

Abstract
Combination antiretroviral therapy (cART) has transformed HIV from a deadly to a chronic disease, but HIV patients are still burdened with excess morbidity and mortality, long-term toxicities from cART, stigmatization, and insufficient access to cART worldwide. Thus, a cure for HIV would have enormous impact on society as well as the individual. As the complexity and mechanisms of HIV persistence during therapy are being unraveled, new therapeutic targets for HIV eradication are discovered. Substances that activate HIV production in the latently infected cells have recently received much attention. By turning on expression of latent HIV proviruses, reactivation strategies could contribute to the eradication HIV infection. Compounds that are currently being or soon to be tested in clinical trials are emphasized. The results from these trials will provide important clues as to whether or not reactivating strategies could become significant components of a cure for HIV.
Keywords: HIV, immune modulation, histone deacetylase inhibitors, experimental research, cure
Introduction
The realization that prolonged combination antiretroviral treatment (cART) did not lead to eradication of HIV infection has spurred an impressive scientific effort in characterizing latent HIV reservoirs and understanding the intricate mechanisms that establish HIV latency and enable the virus to persist for decades evading host immune responses and potent cART. In terms of defining latent HIV reservoirs it is useful to distinguish between proviral latency, referring to the presence of replication competent but transcriptionally silent provirus within resting cells,1 and residual viremia, referring to the continuous existence of trace levels of extracellular HIV-RNA in plasma during suppressive cART.2,3
Whereas the pool of latently infected memory CD4+ T-cells is now the most well-defined latent HIV reservoir and presumably the primary obstacle to the eradication of HIV infection,4,5 the origin and significance of the residual viremia, in particular whether this is caused by on-going replication, is still debated. Yet, the lack of genetic evolution6,7 and absence of resistance development strongly suggest that effective rounds of new replication are not occurring in patients on suppressive cART. Finally, other cellular reservoirs have been suggested to persist in monocytes,8 macrophages,9,10 astrocytes,11 hematopoietic stem cells,12 naïve T cells,13 and regulatory T cells,14 but opposing findings are also reported.15 Importantly, contrary to latently infected memory CD4+ T cells, these cellular reservoirs have not been longitudinally quantified and, therefore, it remains uncertain whether these cells carry inducible replication competent virus for prolonged periods in vivo.
Several therapeutic strategies are pursued to achieve a cure for HIV (Table 1). First, intensification studies have explored whether adding an extra antiretroviral drug to an already suppressive cART regimen can reduce the residual viremia or the latent HIV reservoir. Overall, there seems to be little or no effect from these interventions,16-21 but there are conflicting results.22,23 Second, the remarkable case report of an HIV-infected patient who was cured for HIV after receiving bone marrow transplantation containing a 32 base pair deletion in the HIV co-receptor CCR5 gene24 has inspired studies entailing infusion of autologous CCR5-deleted CD4+ T cells25,26 and studies of chemotherapy in HIV infected patients with lymphoma.27 Third, elimination of latently infected T cells by reactivating HIV-1 expression using agents like histone deacetylase inhibitors (HDACi),28-40 IL-7,41,42 disulfiram43 or prostratin31,44-46 have been investigated in numerous studies in vitro, ex vivo and in vivo. Finally, as reactivation of HIV-1 expression in latently infected cells may be insufficient to ensure the removal of these cells,47 immunotherapy to enhance HIV specific immunity are continuously being developed and tested.48
Table 1. Recent or ongoing clinical trials for HIV eradication.
| Therapeutic Strategy | Mechanism | Trials performed/intervention | Principal investigators | Outcome measure | Main results |
|---|---|---|---|---|---|
| Eliminating residual viremia and/or residual viral replication by treatment intensification | Suppression of residual viral activity by adding an extra anti-retroviral drug to an already suppressive regimen | Enfuvirtide, 2 NRTI and boosted PI | Joseph J. Eron, Jr. | IUPM | No decay of the latent reservoir during 48 weeks |
| Raltegravir | Joseph J. Eron, Jr. | Plasma HIV-RNA | No decrease in plasma HIV-RNA | ||
| Raltegravir | Deborah McMahon | Plasma HIV-RNA | No decrease in plasma HIV-RNA | ||
| Efavirenz, atazanavir/r or lopinavir/r | Frank Maldarelli | Plasma HIV-RNA | No decrease in plasma HIV-RNA | ||
| Raltegravir, raltegravir/efavirenz or raltegravir/darunavir | Diane Havlir, Joseph K Wong, Steven Yukl | Plasma HIV-RNA, cell-associated HIV-RNA and HIV-DNA from PBMCs and 4 gut sites | No consistent decrease in plasma HIV-RNA, cell-associated HIV-RNA or HIV-DNA | ||
| Abacavir | Scott Hammer | Plasma HIV-RNA and HIV-DNA in PBMCs | No decrease in in HIV-DNA or plasma HIV-RNA | ||
| Raltegravir | Javier Martínez-Picado | HIV-DNA and episomal HIV-1 cDNA | No decrease in HIV-DNA; increase in episomal HIV-1 cDNA in 13 of 45 subjects | ||
| Raltegravir | Santiago Moreno | IUPM, plasma HIV-RNA, episomal HIV-1 cDNA | Decrease in IUPM in all 9 subjects, no decrease in plasma HIV-RNA | ||
| Maraviroc | Martin Markowitz | Mucosal cell-associated HIV- RNA | Ongoing | ||
| Host modification to confer resistence to HIV-infection | Infusion of autologous CD4+ T cells with zinc finger nuclease-mediated disruption of CCR5 expression | SB-728-T | Winson Tang | Plasma HIV-RNA during cART interruption | Ongoing |
| SB-728-T | Winson Tang | Persistence and activity of CCR5 ZFN-modified autologous T-Cells | Ongoing | ||
| Chemotherapy for lymphoma | Elimination of latently infected cells through chemotherapy for AIDS-related lymphoma | Chemotherapy for AIDS-related lymphoma | John Mellors | Plasma HIV-RNA, HIV-DNA | No significant effect on plasma HIV-RNA or HIV-DNA |
| Allogeneic hematopoietic cell transplantation for hematological malignancies | Joseph Alvarnas, Richard Ambinder | Plasma HIV-RNA, HIV-DNA | Ongoing | ||
| Autologous hematopoietic stem cell transplantation for lymphoma | Amrita Krishnan | Plasma HIV-RNA | Ongoing | ||
| Eliminating latently infected cells by reactivating HIV-1 expression | Induction of HIV-1 expression in latently infected cells to eradicate the latent reservoir, as these cells could be eliminated due to viral cytopathic effects or immune mediated killing | Vorinostat | David Margolis | Cell-associated HIV-RNA, IUPM | Significant increases in cell-associated HIV-RNA in 8 of 8 subjects receiving a single 400 mg dose |
| Vorinostat | Sharon Lewin | Cell-associated HIV-RNA | Ongoing | ||
| Panobinostat | Thomas Rasmussen | Cell-associated HIV-RNA, HIV-DNA, IUPM | Ongoing | ||
| Disulfiram | Steven Deeks, Adriana Andrade | IUPM, plasma HIV-RNA | No consistent significant decrease in plasma HIV-RNA or HIV-DNA | ||
| Enhance innate immunity | Suppression of viral replication by administering cytokines that are part of the host's innate antiviral reponse | Interferon α2A | Luis Montaner | Viral rebound during cART interruption | Lower proportions of viral rebound than a historical cohort |
| Interferon α2B | Frank Maldarelli | Plasma HIV-RNA | Ongoing | ||
| Enhance HIV-specific immunity and combination approaches | Combining therapeutic HIV vaccination with other therapeutic strategies to enhance the host's adaptive HIV-specific immunity | IL-7 + HIV vaccine + intensification | Christine Katlama (Eramune 1) | HIV-DNA | Ongoing |
| HIV vaccine + intensification | Robert Murphy (Eramune 2) | HIV-DNA | Ongoing |
cART, combination antiretroviral therapy; NRTI, nucleoside reverse transcriptase inhibitor; IUPM, infectious units per million.
