Table 1 (entry 3)
To a solution of 4 (25 mg, 0.0459 mmol) in 0.5 ml of DCM in a flame dried (under vacuum) round-bottom flask, pyrrolidine (1.9 μl, 1.63 mg, 0.023 mmol) was added dropwise at r.t. The resulting mixture was stirred at that temperature for 24 hours, at which time the solvent was removed under vacuum and the crude mixture of products was separated by column chromatography on silica without additional work-up, to give 8 (20 mg, 80%) and 10 (2 mg, 8%).
General procedure for reaction of 4 with amide bases
Solution of a secondary amine (1.1 eq.) in dry THF (0.027M in amine) in a flame-dried (under vacuum) round-bottom flask was cooled to -78 °C, followed by a dropwise addition of n-butyl lithium (2.5M in hexanes, 1.1 eq.). The reaction mixture was warmed up to 0 °C, stirred at this temperature for 5 min and subsequently cooled to -78 °C. A solution of 4 (1 eq.) in dry THF (0.07M in 4) was then added dropwise to the solution of lithium amide by syringe at -78 °C. The reaction mixture was allowed to warm up to r.t overnight and stirred at r.t. for the total of 24 hours. After removing solvent in vacuo the residue was taken up in DCM, washed with water and brine, the organic layer was dried over Na2SO4. Evaporation of the solvent in vacuo gave crude product mixtures that were further separated by chromatography.
Table 1 (entry 4)
Following the general procedure, the use of diisopropylamine (20 mg, 29 μl, 0.202 mmol), n-butyl lithium (0.202 mmol, 81 μl), 4 (100 mg, 0.1836 mmol) and THF (10 ml) gave, after column chromatography on silica, 8 (71 mg, 71%) and 10 (22 mg, 22%).
Table 1 (entry 5)
Following the general procedure, the use of hexamethyldisilazane (6.5 mg, 8.5 μl, 0.0404 mmol), n-butyl lithium (0.0404 mmol, 16.2 μl), 4 (20 mg, 0.0367 mmol) and THF (2 ml) gave, after column chromatography on silica, 8 (13 mg, 65%) and 10 (5 mg, 25%).
Table 1(entry 6)
Following the general procedure, the use of 2,2,6,6-Tetramethylpiperidine (5.7 mg, 0.0404 mmol), n-butyl lithium (0.0404 mmol, 16.2 μl), 4 (20 mg, 0.0367 mmol) and THF (2 ml) gave, after column chromatography on silica, 8 (9.6 mg, 48%) and 10 (8.6 mg, 43%).
Table 1 (entry 7)
Following the general procedure for amide bases, the use of Ph3CH (9.9 mg, 6.9 μl, 0.0404 mmol), n-butyl lithium (0.0404 mmol, 16.2 μl), 4 (20 mg, 0.0367 mmol) and THF (2 ml) gave, after column chromatography on silica, 8 (16 mg, 79%) and 10 (1 mg, 5%).
Table 1 (entry 8)
A round-bottom flask open to atmosphere was charged with basic alumina (1.9 g, 18.4 mmol), followed by the addition of DCM (1 ml). A solution of 4 (100 mg, 0.184 mmol) in DCM (1 ml) was then added to the resulting suspension. The flask was sealed with a glass stopper and the reaction mixture was vigorously stirred at r.t. for 16 hours, at which time the mixture was filtered over a fine sinter funnel and washed successively with ethyl acetate. After the removal of the solvent in vacuo, the column chromatography on silica yielded an inseparable mixture (90 mg) of 8 and 9. Rf = 0.4 (EA/hex = 25/75). 8 and 9 were further separated by semi-prep HPLC (Agilent C18 column 21.2×250 mm, isocratic elution CH3CN/H2O = 95/5, flow rate 5 ml/min) to yield 8 (81 mg, 81%), Rt = 53 min; and 9 (7 mg, 7%). Rt = 68 min.
Table 1 (entry 9)
Dry DCM (1 ml) was added to a flame-dried (under vacuum) round-bottomed flask. The flask was cooled to -78 °C and TiCl4 (41.8 mg, 0.22 mmol, 24.2 μl) was added at that temperature by a quick syringe transfer, followed by a dropwise addition of diisopropyl ethyl amine (33.2 mg, 0.257 mmol, 44.8 μl). A solution of 4 (100 mg, 0.1836 mmol) in dry DCM (1 ml) was added to the reaction mixture at -78 °C, and the reaction mixture was allowed to warm to r.t and stirred at r.t. for 24 hours, at which time water (2 ml) was added to the solution, layers were separated and the aqueous layer was extracted with DCM. The organic layer was dried with Na2SO4, concentrated under vacuum and the crude mixture of products was separated by column chromatography on silica to give 8 (49.5 mg, 50%) and 10 (31.5 mg, 32%).
