

Abstract
Multi-level heterogeneity is a fundamental but underappreciated feature of cancer. Most technical and analytical methods either completely ignore heterogeneity or do not fully account for it, as heterogeneity has been considered noise that needs to be eliminated. We have used single-cell and population-based assays to describe an instability-mediated mechanism where genome heterogeneity drastically affects cell growth and cannot be accurately measured using conventional averages. First, we show that most unstable cancer cell populations exhibit high levels of karyotype heterogeneity, where it is difficult, if not impossible, to karyotypically clone cells. Second, by comparing stable and unstable cell populations, we show that instability-mediated karyotype heterogeneity leads to growth heterogeneity, where outliers dominantly contribute to population growth and exhibit shorter cell cycles. Predictability of population growth is more difficult for heterogeneous cell populations than for homogenous cell populations. Since “outliers” play an important role in cancer evolution, where genome instability is the key feature, averaging methods used to characterize cell populations are misleading. Variances quantify heterogeneity; means (averages) smooth heterogeneity, invariably hiding it. Cell populations of pathological conditions with high genome instability, like cancer, behave differently than karyotypically homogeneous cell populations. Single-cell analysis is thus needed when cells are not genomically identical. Despite increased attention given to single-cell variation mediated heterogeneity of cancer cells, continued use of average-based methods is not only inaccurate but deceptive, as the “average” cancer cell clearly does not exist. Genome-level heterogeneity also may explain population heterogeneity, drug resistance, and cancer evolution.
Keywords: genomic instability, genome theory, punctuated cancer evolution, tumor heterogeneity, nonclonal chromosomal aberration
Introduction
Single-cell variation mediated heterogeneity presents a dilemma for biological research, especially for cancer research.1-3 While most molecular biological dogma is based on experimental measures conducted at the cellular population level that utilize statistical averages, single cells produce a high level of variability that does not equate to the average cell.4 For example, individual cells may employ different genetic mechanisms for the same cellular process.2,5 This heterogeneity is fundamental for robust systems, evolution, and integral to understanding somatic cell-mediated disease progression and its stochastic response to medical intervention.6,7
Single-cell heterogeneity has mainly been used to understand microbial resistance and evolution. Studies reveal that single cells exist in diverse phenotypic states that manifest in multimodal distributions at the population level. These studies illustrate interesting findings: stochastic fluctuations contribute to the formation of distinct cell states;8,9 individual cell states diversify populations that are important for cellular evolution;10-12 and paradoxically, while deterministic relationships can be established in cell populations under defined experimental conditions, causal relationships are not usually found in natural settings where stochasticity is dominant.1,13
It was reasoned that novel regulatory mechanisms exist that lead to single-cell heterogeneity, given that cells are karyotypically identical.4 Similar assumptions have been shared among many who study the status of cell populations and how genetic and non-genetic components contribute to cell population dynamics.3,14 However, recent studies challenge the assumption of karyotypic homogeneity in at least 3 ways. First, most cancer cell populations are highly heterogeneous, displaying different genomic alterations, especially at the karyotypic level.2,15,16 Second, since somatic cell reproduction applies less genomic constraint compared to sexual reproduction, the assumption that “isogenic” cell populations that undergo asexual reproduction (including somatic cell division) are purely clonal is actually not correct.17-19 There are increased numbers of studies that link somatic genome variations, termed somatic cell chimerism, to various physiological conditions and diseases.20-27 The widespread penetrance of somatic chimerism challenges the commonly held view that cells from normal tissue should only display normal karyotypes. Thrid, genome theory suggests that the unit of evolutionary selection is the aggregate genome system, where genes serve as individual parts.1,2,13,16,21 Cancer evolution is mainly mediated by genome replacement (macrocellular evolution) illustrated by karyotype dynamics (the cycles of nonclonal chromosomal aberrations [NCCAs] and clonal chromosome aberrations [CCAs]). Since altered karyotypes change the genome context by introducing new genomic topology, karyotype changes create new systems by changing system inheritance. In contrast, during microcellular evolution, or the Darwinian evolution phase, gene-level and epigenetic change yields gradual genetic/non-genetic alterations when karyotypes are clonal.1,2,7,16
Taken together, genome-level heterogeneity may represent the most significant variation among cancer cells. Therefore, validation of genome heterogeneity becomes our first step in single-cell research. Analysis of genomic heterogeneity may resolve why natural settings are more different than controlled experimental settings, where more genomically homogenous cell populations are used. Recently, multi-level landscape models have been proposed to address the relationship among multiple types of heterogeneity in cancer.2,15,28
Here, we investigated the degree of genome heterogeneity in ovarian surface epithelial cells deficient in Brca1 and p53 and in spontaneously transformed mouse ovarian cancer cells to illustrate the importance of single-cell genomic study, how it is correlated with growth heterogeneity, and its implications on average-based technical and analytical methods routinely used in cancer research. Using single-cell culture and spectral karyotyping, we determined the degree of genome heterogeneity in different cell populations and compared karyotypic and growth profiles. Our data illustrate that genomically unstable cancer cell populations exhibit a large degree of heterogeneity, where single cells are characterized by high karyotypic heterogeneity, which subsequently leads to growth heterogeneity. The statistical average is inconsistent with single-cell contributed population dynamics, and thus represents a poor and misleading measure of the actual population. The instability-mediated mechanism of genome heterogeneity represents a key feature of cancer cell populations, especially when chromosome instability (CIN) is higher, and outliers play an important role for population growth.