This review will be focused on reactivation strategies describing compounds that are being considered for the eradication of HIV infection by turning on expression of latent HIV proviruses with emphasis on agents that are currently being or soon to be tested in clinical trials. Immunotherapy and immunomodulatory effects will be dealt with in detail. The majority of HIV infected individuals reside in areas with deprived health care systems and inadequate infrastructure, and, therefore, the development of an HIV cure will ultimately be faced with the challenge of global accessibility and low cost. As most currently investigated reactivation compounds can be produced on a large scale and administered irrespective of HIV-subtype and HLA-profile, they do seem to possess this potential.
HDAC inhibitors
Role of histone deacetylases and HDACi in HIV Latency
There are 11 known histone deacetylase (HDAC) metalloenzymes, which are classified into class I (HDAC 1, 2, 3, and 8), class IIa (HDAC 4, 5, 7, and 9), class IIb (HDAC 6 and 10), and class IV (HDAC 11).49 The counteracting mechanisms of HDACs and histone acetyl transferases (HAT) exert a key function in regulating gene expression by controlling the degree of acetylation/deacetylation of histone tails, which in turn influences chromatin condensation. The HIV 5′ long-terminal repeat (LTR) that contains promoter and enhancer elements and has binding sites for several transcription factors is arranged in two nucleosomes, nuc-0 and nuc-1.50 In the transcriptionally silent state of HIV latency various transcription factors recruit HDACs to the HIV-1 5′ LTR where they induce chromatin condensation by promoting deacetylation of lysine residues on histones51-57 keeping nuc-1 in the hypoacetylated state and preventing HIV transcription. HDACi offsets these mechanisms by inhibiting HDACs (Fig. 1). Chromatin immunoprecipitation assays have shown that the class I HDACs, HDAC1, 2 and 3, may be particularly important to maintaining latency.53,58 Notably, a recent study correlating HDACi isoform specificity with the ability to reactivate latent HIV-1 expression, showed that potent inhibition or knockdown of HDAC1 was not sufficient to disrupt HIV latency. Instead, HDAC3 inhibition was found to be essential for reactivating viral expression.59 Class I HDACs are ubiquitously expressed60 and deacetylation of lysine residues on histones is a key function of class I HDACs. However, recent data suggest that they may deacetylate more than 1750 non-histone proteins.61 To which degree, if any, the non-histone effects of HDACi contribute to the desired circumvention of HIV latency is largely unknown.

Figure 1. Disruption of HIV latency by HDAC Inhibitors. In the latent state HDACs suppresses HIV-1 expression by catalyzing deacetylation of histone tails and keeping the chromatin in a compacted state. Inhibition of HDACs by HDACi promotes histone acetylation by HATs leading to relaxation of the chromatin and initiation of transcription. HDACs: histone deacetylases; HDACi: histone deacetylase inhibitors; HATs: histone acetyl transferases; LTR: long-terminal repeat.
The HDACi acting on HDAC metalloenzymes may be categorized according to their chemical structure into short chain fatty acids, hydroxamic acids and cyclic tetrapeptides,62 and are further characterized as selective or pan-inhibitors according to their spectrum of action. Consistent with the role histone deacetylases play in repressing transcription, HDAC inhibitors have been shown to disrupt HIV-latency and induce virus HIV-1 expression in latently infected cell lines, latently infected primary T-cells, resting CD4+ T-cells isolated from HIV-infected donors and, recently, in vivo.28-36,40
Valproic acid and vorinostat
Valproic acid (VPA), a known anticonvulsant that also exerts weak HDAC inhibition, was the first HDACi to be tested in a clinical study with the objective of depleting the latent reservoir of HIV-1 infection. Whereas a substantial decline was seen in the frequency of replication competent HIV in circulating resting CD4+ T cells in the initial study,37 additional studies failed to demonstrate any effect of VPA, even in the setting of intensified cART.38,39,63 HDACi with much higher potency are now being investigated. Vorinostat is a hydroxamic acid containing pan-HDACi with activity against class I and II HDACs.64 Having received FDA-approval in 2006 for the treatment of cutaneous T cell lymphoma there is now considerable clinical experience with the use of this drug. It is the most extensively investigated HDACi in HIV context having consistently shown the ability to reactivate HIV-1 expression at therapeutic concentrations in latently infected cell lines, latently infected primary cells, and resting CD4+ T-cells from HIV-infected patients on suppressive HAART.28,29,35,65 In contrast, a recent study, investigating the HDACi vorinostat, VPA and oxamflatin, found that the levels of HIV production by HDAC inhibitor stimulated resting CD4+ T-cells from aviremic donors were not significantly different from those of cells treated with media alone.66 Of note, in this study virion-associated (extracellular) HIV-RNA rather than cell-associated HIV-RNA was quantified. Two clinical trials are currently undertaken to evaluate whether vorinostat can reactivate latent HIV in vivo. The first data from one these trials was recently published showing that a single dose of 400 mg vorinostat significantly increased expression of HIV-RNA in isolated resting CD4+ T cells in 8 of 8 evaluated subjects without any safety issues.40 This is a very important result establishing proof-of-concept for the use vorinostat to reactivate latent HIV. However, as the 8 evaluated subjects were selected from a total of 16 based upon demonstrable virus production following 335nM vorinostat ex vivo stimulation, the effect on a non-selected study group may be of less magnitude. The results from a clinical study (NCT01365065) conducted in Melbourne, Australia in which HIV infected patients on suppressive cART receive 400 mg vorinostat daily for 14 consecutive days are awaited with much anticipation.