(2R,4aS,6aS,6bR,8aS,10S,12aS,13aS,14aS,14bR)-methyl 10-acetoxy-6b-hydroxy- 2,4a,6a,9,9,12a,14a-heptamethyl-13-oxodocosahydrobenzo[f]naphtho[2,1-a]azulene-2-carboxylate (8)
white solid. mp 235-238 °C. Rf = 0.53 (EA/hex = 3/7). Rf = 0.4 (EA/hex = 25/75). 1H NMR (600 MHz, CDCl3, δ): 4.45 (m, 1H), 3.62 (s, 3H), 3.57 (dd, J1 = J2= 12 Hz, 1H), 2.43 (dd, J1 = 12.6 Hz, J2 = 4.2 Hz, 1H), 2.17 (2H), 2.04 (s, 3H), 1.18 (s, 3H), 1.16 (s, 3H), 1.08 (s, 3H), 1.05 (s, 3H), 0.96 (s, 3H), 0.95 (s, 3H), 0.59 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 215.4 (CO), 179.3 (CO), 171.0 (CO), 84.5 (COH), 80.2 (CH), 56.8 (CH), 52.8 (C), 51.8 (CH3), 49.6 (C), 46.8 (C), 46.0 (CH), 44.1 (CH), 40.7 (C), 38.5 (CH2), 37.8 (C), 37.4 (CH2), 35.9 (CH2), 34.9 (CH2), 33.0 (CH2), 32.7 (CH3), 31.4 (C), 31.1 (CH3), 30.6 (CH2), 30.3 (CH2), 27.8 (CH3), 24.0 (CH2), 23.7 (CH2), 21.4 (CH3), 21.2 (CH3), 19.7 (CH2), 17.8 (CH3), 16.6 (CH3), 16.1 (CH3). HRMS (ESI) m/z calcd for C33H52O6Na+ [M+Na]+ 567.3661, found: 567.3660.
Preparation of 8, 9 and 10. (2R,4aS,6aS,6bR,8aS,10S,12aS,13aR,14aS,14bR)-methyl 10-acetoxy-6b-hydroxy-2,4a,6a,9,9,12a,14a-heptamethyl-13-oxodocosahydrobenzo[f]naphtho[2,1-a]azulene-2-carboxylate (9)
white foam. Rf = 0.4 (EA/hex = 25/75). 1H NMR (600 MHz, CDCl3, δ): 4.48 (dd, J1 =17.4 Hz, J2 = 6 Hz, 1H), 3.70 (s, 3H), 3.60 (dd, J1 =16.2 Hz, J2 = 4.2 Hz, 1H), 2.35 (m, 1H), 2.04 (s, 3H), 1.18 (s, 3H), 1.10 (s, 3H), 1.07 (s, 3H), 1.04 (s, 3H), 0.91 (s, 3H), 0.90 (s, 3H), 0.89 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 212.4 (CO), 179.1 (CO), 170.8 (CO), 87.7 (COH), 80.4 (C), 53.3 (CH), 53.0 (C), 52.1 (CH3), 49.5 (C), 48.5 (C), 47.1 (CH), 44.2 (CH), 40.7 (C), 40.0 (C), 37.6 (CH2), 37.4 (CH2), 35.8 (CH2), 35.7 (CH2), 32.8 (CH2), 32.6 (CH3), 32.3 (CH2), 31.5 (C), 31.3 (CH3), 30.2 (CH2), 28.0 (CH3), 24.2 (CH2), 23.7 (CH2), 23.5 (CH2), 21.7 (CH3), 21.5 (CH3), 21.2 (CH3), 18.1 (CH3), 16.4 (CH3). HRMS (ESI) m/z calcd for C33H52O6Na+ [M+Na]+ 567.3661, found: 567.3654.
(2R,4aS,6aS,7aR,8aS,10S,12aS,12bS,14aS,14bR)-methyl 10-acetoxy-12b-hydroxy- 2,4a,6a,9,9,12a,14a-heptamethyl-7-oxodocosahydrobenzo[a]naphtho[2,1-f]azulene-2-carboxylate (10)
white solid. mp 187-190 °C. Rf = 0.46 (EA/hex = 3/7). 1H NMR (600 MHz, CDCl3, δ): 4.45 (dd, J1 = 12 Hz, J2 = 4.8 Hz, 1H), 3.64 (s, 3H), 3.14 (d, J = 9.6 Hz, 1H), 2.42-2.38 (2H), 2.22 (d, J = 14.4 Hz, 1H), 2.16 (ddd, J1 = J2 = 14.4 Hz, J3 = 4.8 Hz, 1H), 2.06 (ddd, J1 = J2 = 14.4 Hz, J3 = 4.8 Hz, 1H), 2.05 (s, 3H), 1.84 (dd, J1 = J2 = 13.2 Hz, 1H), 1.75-1.47 (12H), 1.45 (s, 3H), 1.42-1.27 (4H), 1.18 (s, 3H), 1.13 (dd, J1 = 13.8 Hz, J2 = 6 Hz, 1H), 1.08 (s, 3H), 1.04 (s, 3H), 0.96 (s, 3H), 0.92 (s, 3H), 0.64 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 214.6 (CO), 179.2 (CO), 171.2 (CO), 82.1 (C), 81.0 (CH), 58.5 (CH), 56.3 (C), 52.0 (CH3), 50.5 (CH), 49.7 (C), 45.7 (CH), 40.8 (C), 39.1 (C), 37.5 (C), 36.5 (CH2), 34.0 (CH2), 33.0 (CH3), 32.8 (CH2), 31.8 (C), 31.40 (CH3), 31.35 (CH2), 31.0 (CH2), 30.8 (CH2), 29.8 (CH2), 29.0 (CH3), 27.6 (CH2), 25.0 (CH2), 22.8 (CH2), 21.4 (CH3), 18.0 (CH3), 17.8 (CH3), 17.0 (CH3), 16.1 (CH3). HRMS (ESI) m/z calcd for C33H52O6Na+ [M+Na]+ 567.3661, found: 567.3647.