Results
Highly unstable cell populations are not clonable
Multi-level heterogeneity exists in most cancer types.29 Heterogeneity, specifically karyotypic heterogeneity, is a common feature of cancer cell populations.16,30 However, the degree of heterogeneity in cancer cells has been downplayed, as focus has been toward the pattern of clonal evolution. For example, multiple cancer models indicate that clonal evolution is prominent in tumors, where specific and sequential mutations among a population or subpopulation of cells result in tumor growth.31-33 However, multiple single-cell approaches indicate that punctuated evolution is prominent in tumors, making common markers/mutations difficult to identify, as each cell exhibits distinctly different genetic and genomic profiles.34,35 Because most of the heterogeneity is nonclonal, it is largely ignored by most molecular methods that generate average profiles. Therefore, to paint an accurate picture of cell population heterogeneity between cell generations, and to understand how the degree of genome heterogeneity affects cell growth and evolution, we performed serial dilutions to isolate single cells and generated pure populations from wild-type mouse ovarian surface epithelial (MOSE) cells that have spontaneously transformed in continuous cell culture. Spectral karyotyping (SKY) is then used to characterize the degree of karyotypic heterogeneity. As expected the parent population, which has been in culture for 365 d, exhibited a large degree of karyotype heterogeneity. In contrast, the subclones are not truly clonal, as they exhibit a high level of karyotype diversity. After 3 weeks in culture, all isolated single-cell-derived subpopulations similarly exhibited the same degree of karyotypic heterogeneity (Figs. 1A–E and 2A–E). Analysis of karyotypic abnormalities in subpopulations indicated that nonclonal chromosomal aberrations or NCCAs greatly outnumbered clonal chromosomal aberrations or CCAs. De novo karyotypic intermediates common to both subpopulations were not found, suggesting that evolution is punctuated. Furthermore, analysis at subsequent time points demonstrated that karyotypic heterogeneity actually increased with time.

Figure 1. Genomic heterogeneity of heterogeneous cell populations. (A) Heatmap karyotype of early passage (day 2) wild-type mouse ovarian surface epithelial cells. Most cells have a normal karyotype. (B) Heatmap karyotype of spontaneously transformed parent cell population after 1 y in culture. Cell populations exhibit high aneuploidy and high NCCA frequency. (C and D) Heatmap karyotype of spontaneously transformed single cell-derived subpopulation S1 (C) 23 d after single cell isolation and subpopulation S2 (D) 40 d after single cell isolation. Both subpopulations exhibit a high degree of karyotypic heterogeneity. No direct intermediates were identified between both subpopulations and parent population. (E) Heatmap karyotype of subpopulation S1 117 d after single-cell isolation demonstrates increase in NCCA frequency. (F) Determination of sample size. Each series on the graph represents variation of a single chromosome, where sample size is plotted on the x-axis and standard deviation on the y-axis. Variation among all chromosomes decrease with increasing sample size, and begin to level off at approximately 15 cells. At least 30 cells/sample were analyzed.
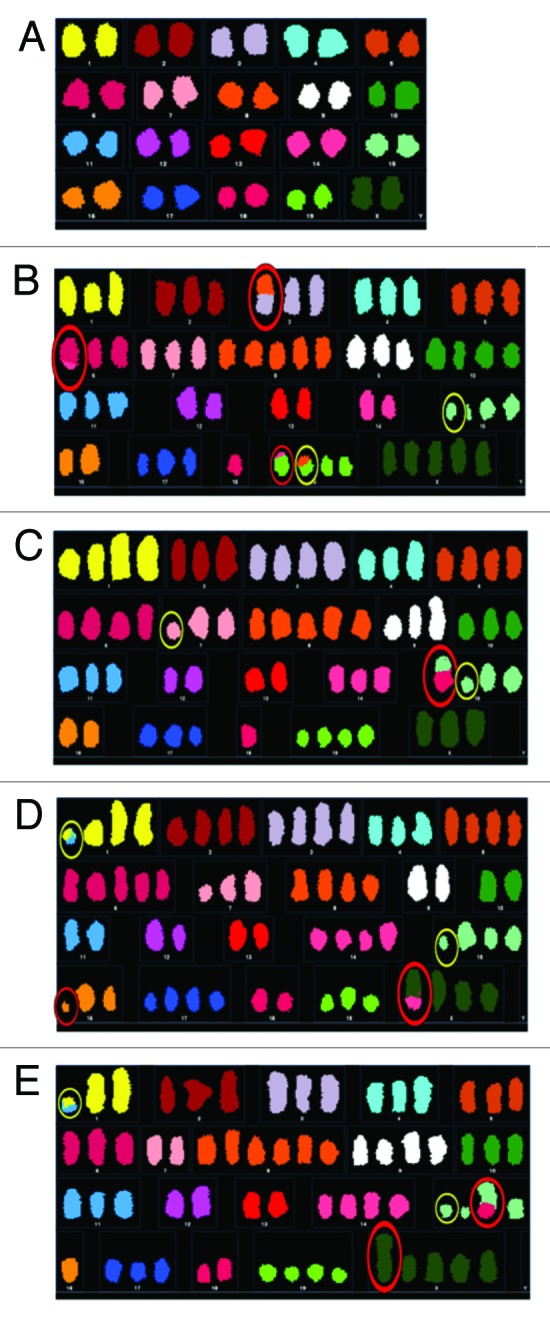
Figure 2. Representative karyotypes of cells from each cell population. Representative karyotypes of early passage MOSE cells (A), parent population after one year in culture (B), single cell derived subpopulation 1, 23 d post-single-cell isolation (C), single-cell-derived subpopulation 2, 40 d post-single-cell isolation (D), and single-cell-derived subpopulation 1, 117 d post-single-cell isolation (E). Structural NCCAs are circled in red; structural CCAs are circled in yellow.