Givinostat, panobinostat and belinostat
Givinostat, panobinostat and belinostat are all hydroxamic acid containing pan-HDACi. Givinostat was initially compared with VPA in an in vitro study employing the latently infected cell lines, ACH2 and U1. Robust induction of HIV-1 expression was shown, approximately 10 times more efficient than VPA at clinically relevant concentrations.30 These results were confirmed recently in the same cell lines showing higher potency for HIV reactivation than vorinostat.67 In addition, givinostat was shown to decrease CXCR4 and CCR5 expression,30 which is probably owing to its anti-inflammatory properties. At nanomolar concentrations, this compound inhibits production of pro-inflammatory cytokines and reduces systemic inflammation.68,69 Furthermore, givinostat was used in a clinical study to treat systemic-onset juvenile arthritis with an acceptable safety profile at a therapeutically effective dose of 1.5 mg/kg.70 Chronic immune activation as evidenced by higher levels of pro-inflammatory biomarkers and T-cell activation is a hallmark of HIV infection and contributes to HIV disease progression,71-74 but may also promote HIV persistence by inducing homeostatic proliferation of latently infected cells5 and inhibiting the function of HIV-specific effector T-cells. Whether givinostat has any effect on these HIV-related pathological processes is unknown, but would be important to explore in future HIV-related trials. In the latently infected cell lines, ACH2 and U1, belinostat has activity against class I and II HDACs with similar potency to givinostat36,67 and also displayed ability to induce HIV production at therapeutic concentrations in a primary CD4+ T cell model of latency (Rasmussen et al., unpublished). However, as belinostat has primarily been used intravenously, published pharmacokinetic information on the oral formulation of belinostat is limited.
Panobinostat has recently displayed considerable potency in reactivating HIV-1 expression in latently infected cell lines and primary resting CD4+ T cells as compared with other HDACi in clinical development.67 In this study, panobinostat reactivated HIV-1 expression at concentrations as low as 8–32nM – well below the levels obtained with oral clinical dosing. Panobinostat is likely one of the most potent pan-HDAC inhibitors in clinical development and as the elimination time of panobinostat is relatively long, prolonged histone hyper acetylation can be observed 7 d after a second dose with this compound.75 This allows for dose reductions or intermittent dosing schedules to diminish the problematic thrombocytopenia seen with all HDAC inhibitors. A clinical trial to investigate the in vivo effect of panobinostat on HIV-1 expression and HIV reservoir size has been initiated by our group at Aarhus University Hospital, Denmark (NCT01680094). This study entails 8 week of cyclic panobinostat therapy with a primary endpoint of change from baseline in cell-associated unspliced HIV-RNA and will also provide a unique opportunity for studying the effect on host immune responses.
Other HDAC inhibitors
An increasing number of other HDACi have been tested in different models for the ability to reactivate HIV-1 expression in latently infected cells, but most of these compounds have never been administered to humans. These investigations include sodium butyrate (cell lines),31 entinostat (cell lines and primary T cells),31,65 trichostatin A (cell lines and primary T cells),31,59,76,77 oxamflatin (cell lines and patient cells),34,66 apicidin (cell lines)78,79, NCF-51 (cell lines)80 and scriptaid (cell lines).76 Also, explorations of HDACi generated by Merck (MRK1, MRK4, MRK10–14) with varying degree of HDAC selectivity showed that inhibitors of class I HDACs were more efficient inducers of HIV-1 expression than inhibitors of class II HDACs in cell lines and resting CD4+ T cells from patients on cART.78 Similarly, givinostat analogs ITFa, ITFb and ITFc increased HIV-1 expression in latently infected cell lines and higher levels of virus production was seen with compounds that exhibited the highest inhibitory potential for class I HDACs.30 Romidepsin (Istodax®), like vorinostat, is an FDA-approved HDACi for the treatment of cutaneous T cell lymphoma. Romidepsin has high potency specificity for HDAC1 and HDAC2,81 but there is no published data on the effect on latent HIV. The Aids Clinical Trial Group (ACTG) is reportedly making preparations toward a romidepsin ascending single dose study to investigate the in vivo effect on virus production.
Immune modulatory effects of HDAC inhibitors
While HDACi initially attracted attention in the oncology field due to their proapoptotic and cell cycle arrest actions on malignant cells, their potential as immunotherapy is now also being intensively tested focusing on anti-inflammatory effects. Clinical and experimental studies have identified a range of immune modulatory effects of HDACi involving both specific inflammation signaling pathways (e.g., regulation of NF-κB via IκBα or p65) as well as epigenetic mechanisms.82,83 Most of these effects are anti-inflammatory but the biologic roles of individual HDAC isoforms and their corresponding selective inhibitors are complex and show great diversity.
In the context of HIV, HDACi's action on T cells and regulatory T cells (Tregs) in particular is highly relevant. Therapy with a pan-HDACi (e.g., SAHA or panobinostat) can stimulate thymic production of Foxp3+ Tregs, promote conversion of T cells into Tregs, and enhance the immune suppressive function of human Tregs. In addition, HDACi increase Foxp3 acetylation thereby protecting it from proteasomal degradation.84 Thus, HDACi induced immune suppression via Tregs may impact the course of HIV infection given the fact that the virus induces excess inflammation that drives disease progression in untreated HIV infection and causes premature immunosenescence and morbidity in persons on HAART.71 In HIV eradication, the consequences of HDACi induced Treg expansion and/or function, could be either beneficial, by suppressing generalized T-cell activation, or detrimental, by weakening HIV-specific responses and thereby hindering immune-mediated clearance of latently infected reactivated CD4+ T cells. However, predicting different HDACi's in vivo anti- or pro-inflammatory effects in HIV may prove challenging since even structurally related compounds have been shown to have opposing actions. For example, in a rodent model of graft-vs.-host disease (GVHD) vorinostat reduced inflammation and GVHD-related mortality85 while Wang et al. found that panobinostat induced a Th1-directed pro-inflammatory response and augmented GVHD progression.86 This divergent effect of two hydroxamic acid containing pan-HDACi may be explained by differences in their isotype-specific HDAC inhibitory potential. Knockout of HDAC3 have been shown to induce upregulation of NF-κB, one of the main pro-inflammatory pathways. Panobinostat inhibits HDAC3 at 10-fold lower EC50 concentrations than vorinostat suggesting that panobinostat may have more pronounced pro-inflammatory effects than vorinostat.64 On the other hand, panobinostat's EC50 for inhibition of HDAC9 is approximately 30–40 fold lower than the EC50 for vorinostat.64 Inhibition of HDAC9 leads to enhanced suppressive Tregs' function and proliferation pointing toward a more potent induction of anti-inflammatory T-regs by panobinostat than vorinostat.83
Nevertheless, in oncologic studies various HDACi have repeatedly been shown to enhance anti-tumor effects by stimulating antigen-presenting cell and T cell activity.87 Collectively, the current literature suggest that the anti-inflammatory effects of HDACi in vivo generally tend to target pathologic inflammatory responses while preserving normal immune cell function.