Preparation of 12, 13 and 14
Solution of 4 (30 mg, 0.055 mmol) in dry DCM (1 ml) in a flame-dried (under vacuum) round-bottom flask was cooled to -78 °C, followed by a dropwise addition of BF3·Et2O (purified, redistilled) (8.6 mg, 0.0605 mmol, 7.6 μl) at this temperature. The reaction mixture was allowed to warm up to rt overnight and stirred at this temperature for the total of 4 days, at which time the solvent was removed under vacuum, water (2 ml) was added to the solution, layers were separated and the aqueous layer was extracted with DCM. The organic layer was dried with Na2SO4, concentrated under vacuum and the crude mixture of products was separated by column chromatography on silica to give 13 as transparent oil (2.5 mg, 9%), Rf = 0.3 (EA/hex = 15/85); 12 as white solid (13 mg, 45%), mp 214 °C, Rf = 0.27 (EA/hex = 15/85); and 14 as white solid (10 mg, 35%). mp 198-202 °C. Rf = 0.22 (EA/hex = 15/85).
(2R,4aS,6aS,6bR,8aS,10S,12aS,13aS)-methyl 10-acetoxy-2,4a,6a,6b,9,9,12a-heptamethyl-13-oxo-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,11,12,12a,13,13a,14-icosahydrobenzo[f]naphtha [2,1-a] azulene-2-carboxylate (12)
1H NMR (600 MHz, CDCl3, δ): 4.52 (dd, J1 = 10.2 Hz, J2 = 6 Hz, 1H), 3.65 (dd, J1 = J2 = 9 Hz, 1H), 3.57 (s, 3H), 2.92 (dd, J1 = 18 Hz, J2 = 8.4 Hz, 1H), 2.74 (dd, J1 = 13.8 Hz, J2 = 2.4 Hz, 1H), 2.14 (dd, J1 = 17.4 Hz, J2 = 9.6 Hz, 1H), 2.05 (s, 3H), 1.25 (s, 3H), 1.17 (s, 3H), 0.99 (s, 6H), 0.96 (s, 3H), 0.89 (s, 3H), 0.48 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 217.4 (CO), 177.4 (CO), 171.0 (CO), 138.3 (C), 130.2 (C), 80.1 (CH), 51.5 (C), 51.33 (CH3), 51.26 (CH), 50.7 (CH), 49.8 (C), 49.7 (C), 45.9 (C), 39.7 (CH2), 38.8 (C), 36.8 (CH2), 36.6 (CH2), 36.1 (CH2), 35.7 (CH2), 34.1 (C), 32.2 (CH2), 28.2 (CH3), 28.1 (CH3), 26.8 (CH2), 25.1 (CH2), 24.8 (CH3), 23.7 (CH2), 22.7 (CH3), 21.8 (CH2), 21.4 (CH3), 19.0 (CH3), 17.5 (CH3), 16.9 (CH3). HRMS (ESI) m/z calcd for C33H50O5Na+ [M+Na]+ 549.3556, found: 549.3551.
(2R,4aS,6aS,7aR,10S,12bR,14aS,14bR)-methyl 10-acetoxy-2,4a,6a,9,9,12b,14a-heptamethyl-7-oxo-1,2,3,4,4a,5,6,6a,7,7a,8,9,10,11,12,12b,13,14,14a,14b-icosahydrobenzo[a]naphtho[2,1-f]azulene-2-carboxylate (13)
1H NMR (600 MHz, CDCl3, δ): 4.75 (ddd, J1 = 10.2 Hz, J2 = 3 Hz, J3 = 1.8 Hz, 1H), 3.58 (s, 3H), 3.32 (ddd, J1 = 9 Hz, J2 = 3 Hz, J3 = 1.2 Hz, 1H), 2.96 (m, 1H), 2.42 (d, J = 15.6 Hz, 1H), 2.05 (s, 3H), 1.32 (s, 3H), 1.25 (s, 3H), 1.17 (s, 3H), 1.07 (s, 6H), 1.00 (s, 3H), 0.67 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 214.8 (CO), 179.1 (CO), 171.2 (CO), 137.5 (C), 137.3 (C), 78.2 (CH), 55.5 (C), 54.5 (CH), 51.8 (CH3), 50.9 (C), 45.3 (CH), 40.7 (C), 39.4 (C), 36.7 (CH2), 36.2 (C), 34.9 (CH2), 32.9 (CH3), 32.2 (CH2), 31.8 (CH2), 31.7 (CH3), 31.5 (C), 31.4 (CH2), 30.4 (CH2), 29.9 (CH2), 27.0 (CH2), 26.2 (CH3), 25.6 (CH3), 24.5 (CH2), 21.6 (CH3), 21.4 (CH3), 19.7 (CH2), 19.3 (CH3), 17.5 (CH3). HRMS (ESI) m/z calcd for C33H50O5Na+ [M+Na]+ 549.3556, found: 549.3544.