This observation is unexpected, since a clonal population should arise from a single cell, the basis for using cell lines for cancer research. After extended continuous culture, it is common practice to subclone cell culture lines to maintain its purity. It is also well known that subcloning is necessary after extended periods of culture time. Our data demonstrated that when the genome is unstable, cloning at karyotype level is not possible even within a short period of time. Each cell sampled exhibited a unique karyotype within a short time-span, making it clear that these karyotypes are not clonable. To illustrate that this is not a cell culture artifact, the same culture conditions were used for various cell lines with stable genomes, including HCT116 cells and HeLa cells. As expected, these cell lines cells exhibited minimal karyotypic change in continuous culture even over extended periods of time.36,37 Thus, heterogeneity is dominant in genomically unstable cancer cell populations, where cell populations cannot be karyotypically cloned. Our data are consistent with previous studies that high levels of genome instability are associated with the phase of punctuated cancer evolution where stochastic genome alterations dominate.13,34,38
Karyotype heterogeneity leads to growth heterogeneity
Single-cell genome heterogeneity may lead to significant consequences related to system dynamics in a cell population. Genome heterogeneity has been identified as a causal factor for transcriptional,15,39,40 and growth, and survival heterogeneity in various experimental systems.41,42 Therefore, we compared the effects of karyotype heterogeneity on cancer cell growth by tracing clones derived from a single cell. We use karyotypically clonal and nonclonal cell populations to compare how single-cell growth contributes to the population growth profile. Population doubling (PD) time is typically used to assess tumor growth.43 Karyotypically unstable single-cell derived subpopulations all exhibited overall growth that is different between subpopulations (Fig. 3A). We further compared PD times of replicates and observed considerable differences between doubling times (Fig. 3B). Regression analysis of doubling times showed no correlation (Fig. 3C, r2 = 0.0068). Exclusion of some PD values may enhance the relationship to a weak positive correlation. However, this exclusion would not be an accurate assessment of the growth comparison, as each individual subpopulation may be independently evolving and thus exhibiting its own unique PD rate. In addition, it would remove any contribution made by outliers at that particular time-point that may have increased or decreased the overall PD rate. Furthermore PD rates were variable between passages. As karyotype heterogeneity is high, the heterogeneous growth profiles and fluctuating PD rates suggests that cell growth may be highly variable for each single-cell derived clone.

Figure 3: Genome-mediated growth heterogeneity. (A) Population doubling rates of 2 subpopulations isolated from same parent. Each subpopulation exhibits unique growth rate. Variation in PD rates is moderate, as measured by CV (subpopulation 1, 40%; subpopulation 2, 42%) (B) PD rate comparison of 2 independent runs of same subpopulation. Each trial exhibits moderate variation in growth, despite being biological replicates. (Set 1 CV = 44%; Set 2 CV = 45%, n = 2) (C) Regression analysis comparing doubling times of 2 replicates of same subpopulation show no correlation (r2 = 0.0068) (D) In situ single cell growth. Single cells are identified on day 1 and monitored daily. (E) Growth is compared between HCT116 (n = 23) and unstable cells (n = 18). Unstable cells (CV = 200%) display significantly greater growth variation than karyotypically homogeneous HCT 116 cells (CV = 44%). (F-test, P ≤ 1.4 × 10−6) (F and G): Density growth distributions of stable (F) and unstable (G) cell population replicates. Growth distribution of stable cells are unimodal with a narrow distribution, while unstable cells are bimodal and exhibit extremely broad growth distributions.
To determine whether karyotypically unstable cells exhibit a high level of cell growth heterogeneity, we performed daily in situ monitoring of single cell growth (Fig. 3D). Single-cell derived subpopulations from conditionally inactivated Brca1/p53 mouse ovarian surface epithelial cells were thinly plated (400 cells/flask) in gridded flasks. Single cells were identified on day 1, and growth was monitored for 6 d, or until colonies began to merge. Surprisingly, we observed that single-cell proliferation rates of karyotypically unstable cell lines are significantly more variable than karyotypically stable HCT 116 cell lines by almost 3-fold (Fig. 3E–G). While each stable colony exhibited relatively similar proliferation (range 8–82 cells), unstable cells exhibited significantly different growth rates, where cells either did not divide or proliferated at a very fast rate. As an example, a single outlier cell was able to produce 593 cells within 6 d. Interestingly, a majority of unstable cell colonies exhibited moderate to slow growth, while few aggressively proliferative outliers exhibited shorter cell cycle times and drove overall population growth. In contrast, karyotypically stable HCT116 cells all exhibited the same degree of proliferation. The disparity in growth among unstable cells indicates that traditional methods of analysis, such as the statistical average, may be inaccurate at assessing actual population growth.