Immunotherapy: Cytokines
Early studies suggested that interleukin (IL)-2 therapy might impact on the frequency of resting cells harboring replication competent virus,88 but rebound viremia occurred rapidly in these subjects upon interruption of cART.89 Moreover, additional studies could not establish an effect of IL-2 on the pool of latently infected CD4+ T cells or HIV production,90,91 and when IL-2 was used in combination with anti-CD3 antibody OKT3 this led to detrimental T cell activation and irreversible CD4+ T cell depletion.92 Currently, there is more focus on the prospects of the homeostatic cytokine, IL-7. Several studies have shown that IL-7 induce virus outgrowth ex vivo in the resting CD4+ T cells of HIV infected patients on cART.41,42 Two small clinical trials conducted in HIV infected patients reported that IL-7 administration increased CD4+ and CD8+ T cells with a memory phenotype. Furthermore, transient increases in plasma HIV-RNA was seen in 4 of 13 and 6 of 11 study subjects, respectively.93,94 To identify the sources of HIV detected, HIV-RNA and HIV-DNA sequences present before, during and after transient viremic episodes were analyzed and these results indicated that the release of virus originated from a preexisting pool of HIV-RNA rather than activation of silent proviruses.95 Also, a recent study showed that, whereas partial reactivation of latent HIV-1 can be achieved with IL-2 and IL-7 in combination, this does not reduce the pool of latently infected cells.96 Rather, homeostatic proliferation induced by these cytokines may favor the maintenance of the latent HIV-1 reservoir.5,96 Collectively, these findings indicate that the homeostatic proliferation induced by IL-7 therapy could be counterproductive in HIV eradication therapy. A recent clinical study among 32 HIV-infected subjects confirmed that administration of recombinant human IL-7 increases CD4+ T cells of predominantly naïve and central memory phenotype.97 Interestingly, transient low-level viremia was seen in a minority of study subjects and, moreover, levels of total HIV-DNA per milliliter blood, but not per 106 CD4+ T cells, increased suggesting that IL-7 treatment could have induced homeostatic proliferation of latently infected cells. An ongoing clinical trial (ERAMUNE) is currently investigating IL-7 for its effect on the latent HIV reservoir.
Immunotherapy: Toll-like receptor (TLR) agonists
Non-pathogen specific stimulation of the innate immune system via TLRs is used to treat certain viral diseases (e.g., Imiquimod against genital warts) and as adjuvant in immunization. In addition, some TLR ligands appear to modulate latent HIV infection. First, the TLR-5 agonist flagellin results in NF-КB activation and induces expression in latently infected cell lines and resting central memory T-cells transfected with HIV-1, but could not be shown to reactivate HIV-1 in purified resting CD4+ T cells from aviremic HIV-patients.98 Second, the TLR7/8 agonist, R-848, activated HIV from cells of myeloid-monocytic origin through TLR8-mediated NF-κΒ activation.99,100 Finally, synthetic CpG oligodeoxynucleotides (CpG ODNs) that stimulate immune cells via TLR9 induced HIV reactivation in vitro.101,102 We recently conducted a double-blind randomized controlled vaccine trial in which 95 HIV-infected adults were randomized to receive pneumococcal vaccines with or without the synthetic CpG ODN, CPG 7909, as adjuvant.103 This trial provided a unique opportunity to explore whether CpG ODNs might have impacted upon the proviral reservoir in vivo despite the limitations in dosage and sampling inherent to the vaccine trial design. Inclusion into this post hoc analysis was restricted to 54 participants who were on cART, had available sample material and had quantifiable HIV-DNA at the time of immunization. Indeed, we observed a moderate but statistically significant reduction in proviral HIV-DNA among CPG 7909 recipients compared with those receiving placebo adjuvant (p = 0.02) advising that further investigation into the effect of TLR9 agonists on HIV latency is warranted (personal communication). Interestingly, in vitro studies conducted at our laboratory revealed a synergistic effect of CpG ODNs and HDACi in combination. Treating the latently infected cell line U1 with increasing concentrations of CpG ODNs produced marked increases in HIV production following stimulation with low concentrations of the HDACi panobinostat (Fig. 2). Briefly, cells were incubated with indicated drug concentrations for 48 h followed by cell lysis and p24 ELISA enumeration as previously described.104 Combining CpG and Panobinostat induces significantly HIV production than both treatments individually (p < 0.001, ANOVA).
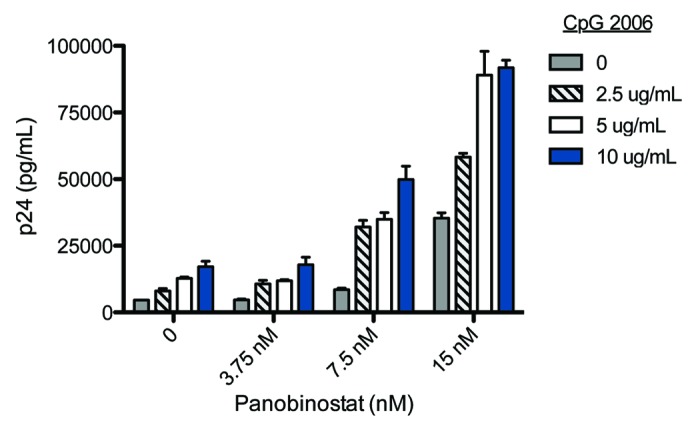
Figure 2. Stimulation of HIV-1 expression by CpG 2006 and panobinostat. HIV-1 expression in the latently infected cell line U1 following treatment for 48 h with combinations of CpG 2006 (0–10 μg/mL) and panobinostat (LBH589; 0–15nM). Virus production was estimated by p24 levels in supernatant; mean +/− SEM shown in figure
Protein kinase C (PKC) activators
Brostatin-1 is a natural occurring PKC activator belonging to the marine macrolide class of molecules. It is isolated from the marine bryozoan Bugula neritina105 and has been administered in numerous clinical trials for its anti-cancer effect. In cell lines bryostatin-1 reactivated HIV-1 expression more potently than vorinostat and prostratin via activation of PKC.106 In addition, lipid nanoparticles with bryostatin-2 incorporated have been developed and were shown to stimulate HIV production in T cell lines in vitro and in latently infected cells ex vivo in a humanized mouse model.107 The limited supply of natural occurring bryostatin has impeded the clinical use of this compound, but production of bryostatin analogs that reactivate latent HIV with similar or higher potencies was described recently.108 Another naturally occurring PKC activator, prostratin, is isolated from the Samoan medicinal plant, Homalanthus nutans. Prostratin induces HIV-1 expression in latently infected cell lines and cells isolated from aviremic HIV-infected patients on cART through PKC-mediated activation of NF-κΒ.45,46,109 However, the in vivo toxicity and safety of prostratin is unknown and advancing this compound to the level of clinical testing, if feasible, will take some time. Notably, synergistic effects of activating virus expression have been described for both bryostatin-1110 and prostratin31 indicating that targeting mechanistically different pathways implicated in silencing HIV transcription is desirable for breaking latency.