(2R,4aS,6aS,8aR,10S,12aS,14aS,14bR)-methyl 10-acetoxy-2,4a,6a,9,9,12a,14a- heptamethyl-7-oxo-1,2,3,4,4a,5,6,6a,7,8,8a,9,10,11,12,12a,13,14,14a,14b-icosahydrobenzo[a]naphtho[2,1-f]azulene-2-carboxylate (14)
1H NMR (600 MHz, CDCl3, δ): 4.52 (dd, J1 = 11.4 Hz, J2 = 4.8 Hz, 1H), 3.59 (s, 3H), 2.68 (dd, J1 =15 Hz, J2 = 6.6 Hz, 1H), 2.35 (d, J = 16.2 Hz, 1H), 2.05 (s, 3H), 1.19 (s, 3H), 1.08 (s, 3H), 1.01 (s, 3H), 0.98 (s, 3H), 0.97 (s, 3H), 0.89 (s, 3H), 0.77 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 206.9 (CO), 179.4 (CO), 171.1 (CO), 158.9 (C), 135.5 (C), 80.9 (CH), 56.2 (CH), 55.6 (C), 51.8 (CH3), 50.8 (C), 46.1 (CH), 40.8 (C), 39.3 (C), 37.2 (C), 36.4 (CH2), 34.0 (CH2), 33.0 (CH3), 32.7 (C), 32.6 (CH2), 32.5 (CH2), 32.0 (CH2), 31.3 (CH3), 29.6 (CH2), 29.3 (CH2), 28.5 (CH3), 26.1 (CH2), 24.9 (CH2), 24.0 (CH2), 21.9 (CH3), 21.4 (CH3), 17.2 (CH3), 17.1 (CH3), 16.7 (CH3). HRMS (ESI) m/z calcd for C33H51O5+ [M+H]+ 527.3737, found: 527.3736.
General procedure for reaction via pathway d
A flame-dried (under vacuum) round-bottom flask was cooled to -78 °C and charged with BF3·Et2O (purified, redistilled) (10 eq.) followed by the addition of DCM (0.14M in starting material) The solution of aldol adduct (1 eq.) in DCM (0.14M in starting material) was then added dropwise to the reaction mixture at -78 °C, allowed to warm up to r.t. overnight and stirred at this temperature for the total of 18 hours, at which time the solvent was removed under vacuum, water (2 ml) was added to the solution, layers were separated and aqueous layer extracted with DCM. The organic layer was dried with Na2SO4, concentrated under vacuum and the crude mixture of products was separated by column chromatography on silica.
Reaction of 8 via pathway d
Following the general procedure for reaction via pathway d, the use of 8 (75 mg, 0.1377 mmol), BF3·Et2O (195.4 mg, 1.377 mmol, 173 μl) and DCM (2 ml) gave, after column chromatography, 12 (67 mg, 92%). Rf = 0.58 (EA/hex = 3/7).
Reaction of 10 via pathway d
Following the general procedure for reaction via pathway d, the use of 10 (37 mg, 0.068 mmol), BF3·Et2O (96.5 mg, 0.68 mmol, 85 μl) and DCM (1.4 ml) gave, after column chromatography, 13 (19 mg, 53%), Rf = 0.5 (EA/hex = 25/75); and 14 (8 mg, 22%), Rf = 0.43 (EA/hex = 25/75).
(2R,4aS,6aS,7aS,8aS,10S,12aS,13aS,14aS,14bR)-methyl 10-acetoxy-7a-hydroxy-2,4a,6a,9,9,12a,14a-heptamethyl-7,13-dioxodocosahydrobenzo[a]tetracene-2-carboxylate (15)
A round-bottom flask open to atmosphere was charged with basic alumina (11 g, 107.4 mmol), followed by the addition of DCM (5.7 ml). A solution of 6 (600 mg, 1.074 mmol) in DCM (5 ml) was then added to the resulting suspension. The flask was sealed with a glass stopper and the reaction mixture was vigorously stirred at r.t. for 24 hours, at which time the mixture was filtered on a fine sinter funnel and washed successively with ethyl acetate. After the removal of the solvent in vacuo, column chromatography on silica yielded 15 as white solid (480 mg, 80%). mp 292 °C. Rf = 0.37 (EA/hex = 3/7). 1H NMR (600 MHz, CDCl3, δ): 4.52 (m, 1H), 3.59 (s, 3H), 2.90 (m, 1H), 2.61(dd, J1 = 16 Hz, J2 = 8 Hz, 1H), 2.43 (m, 1H), 2.15 (m, 1H), 2.05 (s, 3H), 1.96 (ddd, J1 = J2 = 16 Hz, J3 = 4 Hz, 1H), 1.31 (s, 3H), 1.20 (s, 3H), 1.15 (s, 3H), 1.06 (s, 3H), 0.97 (s, 3H), 0.91 (s, 3H), 0.62 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 216.6 (CO), 213.8 (CO), 179.1 (CO), 171.0 (CO), 80.2 (CH), 75.5 (COH), 52.7 (C), 51.9 (CH3), 47.3 (CH), 46.3 (C), 45.9 (CH), 43.8 (CH), 40.7 (C), 39.7 (C), 38.2 (C), 35.6 (CH2), 35.4 (CH2), 33.9 (CH2), 32.7 (CH3), 31.8 (CH3), 30.9 (CH2), 30.8 (C), 30.4 (CH2), 29.9 (CH2), 28.1 (CH2), 27.7 (CH3), 24.7 (CH2), 23.8 (CH2), 21.3 (CH3), 21.0 (CH3), 20.3 (CH3), 19.8 (CH3), 16.7 (CH3). HRMS (EI) m/z calcd for C33H50O7 [M]+ 558.35565, found: 558.35386.