Arithmetic mean is not a representative measure of unstable cell subpopulations
Genome instability-mediated growth heterogeneity has obvious biological significance. The highly dynamic evolutionary potential of unstable cell populations is represented through heterogeneous growth and transcriptome dynamics. However, the overwhelming level of heterogeneity in cell populations with unstable genomes deserves close attention, as it directly challenges most current strategies to profile these cell populations. For example, use of average-based technical and analytical methods for most cancer cell populations where genome instability is high will yield inaccurate results.
To quantitatively demonstrate inefficiency of average-based measures for unstable cell populations, single colony proliferation of single-cell-derived subpopulations of Brca1/p53 knockouts and stable HCT116 controls are compared with their averages (Fig. 4B and C). Unstable cell populations displayed a non-normal growth distribution, while stable cells exhibited a normal distribution (n = 18; n = 24, Shapiro-Wilkes normality test, P ≤ 1.0−5; P ≤ 0.5, respectively); however, growth among unstable cells were drastically more diverse, as single colony proliferation had a much broader range than stable cells. Among the stable HCT116 cells, each colony contributed the same proportion of cells to the overall population total. In contrast, the unstable cell subpopulation exhibited widely different dynamics, as few cells were responsible for generating most of the population growth. For example, one single colony comprised over 70% of cell growth among unstable cells, while each stable cell colony contributed no more than 10% of growth, indicating that average profiles are not suitable for cell populations with high genome heterogeneity (Fig. 4C). Use of the arithmetic mean (AM) in unstable cells estimated 73 cells per colony, where actual proliferation ranged between 1–593 cells per colony. The 73-cell average fell well above a majority of the population, because highly proliferative outliers drove population growth, i.e., the arithmetic mean is much greater than the median. In contrast, for the stable cell population, the AM estimated 41 per colony, where actual number ranged between 8–82 cells.

Figure 4. Arithmetic mean is a poor measure for genomically heterogeneous populations. (A) Composite heatmap karyotype of parent population is completed by averaging chromosome and NCCA frequencies. The composite cell does not contain any NCCAs that are present in individual cells and does not reflect range of aneuploidy observed in all cells. (B) Schematic of single-colony growth. Daily proliferation was counted, averaged, and compared with high and low proliferating colonies. (C) Comparison of single cell colony proliferation to population average in Brca1/p53 unstable cell subpopulation and stable HCT116 cells. Average colony size is calculated at 73 cells in Sub1 and 41 cells in HCT116 (indicated by black columns on far right). Most colonies among the unstable cell population are well under the average, indicating that outliers dominate population behavior and the average is not a representative measure for genomically unstable cell population
Discussion
A high degree of heterogeneity is a well-accepted feature of cancer cells. While this feature is generally accepted, it is rarely documented and discussed much less. Here, we demonstrated that karyotypic heterogeneity exists at an alarming degree, where single cells of unstable cell populations are extremely difficult to clone. Furthermore, elevated karyotype heterogeneity drastically affects other features of the cell system, such as cell growth. Karyotype heterogeneity results in extreme growth heterogeneity. In contrast to the notion that all cells in a single population are believed to divide at relatively the same rate, single cells in unstable cell populations have unique doubling times. Furthermore, while most cells exhibit slow-to-moderate growth, it is the outlier cells that contribute the most to overall population growth. These findings have significant research and clinical significance, and require a reevaluation of fundamental biological principles.
Genomic heterogeneity among cancer cells is common, but is usually not measured. Too often, studies report the arithmetic mean, and occasionally the geometric mean. Yet means and other statistics related to the first moment
do not quantify heterogeneity. In fact, means smooth through heterogeneity, making it invisible. Heterogeneity is quantified by the second moment
and its modifications, such as variance, standard deviation, coefficient variation, and average variation. While many of these indices incorporate both the first and second moment, means only include the first moment. Therefore, using means to study genomically heterogeneous cell populations compromises results, as average-based measures that exclude outliers are commonly used in biomarker discovery and drug design. Furthermore, while the standard deviation is often included in the graphical representation of data, it is not scale-free and thus cannot be used to compare against variation in other samples. To demonstrate, we generate a composite karyotype by averaging the frequencies of all chromosomal structures (Fig. 4A), where the composite karyotype renders heterogeneity invisible. Problems with only reporting means are also observed in cancer cell growth heterogeneity (Fig. 4B and C). While more appropriate averages, such as the geometric mean, can be used, these are also poor indicators of population growth heterogeneity. Geometric mean yields a 19 cell/colony average that is more representative of individual colonies; however, it is not representative of the total population and does not highlight outliers that drive population growth (Table 1). It is worth mentioning, however, that statistical outliers are not frequent events, or their occurrence is subject to probability. Despite this, multi-modal variation is present in unstable cell populations (Fig. 3G). This underscores the contribution of both outliers and variation to population growth and indicates that means cannot characterize genome-mediated growth heterogeneity in genomically unstable cell populations.