Other activators of HIV
Recently, using a primary CD4+ T cell model, in which HIV-1 latency was established by transducing primary human CD4+ T cells with the prosurvival gene bcl-2 and infecting them with HIV-1 before allowing the cells to return to a resting state, drug libraries were screened for compounds that reverse HIV-1 latency in vitro without cellular activation.111 Disulfiram, an inhibitor of acetaldehyde dehydrogenase used to treat alcoholism, was identified as a potential re-activator of latent HIV-1,43 presumably owing to depletion of the phosphatase and tensin homolog (PTEN) resulting in activation of the Akt signaling pathway.112 A single arm pilot study has been undertaken to evaluate whether adding 500 mg disulfiram daily for 2 weeks to stable cART will increase HIV production and decrease the HIV reservoir in vivo. Preliminary results showed no significant effect of disulfiram on these endpoints. However, increases in plasma HIV-RNA were observed among study subjects with available blood samples 1–2 h after the first dose.113 Full results from this study are expected in the near future.
In addition to the actions of HDACs that repress HIV-1 transcription by keeping nuc-1 in the hypoacetylated state, histone methyltransferases (HMTs) have been shown to inhibit viral expression by promoting histone H3 methylation in nuc-1.114 Two inhibitors of HMT, chaetocin and BIX-01294, have been described so far. These compounds induced virus outgrowth in resting CD4+ T cells from aviremic HIV infected donors on cART, but cannot be administered safely to humans.115 Hexamethylbisacetamide (HBMA) is a kinase agonist that was tested in a few clinical studies more than 20 y ago for its effect on hematologic malignancies.116 It has been shown to promote HIV-1 expression in latently infected cell lines in a Tat independent manner117 and induce outgrowth of HIV-1 from resting CD4+ T cells recovered from aviremic patients on cART.118 While thrombocytopenia appears to limit the clinical use of HMBA,116 findings that HMBA mediates its effect on HIV latency through signaling via both protein kinase C (PKC) μ and phosphatidylinositol 3–kinase, reveal cellular kinases that may be therapeutically exploited.118
Discussion
Thus, while an increasing number of substances that could reactivate HIV-1 from latency are being described, there are limitations to this approach and significant gaps in knowledge. Ongoing viral replicative activity or cell-to-cell spread119 is not targeted by reactivation approaches and, to the extent that is at all occurring during suppressive therapy, must be addressed by improvements in drug delivery to tissues of residual HIV exchange. Moreover, for reactivating strategies to be successful, the induced HIV-1 expression in latently infected cells must be followed by the removal of these cells by viral cytopathic effects or immune mediated mechanisms. It is currently unknown to which extend this occurs in vivo as chronic HIV-infection is characterized by an impaired cytolytic capacity of CD8+ T cells, which is not restored by cART.120 Notably, a recent in vitro study showed that reactivation of virus production in latently infected resting cells was insufficient to eliminate these cells; only after stimulation of HIV-1 specific cytolytic T cells was efficient killing of latently infected cells achieved.47 This suggests that combining pharmacological reactivation of HIV-1 from latency with therapies designed to improve the killing capacity of cytolytic T cells could be needed and would be a logical next step once HDACi induced HIV reactivation in vivo is described in more detail. In addition, there are several unique challenges related to testing strategies in clinical trials that require careful consideration. While eradication therapies that could significantly impact the latent reservoir may also have associated toxicities or unknown long-term effects, there is little chance of a health benefit for study participants in the initial trials. Thus, careful consideration of acceptable risks weighed against possible long-term benefits is necessary and poses challenges for investigators and regulatory authorities. Also, difficulties in measuring the effects on HIV transcription or the latent reservoir are significant barriers to expanding clinical trial strategies. Large cell numbers and complex assays are currently applied and these methods, and the inherent high costs, will be difficult to operate in larger clinical trials. In the end viral rebound parameters during cART interruption will be the most relevant clinical endpoint, but will require careful consideration of when this is justified and which efficacy criteria should be met.
The development and implementation of cART has been a major medical achievement that has transformed HIV from a deadly to a chronic disease, but HIV infected patients are still burdened with excess morbidity and mortality, long-term toxicities from cART, stigmatization and, finally, insufficient access to cART worldwide. Thus, a cure for HIV would have a substantial impact on society as well as the individual and continues to be a high research priority. As the complexity and mechanisms of HIV persistence during therapy are unraveled, new therapeutic targets are discovered. A growing number of substances that could promote the eradication of HIV through activating HIV production in latently infected cells are now being described. Whether or not reactivating strategies will prove to be a significant component of a cure for HIV is a key question within this field of research. The first indications of what the answer will be will come from clinical trials currently conducted or underway.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/vaccines/article/23202
References
- 1.Margolis DM. Eradication therapies for HIV Infection: time to begin again. AIDS Res Hum Retroviruses. 2011;27:347–53. doi: 10.1089/aid.2011.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palmer S, Maldarelli F, Wiegand A, Bernstein B, Hanna GJ, Brun SC, et al. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci U S A. 2008;105:3879–84. doi: 10.1073/pnas.0800050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palmer S, Wiegand AP, Maldarelli F, Bazmi H, Mican JM, Polis M, et al. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol. 2003;41:4531–6. doi: 10.1128/JCM.41.10.4531-4536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5:512–7. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 5.Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hermankova M, Siliciano JD, Zhou Y, Monie D, Chadwick K, Margolick JB, et al. Analysis of human immunodeficiency virus type 1 gene expression in latently infected resting CD4+ T lymphocytes in vivo. J Virol. 2003;77:7383–92. doi: 10.1128/JVI.77.13.7383-7392.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kieffer TL, Finucane MM, Nettles RE, Quinn TC, Broman KW, Ray SC, et al. Genotypic analysis of HIV-1 drug resistance at the limit of detection: virus production without evolution in treated adults with undetectable HIV loads. J Infect Dis. 2004;189:1452–65. doi: 10.1086/382488. [DOI] [PubMed] [Google Scholar]