Preparation of 16 anf 17
A round-bottom flask open to atmosphere was charged with RuCl3 (161 mg, 0.777 mmol) and NaIO4 (1.66 g, 7.77 mmol), followed by the addition of H2O (11.7 ml) and CH3CN (7.8 ml). To the resulting dark suspension, a solution of 7 (420 mg, 0.777 mmol) in CCl4 (7.8 ml) was added by a quick syringe transfer. The flask was sealed with a glass stopper and the resulting biphasic mixture was vigorously stirred for 7 days, at which time 5 ml of ethanol was added to the solution. Subsequently, layers were separated and aqueous layer was extracted with DCM. The organic layer was dried with Na2SO4, concentrated under vacuum and the crude mixture of products was separated by flash column chromatography with gradient elution to give 16 as a yellow solid (75 mg, 18%), mp 186-188 °C, Rf = 0.76 (EA/hex = 1/1); unreacted 7 (18 mg), Rf = 0.76 (EA/hex = 1/1); 19 as a transparent oil (26 mg, 6%), Rf = 0.56 (EA/hex = 1/1); 17 as a yellow solid (92 mg, 21%). mp 93-96 °C. Rf = 0.1 (EA/hex = 1/1).
(2R,4aS,6aS,9aS,11S,13aS,16aS,16bR)-methyl 11-acetoxy-2,4a,6a,10,10,13a,16a- heptamethyl-7,8,14,15-tetraoxodocosahydrobenzo[6,7]cyclodeca[1,2-a]naphthalene-2-carboxylate (16)
1H NMR (600 MHz, CDCl3, δ): 4.74 (dd, J1 =12 Hz, J2 = 3.6 Hz, 1H), 3.74 (s, 3H), 3.51 (d, J = 13.2 Hz, 1H), 3.28 (dd, J1 =14.4 Hz, J2 = 4.2 Hz, 1H), 2.76 (ddd, J1 = J2 = 13.8 Hz, J3 = 3.6 Hz, 1H), 2.59 (dd, J1 = 10.2 Hz, J2 = 3.6 Hz, 1H), 2.34 (d, J = 16.2 Hz, 1H), 2.28 (d, J = 14.4 Hz, 1H), 2.07 (s, 3H), 1.48 (s, 3H), 1.26 (s, 3H), 1.22 (s, 3H), 1.05 (s, 3H), 1.01 (s, 3H), 1.00 (s, 3H), 0.96 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 207.4 (CO), 205.9 (CO), 203.6 (CO), 201.1 (CO), 178.7 (CO), 170.6 (CO), 79.1 (CH), 53.3 (C), 52.1 (CH3), 51.1 (C), 45.8 (CH), 44.0 (C), 42.5 (CH2), 41.6 (CH), 40.6 (C), 38.7 (C), 38.5 (CH2), 35.1 (CH2), 34.7 (CH2), 33.7 (CH2), 32.7 (CH3), 32.0 (C), 31.6 (CH2), 31.4 (CH3), 29.69 (CH2), 29.67 (CH2), 27.7 (CH3), 23.4 (CH2), 21.4 (CH3), 18.0 (CH3), 17.4 (CH3), 17.2 (CH3), 16.5 (CH3). HRMS (ESI) m/z calcd for C33H48O8Na+ [M+Na]+ 595.3247, found: 595.3256.
(1S,2S,4aR,7R,8aR)-1-(3-((1S,2S,4S)-4-acetoxy-2-(carboxymethyl)-1,3,3-trimethylcyclohexyl)-2,3-dioxopropyl)-7-(methoxycarbonyl)-1,2,4a,7-tetramethyldecahydronaphthalene-2-carboxylic acid (17)
1H NMR (600 MHz, CDCl3, δ): 9.65 (br. s), 4.73 (dd, J1 =11.4 Hz, J2 = 4.2 Hz, 1H), 3.62 (s, 3H), 3.52 (d, J = 19.8 Hz, 1H), 2.80 (dd, J1 = J2 = 4.8 Hz, 1H), 2.64 (d, J = 19.8 Hz, 1H), 2.47 (dd, J1 =17.4 Hz, J2 = 5.4 Hz, 1H), 2.18 (dd, J1 =17.4 Hz, J2 = 5.4 Hz, 1H), 2.07 (s, 3H), 1.32 (s, 3H), 1.23 (s, 3H), 1.20 (s, 3H), 1.13 (s, 3H), 0.97 (s, 3H), 0.94 (s, 3H), 0.94 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 204.6 (CO), 199.7 (CO), 183.9 (CO), 179.5 (CO), 179.5 (CO), 170.8 (CO), 79.1 (CH), 51.8 (CH3), 50.4 (C), 47.2 (C, br.), 45.8 (CH2, br.), 43.6 (CH), 42.9 (C, br.), 42.6 (C, br.), 40.4 (CH, br.), 38.9 (C), 37.9 (CH2, br.), 33.7 (C), 32.9 (CH2, br.), 32.4 (CH2), 32.2 (CH2, br.), 30.5 (CH3), 29.8 (CH2, br.), 29.4 (CH2), 28.6 (CH2, br.), 27.7 (CH3), 24.5 (CH3, br.), 23.1 (CH2), 22.7 (CH3, br.), 21.4 (CH3), 20.5 (CH3, br.), 17.6 (CH3, br.), 17.3 (CH3, br.). HRMS (ESI) m/z calcd for C33H50O10Na+ [M+Na]+ 629.3301, found: 629.3299.