Table 1. Inefficiency of statistical means to describe population growth.
| Mean colony size | Est total population | Difference from actual population | |
|---|---|---|---|
| Arithmetic mean, Heterogeneous subpopulation | 73 | 1314 cells | 8 cells |
| Arithmetic, HCT 116 cells | 41 | 943 cells | 7 cells |
| Geometric mean, Heterogeneous subpopulation | 19 | 342 cells | 980 cells |
| Geometric mean, HCT 116 cells | 37 | 806 cells | 99 cells |
Genome and growth heterogeneity have significant research and clinical implications that are related to cancer evolution and drug resistance. While the statistical mean can be reliably used to characterize genomically homogenous cell populations, like normal physiological or developmental conditions, this method is ill suited for profiling conditions of the pathological context with elevated genome heterogeneity. During the macro-evolutionary phase, the “average cancer cell” is nonexistent, as system heterogeneity is dominant. Use of the average is incorrect during this phase, because averages eliminate diversity, the very entity that defines cancer, which is reflected by the level of cells with NCCAs, and the dynamic relationship of the NCCA/CCA cycle. However, when cancer enters the micro-evolutionary phase, statistical averages can faithfully be employed, because system heterogeneity is low, and most change predominantly occurs at the gene-level. It is for this reason that averages work within specific stages for linear cancer models.19 Inappropriate use of statistical means is thus a significant issue in modeling and drug design, where improper target selection based on the average may lead to increased resistance and off-target effects.2,19,44 For example, PD rates are used in clinical settings to measure growth rates of tumors, despite its noted inaccuracies.43 A number of growth models that use alternative indices to measure tumor growth have been proposed. While some of these proxies may be superior to doubling rates, their use of averaging and stationary growth models make them poor at capturing the full spectrum of growth heterogeneity.43 Instead, variance and coefficient of variation are natural indices of heterogeneity and should be used more often when genome instability is high. Furthermore, average-based technical methods that use whole-cell lysate, such as genome sequencing, RNA-seq, expression profiling, etc. to generate an average nucleotide sequence or expression level based on the entire cell population are common for biomarker and drug target identification. While an increased number of studies are moving to a single-cell platform, efforts like the TCGA and other clinical sequencing projects maintain their use of an entire population of cells to characterize a tumor. While these strategies initially work, tumor growth almost always recurs, as these methods may only reflect more dominant subpopulations and do not target outlier cells that play a more significant role in cancer evolution.44
In vitro systems provide an excellent opportunity to systematically investigate how single-cell systems function under various conditions. Regarding the notion that heterogeneity might be a cell culture artifact, studies using human cancer samples indicate otherwise. For example, Navin et al. demonstrate through single-cell sequencing of breast cancer cells that cancer evolves stochastically.35 Individual cells exhibited a large degree of mutations and copy number changes that were not conserved among cells. Baca et al. describe punctuated evolution of prostate cancer cells through a process termed chromoplexy, where chromosome regions interact in a stochastic fashion to drive cancer evolution.45 Both cancer genome sequencing studies have confirmed the 2 phases of cancer evolution (punctuated phase and stepwise phase) initially proposed based on karyotype analysis.2,17,22,34 Furthermore, genome heterogeneity is a common feature observed in healthy tissue. For example, healthy mammalian liver cells exhibit a greater degree of polyploidy than in vitro systems, ranging from 2N-16N.23,46 In addition, an increased number of studies link the chimeric genome to various human diseases.20,24,27 Despite normal tissue having a relatively low frequency of genome level alterations as reflected by the low frequency of NCCAs (for normal individuals, the frequencies of NCCAs detected from short term blood culture is only a few percent), NCCAs are elevated under disease conditions. For example, NCCA frequency in lymphocytes of individuals with various illnesses including cancer can reach up to 20–40% (Heng et al., in preparation). In addition, we traced NCCA frequency in tumor growth of multiple mouse models and found a high degree of genome heterogeneity in each case.47 In cases where NCCA frequency is low, the statistical average will give an appropriate measure of population dynamics. However, the average will be an inefficient measure with elevated NCCA frequency in patient tumors and other pathological conditions.
Finally, we address the issue of “isogenic cells” with multi-level heterogeneity, especially at the genome level. Most cancer cell populations are not isogenic. Multi-level heterogeneity in fact is a key feature of cancer that plays a significant role in population dynamics and cancer evolution. A direct relationship may link genome heterogeneity to growth and other systems-based heterogeneity. For example, high degrees of heterogeneity have been observed in gene mutations,48 transcription,39,49 biochemical signaling pathways,50,51 the tumor microenvironment, and response to drug treatment due to differences in cell proliferation.52 Currently, it is challenging to study the relationship among them, as most studies that report heterogeneity do not address genome level heterogeneity. Since the genome is the highest level of genetic organization, and the genome package is responsible for the macrocellular evolution of cancer, our effort to unify genome heterogeneity with other types of heterogeneity is highly significant. Such an analysis can also be used to profile the heterogeneity of dominant proliferating cells for drug treatment.52
Genome-based systems heterogeneity can explain why different labs often cannot validate published findings, as the cell lines or samples used might exhibit altered genomes.53 We demonstrated this in a recent study in human fibroblasts, where increased karyotypic diversity results in high expression variation.39,49 Multiple mouse models have been developed to understand the specific roles of deregulated genes and pathways.54 While deregulation of a particular pathway can be highly penetrant (owing to any member of that pathway being mutated), pathway switching is a dominant feature of heterogeneous cancers, like high-grade astrocytoma, that cancer cells readily exploit to acquire resistance.15,39,49 Interestingly, genome-based heterogeneity can also explain the gap between experimental systems and clinical samples, as clinical samples are often more complicated due to increased heterogeneity when compared to highly homogeneous inbred mouse strains. Together, genome heterogeneity is extremely significant as genome replacement is the key feature of cancer cell populations that is responsible for cancer initiation, progression, metastasis, and drug resistance. In particular, many firmly held beliefs regarding cancer cells that were deduced based on average-based methods require reexamination and reinterpretation, as the average cancer cell clearly is nonexistent.