- 8.Ellery PJ, Tippett E, Chiu YL, Paukovics G, Cameron PU, Solomon A, et al. The CD16+ monocyte subset is more permissive to infection and preferentially harbors HIV-1 in vivo. J Immunol. 2007;178:6581–9. doi: 10.4049/jimmunol.178.10.6581. [DOI] [PubMed] [Google Scholar]
- 9.Collman R, Hassan NF, Walker R, Godfrey B, Cutilli J, Hastings JC, et al. Infection of monocyte-derived macrophages with human immunodeficiency virus type 1 (HIV-1). Monocyte-tropic and lymphocyte-tropic strains of HIV-1 show distinctive patterns of replication in a panel of cell types. J Exp Med. 1989;170:1149–63. doi: 10.1084/jem.170.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sonza S, Mutimer HP, Oelrichs R, Jardine D, Harvey K, Dunne A, et al. Monocytes harbour replication-competent, non-latent HIV-1 in patients on highly active antiretroviral therapy. AIDS. 2001;15:17–22. doi: 10.1097/00002030-200101050-00005. [DOI] [PubMed] [Google Scholar]
- 11.Churchill MJ, Wesselingh SL, Cowley D, Pardo CA, McArthur JC, Brew BJ, et al. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann Neurol. 2009;66:253–8. doi: 10.1002/ana.21697. [DOI] [PubMed] [Google Scholar]
- 12.Carter CC, Onafuwa-Nuga A, McNamara LA, Riddell J, 4th, Bixby D, Savona MR, et al. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat Med. 2010;16:446–51. doi: 10.1038/nm.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wightman F, Solomon A, Khoury G, Green JA, Gray L, Gorry PR, et al. Both CD31(+) and CD31⁻ naive CD4(+) T cells are persistent HIV type 1-infected reservoirs in individuals receiving antiretroviral therapy. J Infect Dis. 2010;202:1738–48. doi: 10.1086/656721. [DOI] [PubMed] [Google Scholar]
- 14.Tran TA, de Goër de Herve MG, Hendel-Chavez H, Dembele B, Le Névot E, Abbed K, et al. Resting regulatory CD4 T cells: a site of HIV persistence in patients on long-term effective antiretroviral therapy. PLoS One. 2008;3:e3305. doi: 10.1371/journal.pone.0003305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Josefsson L, Eriksson S, Sinclair E, Ho T, Killian M, Epling L, et al. Hematopoietic precursor cells isolated from patients on long-term suppressive HIV therapy did not contain HIV-1 DNA. J Infect Dis. 2012;206:28–34. doi: 10.1093/infdis/jis301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gandhi RT, Bosch RJ, Aga E, Albrecht M, Demeter LM, Dykes C, et al. AIDS Clinical Trials Group A5173 Team No evidence for decay of the latent reservoir in HIV-1-infected patients receiving intensive enfuvirtide-containing antiretroviral therapy. J Infect Dis. 2010;201:293–6. doi: 10.1086/649569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gandhi RT, Zheng L, Bosch RJ, Chan ES, Margolis DM, Read S, et al. AIDS Clinical Trials Group A5244 team The effect of raltegravir intensification on low-level residual viremia in HIV-infected patients on antiretroviral therapy: a randomized controlled trial. PLoS Med. 2010;7 doi: 10.1371/journal.pmed.1000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McMahon D, Jones J, Wiegand A, Gange SJ, Kearney M, Palmer S, et al. Short-course raltegravir intensification does not reduce persistent low-level viremia in patients with HIV-1 suppression during receipt of combination antiretroviral therapy. Clin Infect Dis. 2010;50:912–9. doi: 10.1086/650749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dinoso JB, Kim SY, Wiegand AM, Palmer SE, Gange SJ, Cranmer L, et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc Natl Acad Sci U S A. 2009;106:9403–8. doi: 10.1073/pnas.0903107106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yukl SA, Shergill AK, McQuaid K, Gianella S, Lampiris H, Hare CB, et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS. 2010;24:2451–60. doi: 10.1097/QAD.0b013e32833ef7bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hammer SM, Ribaudo H, Bassett R, Mellors JW, Demeter LM, Coombs RW, et al. AIDS Clinical Trials Group (ACTG) 372A Study Team A randomized, placebo-controlled trial of abacavir intensification in HIV-1-infected adults with virologic suppression on a protease inhibitor-containing regimen. HIV Clin Trials. 2010;11:312–24. doi: 10.1310/hct1106-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buzón MJ, Massanella M, Llibre JM, Esteve A, Dahl V, Puertas MC, et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat Med. 2010;16:460–5. doi: 10.1038/nm.2111. [DOI] [PubMed] [Google Scholar]
- 23.Vallejo A, Gutierrez C, Hernandez-Novoa B, Diaz L, Madrid N, Abad-Fernandez M, et al. The effect of intensification with raltegravir on the HIV-1 reservoir of latently infected memory CD4 T cells in suppressed patients. AIDS. 2012;26:1885–94. doi: 10.1097/QAD.0b013e3283584521. [DOI] [PubMed] [Google Scholar]
- 24.Hütter G, Nowak D, Mossner M, Ganepola S, Müssig A, Allers K, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360:692–8. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 25.Lalezari J, Mitsuyasu R, Wang S, Lee G, Giedlin M, Tang W, et al. A Single Infusion of Zinc Finger Nuclease CCR5 Modified Autologous CD4 T Cells (SB-728-T) Increases CD4 Counts and Leads to Decrease in HIV Proviral Load in an Aviremic HIV-infected Subject. 19th Conference on Retroviruses and Opportunistic Infections Seattle, 2012. [Google Scholar]
- 26.June C, Tebas P, Stein D, Mitsuyasu R, Lalezari J, Wang SS, et al. Induction of Acquired CCR5 Deficiency with Zinc Finger Nuclease-modified Autologous CD4 T Cells (SB-728-T) Correlates with Increases in CD4 Count and Effects on Viral Load in HIV+ Subjects. Conference on Retroviruses and Opportunistic Infections Seattle, 2012. [Google Scholar]