(1S,2S,4aR,7R,8aR)-dimethyl 1-(3-((1S,2S,4S)-4-acetoxy-2-(2-methoxy-2-oxoethyl)-1,3,3-trimethylcyclohexyl)-2,3-dioxopropyl)-1,2,4a,7-tetramethyldecahydronaphthalene-2,7-dicarboxylate (18)
Solution of 17 (30 mg, 0.05 mmol) in diethyl ether (1 ml) in a round-bottomed flask was cooled down to 0 °C, followed by a dropwise addition of solution of diazomethane in ether (about 2 ml total), and the reaction mixture was stirred at 0 °C until full conversion of the starting material was detected by TLC (approximately 15 min). The excess of diazomethane was quenched by a drop of glacial acetic acid, the solvent was removed in vacuo and the crude product was further purified by column chromatography on silica to give trimethyl ester 18 as a white solid (29.5 mg, 93%). mp 114-117 °C. Rf = 0.7 (EA/hex = 1/1). 1H NMR (400 MHz, CDCl3, δ): 4.69 (dd, J1 = 11.6 Hz, J2 = 4 Hz, 1H), 3.66 (s, 3H), 3.63 (s, 3H), 3.62 (s, 3H), 3.51 (d, J = 20 Hz, 1H), 2.77 (dd, J1 = 6.4 Hz, J2 = 5.2 Hz, 1H), 2.72 (d, J = 20 Hz, 1H), 2.44-2.31 (3H), 2.06 (s, 3H), 1.33 (s, 3H), 1.26 (s, 3H), 1.20 (s, 3H), 1.08 (s, 3H), 0.97 (s, 3H), 0.95 (s, 3H), 0.92 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 205.5, 200.9, 179.5, 177.9, 174.0, 170.7, 79.1, 52.0, 46.9, 46.1, 44.2, 43.0, 42.6, 40.1, 38.8, 38.2, 33.7, 33.6 (br.), 32.4, 31.9, 30.7, 30.0, 29.5, 28.4 (br.), 27.7, 24.4, 23.1, 23.0, 21.4, 19.9 (br.), 17.31 (br.), 17.28 (br.). HRMS (EI) m/z calcd for C35H54O10+ [M]+ 634.37170, found: 634.36992.
Preparation of 19 and 20
Solution of diisopropylamine (10.7 mg, 0.1056 mmol, 15 μl) in dry THF (2 ml) in a flame-dried (under vacuum) round-bottom flask was cooled to -78 °C, followed by a dropwise addition of n-butyl lithium (2.5M in hexanes, 0.1056 mmol, 42 μl.). The reaction mixture was warmed to 0 °C, stirred at this temperature for 5 min and then cooled to -78 °C. A solution of 16 (55 mg, 0.096 mmol) in dry THF (3 ml) was then added dropwise to the solution of LDA by syringe at -78 °C. The reaction mixture was stirred at this temperature for 30 min, at which time the reaction was quenched by H2O (D2O) (2 ml). Followed the the addition of DCM (2 ml), the layers were separated and the aqueous layer extracted with DCM. Organic fractions were combined, washed with water and brine and dried over Na2SO4. Evaporation of the solvent in vacuo gave crude product mixtures that were further separated by chromatography. Column chromatography on silica yielded an inseparable mixture of compounds (7 mg) containing 20, Rf = 0.43 (EA/hex = 4/6); and pure 19 as a transparent oil (36 mg, 64%). Rf = 0.31 (EA/hex = 4/6). A mixture of unidentifiable compounds containing 20 was further purified by semi-prep HPLC (Agilent C18 column 21.2×250 mm, isocratic elution CH3CN/H2O = 8/2, flow rate 5 ml/min) to yield 20 as a white foam (5 mg, 9%). Rt = 30 min.
(2R,4aS,6aS,8S,9aS,11S,13aS,14R,16aS,16bR)-methyl 11-acetoxy-8,14-dihydroxy-2,4a,6a,10,10,13a,16a-heptamethyl-7,15-dioxodocosahydro-8,14-epoxybenzo[6,7]cyclodeca[1,2-a]naphthalene-2-carboxylate (19, 6-H)
1H NMR (600 MHz, CDCl3, δ): 4.65 (dd, J1 =11.4 Hz, J2 = 3.6 Hz, 1H), 3.74 (s, 3H), 3.14 (d, J = 12 Hz, 1H), 3.08 (br. s, 1H), 2.91 (br. S, 1H), 2.75 (2H), 2.37 (d, J = 15.6 Hz, 1H), 2.31-2.23 (2H), 2.18 (d, J = 12 Hz, 1H), 2.05 (s, 3H), 1.82 (s, 3H), 1.19 (s, 3H), 1.18 (s, 3H), 1.08 (s, 3H), 0.92 (s, 3H), 0.91 (s, 3H), 0.71 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 207.6 (CO), 205.1 (CO), 178.6 (CO), 170.9 (CO), 100.9 (COH), 99.0 (COH), 79.9 (CH), 55.0 (C), 52.1 (CH3), 44.8 (CH), 44.6 (C), 43.6 (CH2), 40.6 (C), 40.5 (CH), 40.1 (C), 37.3 (C), 35.8 (CH2), 34.7 (CH2), 32.7 (CH3), 31.6 (CH2), 31.54 (CH3), 31.47 (C), 29.8 (CH2), 29.4 (CH2), 29.0 (CH2), 28.7 (CH2), 27.8 (CH3), 23.6 (CH2), 21.4 (CH3), 16.92 (CH3), 16.88 (CH3), 16.5 (CH3), 16.0 (CH3). HRMS (ESI) m/z calcd for C33H50O9Na+ [M+Na]+ 613.3353, found: 613.3352.