Materials and Methods
Cell lines
Spontaneously transformed cell line was isolated as previously described.55 Brca1/p53 conditional knockouts were obtained from the University of Ottawa.56 Single cells were isolated after 1 y (spontaneously transformed) or 60 d (Brca1/p53 conditional knockout) in continuous culture through serial dilutions and identified by microscope. HCT116 cell lines obtained from ATCC.
Cell culture
Standard cell culture was used for all cell lines. All mouse lines were maintained in high-glucose DMEM, supplemented with 4% FBS, antibiotics, and insulin, transferrin, sodium selenite growth supplement (BD Biosciences). HCT116 cells were maintained in RPMI, supplemented with 10% FBS and antibiotics.
Cytogenetic metaphase slide preparation and spectral karyotyping (SKY)
Cytogenetic slides were prepared as described. SKY was completed on metaphase spreads as described.34,38,47
Karyotypic analysis
Karyotypic analysis was completed as previously described.13,34 Chromosomal heterogeneity was visualized on heatmaps. In each heatmap, each series on the y-axis corresponds to one cell. The x-axis represents chromosome structure (normal chromosomes and derivative chromosomes that include translocations, fragments, and other cytogenetic abnormalities). The darkening of color intensity represents an increase in the frequency of a specific chromosome structure. Chromosomal aberrations are considered NCCAs (nonclonal chromosomal aberrations) if they are prevalent in less than 20% of the sample, or not present in the parent population. Chromosomal aberrations that are present in both parent population and single-cell subpopulation and prevalent in over 20% of cells are considered CCAs (clonal chromosomal aberrations).
Population-level counting
Cells were grown in T-75 flasks, passaged, and counted upon ~80% confluency. Cells were re-plated in fresh flasks.
In situ single cell counting
Cells were plated in T-25 flasks, labeled with grids at a concentration of 400 cells/flask. Single cells were identified and growth was measured daily.
Statistical analysis
All chromosome structures within a cell were measured to determine degree of karyotypic heterogeneity. Two methods were used to determine sample size for SKY to ensure statistical robustness and to account for variation. First, a power calculation was completed (α = 0.95, β = 0.9), yielding a sample size of 15 cells. Second, we examine variation of each chromosome by plotting standard deviation as a function of increasing sample size (Fig. 1F). Expectedly, variation among all chromosomes decreases with increasing sample size, where it levels off at approximately 15 cells. Based on these 2 parameters, at least 15 cells were used per sample; at least 15 per sample were required; however, at least 30 cells per sample were analyzed.
Acknowledgments
We would like to thank Dr Jason Caravas for his assistance with the clustering analysis. We would also like to thank Dr Francesca Luca, Dr Ghassan Saed, and Dr Gloria Heppner for their continued support. This manuscript is part of a series of studies entitled "The mechanism of somatic and organismal evolution".
Glossary
Abbreviations:
- NCCA
nonclonal chromosome aberration
- CCA
clonal chromosomal aberration
- CIN
chromosome instability
- MOSE
mouse ovarian surface epithelial
- SKY
spectral karyotyping
- PD
population doubling
- AM
arithmetic mean
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Grant Support
This work was supported by grants to HHQH from the Susan G Komen Breast Cancer Foundation, See DNA Inc, the United States Department of Defense (GW093028), the National Chronic Fatigue and Immune Dysfunction Syndrome Foundation, and the Nancy Taylor Foundation for Chronic Diseases. This work was also supported by the Discovery Grant from the Natural Science and Engineering Research Council of Canada *NSERC), to RG.
Author Contributions
BYA and GL performed all experiments. BCV provided the primary cultures of mouse ovarian surface epithelial cells with inactivated BRCA1 and p53. BYA, HHQH, and RG wrote the manuscript. All authors contributed to experiment design.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26580
References
- 1.Heng HH, Liu G, Stevens JB, Bremer SW, Ye KJ, Abdallah BY, Horne SD, Ye CJ. Decoding the genome beyond sequencing: the new phase of genomic research. Genomics. 2011;98:242–52. doi: 10.1016/j.ygeno.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 2.Heng HH, Stevens JB, Bremer SW, Liu G, Abdallah BY, Ye CJ. Evolutionary mechanisms and diversity in cancer. Adv Cancer Res. 2011;112:217–53. doi: 10.1016/B978-0-12-387688-1.00008-9. [DOI] [PubMed] [Google Scholar]
- 3.Huang S, Ernberg I, Kauffman S. Cancer attractors: a systems view of tumors from a gene network dynamics and developmental perspective. Semin Cell Dev Biol. 2009;20:869–76. doi: 10.1016/j.semcdb.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pelkmans L. Cell Biology. Using cell-to-cell variability--a new era in molecular biology. Science. 2012;336:425–6. doi: 10.1126/science.1222161. [DOI] [PubMed] [Google Scholar]