- 27.Cillo A, Krishnan A, Mitsuyasu R, McMahon D, Li S, Rossi J, et al. Plasma Viremia and Cellular HIV-1 DNA Persist Despite Autologous Hematopoietic Stem Cell Transplantation for AIDS-related Lymphoma. 19th Conference on Retroviruses and Opportunistic Infections Seattle, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis DM. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res Hum Retroviruses. 2009;25:207–12. doi: 10.1089/aid.2008.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Contreras X, Schweneker M, Chen CS, McCune JM, Deeks SG, Martin J, et al. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J Biol Chem. 2009;284:6782–9. doi: 10.1074/jbc.M807898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matalon S, Palmer BE, Nold MF, Furlan A, Kassu A, Fossati G, et al. The histone deacetylase inhibitor ITF2357 decreases surface CXCR4 and CCR5 expression on CD4(+) T-cells and monocytes and is superior to valproic acid for latent HIV-1 expression in vitro. J Acquir Immune Defic Syndr. 2010;54:1–9. doi: 10.1097/QAI.0b013e3181d3dca3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reuse S, Calao M, Kabeya K, Guiguen A, Gatot JS, Quivy V, et al. Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: implications for treatment of latent infection. PLoS One. 2009;4:e6093. doi: 10.1371/journal.pone.0006093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi BS, Lee HS, Oh YT, Hyun YL, Ro S, Kim SS, et al. Novel histone deacetylase inhibitors CG05 and CG06 effectively reactivate latently infected HIV-1. AIDS. 2010;24:609–11. doi: 10.1097/QAD.0b013e328333bfa1. [DOI] [PubMed] [Google Scholar]
- 33.Ylisastigui L, Archin NM, Lehrman G, Bosch RJ, Margolis DM. Coaxing HIV-1 from resting CD4 T cells: histone deacetylase inhibition allows latent viral expression. AIDS. 2004;18:1101–8. doi: 10.1097/00002030-200405210-00003. [DOI] [PubMed] [Google Scholar]
- 34.Yin H, Zhang Y, Zhou X, Zhu H. Histonedeacetylase inhibitor Oxamflatin increase HIV-1 transcription by inducing histone modification in latently infected cells. Mol Biol Rep. 2011;38:5071–8. doi: 10.1007/s11033-010-0653-6. [DOI] [PubMed] [Google Scholar]
- 35.Wightman F, Ellenberg P, Churchill M, Lewin SR. HDAC inhibitors in HIV. Immunol Cell Biol. 2012;90:47–54. doi: 10.1038/icb.2011.95. [DOI] [PubMed] [Google Scholar]
- 36.Matalon S, Rasmussen TA, Dinarello CA. Histone deacetylase inhibitors for purging HIV-1 from the latent reservoir. Mol Med. 2011;17:466–72. doi: 10.2119/molmed.2011.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehrman G, Hogue IB, Palmer S, Jennings C, Spina CA, Wiegand A, et al. Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet. 2005;366:549–55. doi: 10.1016/S0140-6736(05)67098-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Archin NM, Cheema M, Parker D, Wiegand A, Bosch RJ, Coffin JM, et al. Antiretroviral intensification and valproic acid lack sustained effect on residual HIV-1 viremia or resting CD4+ cell infection. PLoS One. 2010;5:e9390. doi: 10.1371/journal.pone.0009390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Archin NM, Eron JJ, Palmer S, Hartmann-Duff A, Martinson JA, Wiegand A, et al. Valproic acid without intensified antiviral therapy has limited impact on persistent HIV infection of resting CD4+ T cells. AIDS. 2008;22:1131–5. doi: 10.1097/QAD.0b013e3282fd6df4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–5. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang FX, Xu Y, Sullivan J, Souder E, Argyris EG, Acheampong EA, et al. IL-7 is a potent and proviral strain-specific inducer of latent HIV-1 cellular reservoirs of infected individuals on virally suppressive HAART. J Clin Invest. 2005;115:128–37. doi: 10.1172/JCI22574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lehrman G, Ylisastigui L, Bosch RJ, Margolis DM. Interleukin-7 induces HIV type 1 outgrowth from peripheral resting CD4+ T cells. J Acquir Immune Defic Syndr. 2004;36:1103–4. doi: 10.1097/00126334-200408150-00015. [DOI] [PubMed] [Google Scholar]
- 43.Xing S, Bullen CK, Shroff NS, Shan L, Yang HC, Manucci JL, et al. Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J Virol. 2011;85:6060–4. doi: 10.1128/JVI.02033-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Biancotto A, Grivel JC, Gondois-Rey F, Bettendroffer L, Vigne R, Brown S, et al. Dual role of prostratin in inhibition of infection and reactivation of human immunodeficiency virus from latency in primary blood lymphocytes and lymphoid tissue. J Virol. 2004;78:10507–15. doi: 10.1128/JVI.78.19.10507-10515.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kulkosky J, Culnan DM, Roman J, Dornadula G, Schnell M, Boyd MR, et al. Prostratin: activation of latent HIV-1 expression suggests a potential inductive adjuvant therapy for HAART. Blood. 2001;98:3006–15. doi: 10.1182/blood.V98.10.3006. [DOI] [PubMed] [Google Scholar]
- 46.Williams SA, Chen LF, Kwon H, Fenard D, Bisgrove D, Verdin E, et al. Prostratin antagonizes HIV latency by activating NF-kappaB. J Biol Chem. 2004;279:42008–17. doi: 10.1074/jbc.M402124200. [DOI] [PubMed] [Google Scholar]
- 47.Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. 2012;36:491–501. doi: 10.1016/j.immuni.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Persaud D, Luzuriaga K, Ziemniak C, Muresan P, Greenough T, Fenton T, et al. Effect of therapeutic HIV recombinant poxvirus vaccines on the size of the resting CD4+ T-cell latent HIV reservoir. AIDS. 2011;25:2227–34. doi: 10.1097/QAD.0b013e32834cdaba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L, de Zoeten EF, Greene MI, Hancock WW. Immunomodulatory effects of deacetylase inhibitors: therapeutic targeting of FOXP3+ regulatory T cells. Nat Rev Drug Discov. 2009;8:969–81. doi: 10.1038/nrd3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verdin E, Paras P, Jr., Van Lint C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993;12:3249–59. doi: 10.1002/j.1460-2075.1993.tb05994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996;15:1112–20. [PMC free article] [PubMed] [Google Scholar]
- 52.Coull JJ, Romerio F, Sun JM, Volker JL, Galvin KM, Davie JR, et al. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol. 2000;74:6790–9. doi: 10.1128/JVI.74.15.6790-6799.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006;25:139–49. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Imai K, Okamoto T. Transcriptional repression of human immunodeficiency virus type 1 by AP-4. J Biol Chem. 2006;281:12495–505. doi: 10.1074/jbc.M511773200. [DOI] [PubMed] [Google Scholar]
- 55.Jiang G, Espeseth A, Hazuda DJ, Margolis DM. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J Virol. 2007;81:10914–23. doi: 10.1128/JVI.01208-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lusic M, Marcello A, Cereseto A, Giacca M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003;22:6550–61. doi: 10.1093/emboj/cdg631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tyagi M, Karn J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007;26:4985–95. doi: 10.1038/sj.emboj.7601928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Keedy KS, Archin NM, Gates AT, Espeseth A, Hazuda DJ, Margolis DM. A limited group of class I histone deacetylases acts to repress human immunodeficiency virus type 1 expression. J Virol. 2009;83:4749–56. doi: 10.1128/JVI.02585-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huber K, Doyon G, Plaks J, Fyne E, Mellors JW, Sluis-Cremer N. Inhibitors of histone deacetylases: correlation between isoform specificity and reactivation of HIV type 1 (HIV-1) from latently infected cells. J Biol Chem. 2011;286:22211–8. doi: 10.1074/jbc.M110.180224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci U S A. 1999;96:4868–73. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 62.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 63.Routy JP, Tremblay CL, Angel JB, Trottier B, Rouleau D, Baril JG, et al. Valproic acid in association with highly active antiretroviral therapy for reducing systemic HIV-1 reservoirs: results from a multicentre randomized clinical study. HIV Med. 2012;13:291–6. doi: 10.1111/j.1468-1293.2011.00975.x. [DOI] [PubMed] [Google Scholar]