Hemiketal (19, 6-D)
2H NMR (600 MHz, CHCl3, δ): 1.35 (br. m). HRMS (ESI) m/z calcd for C33H49DO9Na+ [M+Na]+ 614.34098, found: 614.34077.
(2R,4aS,6aS,8aR,8bS,10S,12aS,13aS,14aS,14bR)-methyl 10-acetoxy-13a-hydroxy-2,4a,6a,9,9,12a,14a-heptamethyl-7,8,13-trioxodocosahydrobenzo[a]naphtho[1,2-f]azulene-2-carboxylate (20)
1H NMR (600 MHz, CDCl3, δ): 4.49 (dd, J1 = 12 Hz, J2 = 4.8 Hz, 1H), 3.62 (s, 3H), 3.40 (d, J = 13.8 Hz, 1H), 2.76 (d, J = 15 Hz, 1H), 2.58 (d, J = 16.2 Hz, 1H), 2.22 (d, J = 13.8 Hz, 1H), 2.06 (s, 3H), 1.33 (s, 3H), 1.24 (s, 3H), 1.22 (s, 3H), 1.09 (s, 3H), 1.05 (s, 3H), 1.02 (s, 3H), 0.51 (s, 3H). 13C NMR (150 MHz, CDCl3, δ): 216.1 (CO), 209.9 (CO), 209.7 (CO), 178.9 (CO), 170.8 (CO), 80.0 (CH), 78.9 (COH), 57.6 (CH), 54.2 (C), 52.1 (CH3), 50.0 (CH), 47.3 (C), 44.5 (CH), 43.6 (CH2), 40.6 (C), 40.0 (C), 39.9 (C), 35.7 (CH2), 33.9 (CH2), 32.5 (CH3), 32.1 (C), 31.52 (CH2), 31.45 (CH3), 30.9 (CH2), 29.8 (CH2), 27.3 (CH3), 23.8 (CH2), 23.7 (CH2), 21.4 (CH3), 21.2 (CH3), 19.1 (CH3), 18.0 (CH3), 17.8 (CH3). HRMS (EI) m/z calcd for C33H48O8+ [M]+ 572.33492, found: 572.33395.
Supplementary Material
Acknowledgments
This work was begun in the laboratory of Stuart L. Schreiber during the postdoctoral tenure of G.P.T. and was supported by a grant from the National Institute of General. Medical Sciences, GM38627. The completion of the work occurred in the laboratory of G.P.T. and was supported by grants from the National Cancer Institute CA157735 & CA132168, NSF grant MCB-084480, Reuter Foundation, and Landon Foundation INNOVATOR award from AACR to G.P.T.
Footnotes
Supporting Information: General experimental methods, mechanistic considerations for the formation of 10 and 11, conformational analysis of 4, 6, 16; complete reference 46; 1H and 13C NMR spectra of all new compounds, as well as HMQC, HMBC, COSY, NOESY NMR spectra and the key COSY, HMBC and NOESY correlations of compounds 6, 8-10, 12-15, 17, 19, 20. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Trish A. Drug Discovery Today. 1999;4:398. doi: 10.1016/s1359-6446(99)01384-7. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. J Comb Chem. 2008;10:345. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]
- 3.Borman STU. Chemical & Engineering News Archive. 1999;77:33. [Google Scholar]
- 4.Newman DJ, Cragg GM. J Nat Prod. 2007;70:461. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 5.Clardy J, Walsh C. Nature. 2004;432:829. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- 6.Newman DJ. J Med Chem. 2008;51:2589. doi: 10.1021/jm0704090. [DOI] [PubMed] [Google Scholar]
- 7.Li JWH, Vederas JC. Science. 2009;325:161. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 8.Henkel T, Brunne RM, Müller H, Reichel F. Angew Chem Int Ed. 1999;38:643. doi: 10.1002/(SICI)1521-3773(19990301)38:5<643::AID-ANIE643>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 9.Feher M, Schmidt JM. J Chem Inf Comput Sci. 2003;43:218. doi: 10.1021/ci0200467. [DOI] [PubMed] [Google Scholar]
- 10.Cohen F, Collins SK, Overman LE. Org Lett. 2003;5:4485. doi: 10.1021/ol0357999. [DOI] [PubMed] [Google Scholar]
- 11.Nicolaou KC, Pfefferkorn JA, Barluenga S, Mitchell HJ, Roecker AJ, Cao GQ. J Am Chem Soc. 2000;122:9968. [Google Scholar]
- 12.Nicolaou KC, Pfefferkorn JA, Mitchell HJ, Roecker AJ, Barluenga S, Cao GQ, Affleck RL, Lillig JE. J Am Chem Soc. 2000;122:9954. [Google Scholar]
- 13.Nicolaou KC, Pfefferkorn JA, Roecker AJ, Cao GQ, Barluenga S, Mitchell HJ. J Am Chem Soc. 2000;122:9939. [Google Scholar]
- 14.Lopez SN, Ramallo IA, Sierra MG, Zacchino SA, Furlan RLE. Proc Natl Acad Sci U S A. 2007;104:441. doi: 10.1073/pnas.0608438104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salazar MO, Ramallo IA, Micheloni O, Sierra MG, Furlan RLE. Bioorg Med Chem Lett. 2009;19:5067. doi: 10.1016/j.bmcl.2009.07.038. [DOI] [PubMed] [Google Scholar]