- 5.Stevens JB, Abdallah BY, Liu G, Ye CJ, Horne SD, Wang G, Savasan S, Shekhar M, Krawetz SA, Hüttemann M, et al. Diverse system stresses: common mechanisms of chromosome fragmentation. Cell Death Dis. 2011;2:e178. doi: 10.1038/cddis.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heng HH. The conflict between complex systems and reductionism. JAMA. 2008;300:1580–1. doi: 10.1001/jama.300.13.1580. [DOI] [PubMed] [Google Scholar]
- 7.Heng HH. Biocomplexity: challenging reductionism. In: Sturmberg JP, Martin CM, ed. Handbook on Systems and Complexity in Health: Springer, 2013:1132. [Google Scholar]
- 8.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305:1622–5. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 9.Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–6. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 10.Acar M, Mettetal JT, van Oudenaarden A. Stochastic switching as a survival strategy in fluctuating environments. Nat Genet. 2008;40:471–5. doi: 10.1038/ng.110. [DOI] [PubMed] [Google Scholar]
- 11.Kussell E, Leibler S. Phenotypic diversity, population growth, and information in fluctuating environments. Science. 2005;309:2075–8. doi: 10.1126/science.1114383. [DOI] [PubMed] [Google Scholar]
- 12.Cohen AA, Geva-Zatorsky N, Eden E, Frenkel-Morgenstern M, Issaeva I, Sigal A, Milo R, Cohen-Saidon C, Liron Y, Kam Z, et al. Dynamic proteomics of individual cancer cells in response to a drug. Science. 2008;322:1511–6. doi: 10.1126/science.1160165. [DOI] [PubMed] [Google Scholar]
- 13.Heng HH, Bremer SW, Stevens J, Ye KJ, Miller F, Liu G, Ye CJ. Cancer progression by non-clonal chromosome aberrations. J Cell Biochem. 2006;98:1424–35. doi: 10.1002/jcb.20964. [DOI] [PubMed] [Google Scholar]
- 14.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–44. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 15.Heng HH, Bremer SW, Stevens JB, Horne SD, Liu G, Abdallah BY, Ye KJ, Ye CJ. Chromosomal instability (CIN): what it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013 doi: 10.1007/s10555-013-9427-7. [DOI] [PubMed] [Google Scholar]
- 16.Heng HH. The genome-centric concept: resynthesis of evolutionary theory. Bioessays. 2009;31:512–25. doi: 10.1002/bies.200800182. [DOI] [PubMed] [Google Scholar]
- 17.Heng HH. Elimination of altered karyotypes by sexual reproduction preserves species identity. Genome. 2007;50:517–24. doi: 10.1139/G07-039. [DOI] [PubMed] [Google Scholar]
- 18.Gorelick R, Heng HH. Sex reduces genetic variation: a multidisciplinary review. Evolution. 2011;65:1088–98. doi: 10.1111/j.1558-5646.2010.01173.x. [DOI] [PubMed] [Google Scholar]
- 19.Horne SD, Stevens JB, Abdallah BY, Liu G, Bremer SW, Ye CJ, Heng HH. Why imatinib remains an exception of cancer research. J Cell Physiol. 2013;228:665–70. doi: 10.1002/jcp.24233. [DOI] [PubMed] [Google Scholar]
- 20.Iourov IY, Vorsanova SG, Yurov YB. Chromosomal mosaicism goes global. Mol Cytogenet. 2008;1:26. doi: 10.1186/1755-8166-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heng HH, Stevens JB, Bremer SW, Ye KJ, Liu G, Ye CJ. The evolutionary mechanism of cancer. J Cell Biochem. 2010;109:1072–84. doi: 10.1002/jcb.22497. [DOI] [PubMed] [Google Scholar]
- 22.Heng HH, Stevens JB, Liu G, Bremer SW, Ye CJ. Imaging genome abnormalities in cancer research. Cell Chromosome. 2004;3:1. doi: 10.1186/1475-9268-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duncan AW, Hanlon Newell AE, Smith L, Wilson EM, Olson SB, Thayer MJ, Strom SC, Grompe M. Frequent aneuploidy among normal human hepatocytes. Gastroenterology. 2012;142:25–8. doi: 10.1053/j.gastro.2011.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heng HH. The contribution of genomic heterogeneity. Preface. Cytogenet Genome Res. 2013;139:141–3. doi: 10.1159/000347035. [DOI] [PubMed] [Google Scholar]
- 25.Hultén MA, Jonasson J, Iwarsson E, Uppal P, Vorsanova SG, Yurov YB, Iourov IY. Trisomy 21 mosaicism: we may all have a touch of Down syndrome. Cytogenet Genome Res. 2013;139:189–92. doi: 10.1159/000346028. [DOI] [PubMed] [Google Scholar]
- 26.Sgaramella V. Variability of our somatic (epi)genomes. Science. 2010;329:32–3. doi: 10.1126/science.329.5987.32-c. [DOI] [PubMed] [Google Scholar]
- 27.Heng HH, Liu G, Stevens JB, Abdallah BY, Horne SD, Ye KJ, Bremer SW, Chowdhury SK, Ye CJ. Karyotype heterogeneity and unclassified chromosomal abnormalities. Cytogenet Genome Res. 2013;139:144–57. doi: 10.1159/000348682. [DOI] [PubMed] [Google Scholar]
- 28.Huang S. Genetic and non-genetic instability in tumor progression: link between the fitness landscape and the epigenetic landscape of cancer cells. Cancer Metastasis Rev. 2013 doi: 10.1007/s10555-013-9435-7. [DOI] [PubMed] [Google Scholar]
- 29.Heppner GH. Tumor heterogeneity. Cancer Res. 1984;44:2259–65. [PubMed] [Google Scholar]
- 30.Heng HH. Missing heritability and stochastic genome alterations. Nat Rev Genet. 2010;11:813. doi: 10.1038/nrg2809-c3. [DOI] [PubMed] [Google Scholar]
- 31.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-I. [DOI] [PubMed] [Google Scholar]
- 32.Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, Paulson TG, Blount PL, Risques RA, Rabinovitch PS, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–73. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- 33.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 34.Heng HH, Stevens JB, Liu G, Bremer SW, Ye KJ, Reddy PV, Wu GS, Wang YA, Tainsky MA, Ye CJ. Stochastic cancer progression driven by non-clonal chromosome aberrations. J Cell Physiol. 2006;208:461–72. doi: 10.1002/jcp.20685. [DOI] [PubMed] [Google Scholar]
- 35.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–4. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knutsen T, Padilla-Nash HM, Wangsa D, Barenboim-Stapleton L, Camps J, McNeil N, Difilippantonio MJ, Ried T. Definitive molecular cytogenetic characterization of 15 colorectal cancer cell lines. Genes Chromosomes Cancer. 2010;49:204–23. doi: 10.1002/gcc.20730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shih IM, Zhou W, Goodman SN, Lengauer C, Kinzler KW, Vogelstein B. Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res. 2001;61:818–22. [PubMed] [Google Scholar]
- 38.Heng HH, Liu G, Bremer S, Ye KJ, Stevens J, Ye CJ. Clonal and non-clonal chromosome aberrations and genome variation and aberration. Genome. 2006;49:195–204. doi: 10.1139/G06-023. [DOI] [PubMed] [Google Scholar]
- 39.Stevens JB, Horne SD, Abdallah BY, Ye CJ, Heng HH. Chromosomal instability and transcriptome dynamics in cancer. Cancer Metastasis Rev. 2013 doi: 10.1007/s10555-013-9428-6. [DOI] [PubMed] [Google Scholar]
- 40.Harewood L, Schütz F, Boyle S, Perry P, Delorenzi M, Bickmore WA, Reymond A. The effect of translocation-induced nuclear reorganization on gene expression. Genome Res. 2010;20:554–64. doi: 10.1101/gr.103622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. England, 2010:321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levy SF, Ziv N, Siegal ML. Bet hedging in yeast by heterogeneous, age-correlated expression of a stress protectant. PLoS Biol. 2012;10:e1001325. doi: 10.1371/journal.pbio.1001325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehrara E, Forssell-Aronsson E, Ahlman H, Bernhardt P. Specific growth rate versus doubling time for quantitative characterization of tumor growth rate. Cancer Res. 2007;67:3970–5. doi: 10.1158/0008-5472.CAN-06-3822. [DOI] [PubMed] [Google Scholar]
- 44.Abdallah BY, Horne S, Kurkinen M, Stevens JB, Liu G, Ye CJ, Barbat J, Bremer SW, Heng HH. Ovarian cancer evolution through stochastic genome alterations: defining the genomic role in ovarian cancer. Systems Biology in Reproductive Medicine. 2013 doi: 10.3109/19396368.2013.837989. In press. [DOI] [PubMed] [Google Scholar]
- 45.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–77. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB, Finegold MJ, Grompe M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467:707–10. doi: 10.1038/nature09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ye CJ, Stevens JB, Liu G, Bremer SW, Jaiswal AS, Ye KJ, Lin MF, Lawrenson L, Lancaster WD, Kurkinen M, et al. Genome based cell population heterogeneity promotes tumorigenicity: the evolutionary mechanism of cancer. J Cell Physiol. 2009;219:288–300. doi: 10.1002/jcp.21663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stevens JB, Liu G, Abdallah BY, Horne SD, Ye KJ, Bremer SW, Ye CJ, Krawetz SK, Heng HH. Unstable genomes elevate transcriptome dynamics. Int J Cancer. 2013 doi: 10.1002/ijc.28531. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chakraborty AK, Roose JP. Biochemical heterogeneity and developmental varieties in T-cell leukemia. Cell Cycle. 2013;12:1480–1. doi: 10.4161/cc.24858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hartzell C, Ksionda O, Lemmens E, Coakley K, Yang M, Dail M, Harvey RC, Govern C, Bakker J, Lenstra TL, et al. Dysregulated RasGRP1 responds to cytokine receptor input in T cell leukemogenesis. Sci Signal. 2013;6:ra21. doi: 10.1126/scisignal.2003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blagosklonny MV. Target for cancer therapy: proliferating cells or stem cells. Leukemia. 2006;20:385–91. doi: 10.1038/sj.leu.2404075. [DOI] [PubMed] [Google Scholar]
- 53.Heng HH. Genomics: HeLa genome versus donor's genome. Nature. 2013;501:167. doi: 10.1038/501167d. [DOI] [PubMed] [Google Scholar]
- 54.Chow LM, Baker SJ. Capturing the molecular and biological diversity of high-grade astrocytoma in genetically engineered mouse models. Oncotarget. 2012;3:67–77. doi: 10.18632/oncotarget.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, Persons DL, Smith PG, Terranova PF. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. 2000;21:585–91. doi: 10.1093/carcin/21.4.585. [DOI] [PubMed] [Google Scholar]
- 56.Clark-Knowles KV, Garson K, Jonkers J, Vanderhyden BC. Conditional inactivation of Brca1 in the mouse ovarian surface epithelium results in an increase in preneoplastic changes. Exp Cell Res. 2007;313:133–45. doi: 10.1016/j.yexcr.2006.09.026. [DOI] [PubMed] [Google Scholar]