- 16.Ramallo IA, Salazar MO, Mendez L, Furlan RLE. Acc Chem Res. 2011;44:241. doi: 10.1021/ar100106n. [DOI] [PubMed] [Google Scholar]
- 17.Kawamura T, Matsubara K, Otaka H, Tashiro E, Shindo K, Yanagita RC, Irie K, Imoto M. Bioorg Med Chem. 2011;19:4377. doi: 10.1016/j.bmc.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 18.Burke MD, Schreiber SL. Angew Chem Int Ed. 2004;43:46. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- 19.Schreiber SL. Science. 2000;287:1964. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 20.Cordier C, Morton D, Murrison S, Nelson A, O'Leary-Steele C. Nat Prod Rep. 2008;25:719. doi: 10.1039/b706296f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barjau J, Schnakenburg G, Waldvogel SR. Angew Chem Int Ed. 2011;50:1415. doi: 10.1002/anie.201006637. [DOI] [PubMed] [Google Scholar]
- 22.Kumar N, Kiuchi M, Tallarico JA, Schreiber SL. Org Lett. 2005;7:2535. doi: 10.1021/ol0504345. [DOI] [PubMed] [Google Scholar]
- 23.Kitayama T. Biosci, Biotechnol, Biochem. 2011;75:199. doi: 10.1271/bbb.100532. [DOI] [PubMed] [Google Scholar]
- 24.de la Torre MC, Garcia I, Sierra MA. J Org Chem. 2003;68:6611. doi: 10.1021/jo034177y. [DOI] [PubMed] [Google Scholar]
- 25.de la Torre MC, Garcia I, Sierra MA. Chem--Eur J. 2005;11:3659. doi: 10.1002/chem.200401220. [DOI] [PubMed] [Google Scholar]
- 26.Balthaser BR, Maloney MC, Beeler AB, Porco JA, Snyder JK. Nat Chem. 2011;3:969. doi: 10.1038/nchem.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Appendino G, Tron GC, Jarevng T, Sterner O. Org Lett. 2001;3:1609. doi: 10.1021/ol0155541. [DOI] [PubMed] [Google Scholar]
- 28.Snatzke G, Nisar A. Justus Liebigs Ann Chem. 1965;683:159. doi: 10.1002/jlac.19656860120. [DOI] [PubMed] [Google Scholar]
- 29.Fox JE, Scott AI, Young DW. J Chem Soc Perkin 1. 1972;6:799. doi: 10.1039/p19720000799. [DOI] [PubMed] [Google Scholar]
- 30.Rodewald WJ, Jagodzinski JJ. Pol J Chem. 1979;53:1203. [Google Scholar]
- 31.Jagodziński JJ, Siciński RR. Tetrahedron Lett. 1981;22:3901. [Google Scholar]
- 32.Castellano EE, Zukerman-Schpector J, Rehder VLG, Marsaioli AJ. Acta Crystallogr, Sect C: Cryst Struct Commun. 1989;C45:966. [Google Scholar]
- 33.Fujiwara FY, Rehder VG, Marsaioli AJ. Magn Reson Chem. 1992;30:500. [Google Scholar]
- 34.Connolly JD, Hill RA. Nat Prod Rep. 2010;27:79. doi: 10.1039/b808530g. [DOI] [PubMed] [Google Scholar]
- 35.Barker EC, Gatbonton-Schwager TN, Han Y, Clay JE, Letterio JJ, Tochtrop GP. J Nat Prod. 2010;73:1064. doi: 10.1021/np1000076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gatbonton-Schwager TN, Letterio JJ, Tochtrop GP. J Nat Prod. 2012;75:591. doi: 10.1021/np200823p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. J Org Chem. 1981;46:3936. [Google Scholar]
- 38.Chandler CL, List B. J Am Chem Soc. 2008;130:6737. doi: 10.1021/ja8024164. [DOI] [PubMed] [Google Scholar]
- 39.Inoue M, Sato T, Hirama M. Angew Chem Int Ed. 2006;45:4843. doi: 10.1002/anie.200601358. [DOI] [PubMed] [Google Scholar]
- 40.Payne AD, Skelton BW, Wege D, White AH. Eur J Org Chem. 2007:1184. [Google Scholar]
- 41.Minnaard AJ, Wijnberg JBPA, de Groot A. Tetrahedron. 1994;50:4755. [Google Scholar]
- 42.Mahrwald R, editor. Modern Aldol Reactions, vol 1: Enolates, Organocatalysis, Biocatalysis and Natural Product Synthesis. Wiley-VCH Verlag GmbH & Co KGaA; 2004. [Google Scholar]
- 43.Mahrwald R, editor. Modern Aldol Reactions, volume 2: Metal Catalysis. Wiley-VCH Verlag GmbH & Co KGaA; 2004. [Google Scholar]
- 44.Zimmerman HE, Traxler MD. J Am Chem Soc. 1957;79:1920. [Google Scholar]
- 45.Chavdarian CG, Chang LL, Onisko BC. Heterocycles. 1988;27:651. [Google Scholar]
- 46.All conformational analyses described herein were carried out using Gaussian 09, Revision A.02, (see Supporting Information for full reference)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.