- 64.Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–9. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 65.Wightman F, Ramanayake S, Saleh S, Solomon A, Dear A, Shebu-Xhilaga M, et al. Potency and Toxicity of HDACi and Other Immune Activators in Inducing HIV Production Using a Primary Resting T Cell Model of HIV Latency. Conference on Retroviruses and Opportunistic Infections Boston, 2011. [Google Scholar]
- 66.Blazkova J, Chun TW, Belay BW, Murray D, Justement JS, Funk EK, et al. Effect of histone deacetylase inhibitors on HIV production in latently infected, resting CD4(+) T cells from infected individuals receiving effective antiretroviral therapy. J Infect Dis. 2012;206:765–9. doi: 10.1093/infdis/jis412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rasmussen TA, Søgaard O, Melchjorsen J, Brinkmann C, Østergaard L, Dinarello CA, et al. HDACi LBH589 Stimulates HIV-1 Expression More Potently than Other HDACi in Clinical Use and Disrupts HIV Latency at Clinically Achievable Concentrations. Conference on Retroviruses and Opportunistic Infections Seattle, 2012. [Google Scholar]
- 68.Furlan A, Monzani V, Reznikov LL, Leoni F, Fossati G, Modena D, et al. Pharmacokinetics, safety and inducible cytokine responses during a phase 1 trial of the oral histone deacetylase inhibitor ITF2357 (givinostat) Mol Med. 2011;17:353–62. doi: 10.2119/molmed.2011.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leoni F, Fossati G, Lewis EC, Lee JK, Porro G, Pagani P, et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med. 2005;11:1–15. doi: 10.2119/2006-00005.Dinarello. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vojinovic J, Dinarello CA, Damjano N, Oldoni T. Safety and Efficacy of Oral ITF2357 in Patients with Active Systemic Onset Juvenile Idiopathic Arthritis (SOJIA) - Results of a Phase II, Open Label, International, Multicentre Clinical Trial. Abstract 2008 Annual Scientific Meeting, American College of Rheumatology 2008. [Google Scholar]
- 71.Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, et al. INSIGHT SMART Study Group Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5:e203. doi: 10.1371/journal.pmed.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neuhaus J, Jacobs DR, Jr., Baker JV, Calmy A, Duprez D, La Rosa A, et al. Markers of inflammation, coagulation, and renal function are elevated in adults with HIV infection. J Infect Dis. 2010;201:1788–95. doi: 10.1086/652749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Phillips AN, Gazzard B, Gilson R, Easterbrook P, Johnson M, Walsh J, et al. UK Collaborative HIV Cohort Study Rate of AIDS diseases or death in HIV-infected antiretroviral therapy-naive individuals with high CD4 cell count. AIDS. 2007;21:1717–21. doi: 10.1097/QAD.0b013e32827038bf. [DOI] [PubMed] [Google Scholar]
- 74.Deeks SG, Phillips AN. HIV infection, antiretroviral treatment, ageing, and non-AIDS related morbidity. BMJ. 2009;338:a3172. doi: 10.1136/bmj.a3172. [DOI] [PubMed] [Google Scholar]
- 75.Prince HM, Bishton MJ, Johnstone RW. Panobinostat (LBH589): a potent pan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol. 2009;5:601–12. doi: 10.2217/fon.09.36. [DOI] [PubMed] [Google Scholar]
- 76.Ying H, Zhang Y, Lin S, Han Y, Zhu HZ. Histone deacetylase inhibitor Scriptaid reactivates latent HIV-1 promoter by inducing histone modification in in vitro latency cell lines. Int J Mol Med. 2010;26:265–72. doi: 10.3892/ijmm_00000461. [DOI] [PubMed] [Google Scholar]
- 77.Sahu GK, Cloyd MW. Latent HIV in primary T lymphocytes is unresponsive to histone deacetylase inhibitors. Virol J. 2011;8:400. doi: 10.1186/1743-422X-8-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Archin NM, Keedy KS, Espeseth A, Dang H, Hazuda DJ, Margolis DM. Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. AIDS. 2009;23:1799–806. doi: 10.1097/QAD.0b013e32832ec1dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lin S, Zhang Y, Ying H, Zhu H. HIV-1 reactivation induced by apicidin involves histone modification in latently infected cells. Curr HIV Res. 2011;9:202–8. doi: 10.2174/157016211796320333. [DOI] [PubMed] [Google Scholar]
- 80.Victoriano AF, Imai K, Togami H, Ueno T, Asamitsu K, Suzuki T, et al. Novel histone deacetylase inhibitor NCH-51 activates latent HIV-1 gene expression. FEBS Lett. 2011;585:1103–11. doi: 10.1016/j.febslet.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 81.Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62:4916–21. [PubMed] [Google Scholar]
- 82.Hancock WW, Akimova T, Beier UH, Liu Y, Wang L. HDAC inhibitor therapy in autoimmunity and transplantation. Ann Rheum Dis. 2012;71(Suppl 2):i46–54. doi: 10.1136/annrheumdis-2011-200593. [DOI] [PubMed] [Google Scholar]
- 83.Akimova T, Beier UH, Liu Y, Wang L, Hancock WW. Histone/protein deacetylases and T-cell immune responses. Blood. 2012;119:2443–51. doi: 10.1182/blood-2011-10-292003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 85.Leng C, Gries M, Ziegler J, Lokshin A, Mascagni P, Lentzsch S, et al. Reduction of graft-versus-host disease by histone deacetylase inhibitor suberonylanilide hydroxamic acid is associated with modulation of inflammatory cytokine milieu and involves inhibition of STAT1. Exp Hematol. 2006;34:776–87. doi: 10.1016/j.exphem.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 86.Wang D, Iclozan C, Liu C, Xia C, Anasetti C, Yu XZ. LBH589 enhances T cell activation in vivo and accelerates graft-versus-host disease in mice. Biol Blood Marrow Transplant. 2012;18:1182–90, e1. doi: 10.1016/j.bbmt.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dawson MA, Kouzarides T, Huntly BJ. Targeting epigenetic readers in cancer. N Engl J Med. 2012;367:647–57. doi: 10.1056/NEJMra1112635. [DOI] [PubMed] [Google Scholar]
- 88.Chun TW, Engel D, Mizell SB, Hallahan CW, Fischette M, Park S, et al. Effect of interleukin-2 on the pool of latently infected, resting CD4+ T cells in HIV-1-infected patients receiving highly active anti-retroviral therapy. Nat Med. 1999;5:651–5. doi: 10.1038/9498. [DOI] [PubMed] [Google Scholar]
- 89.Chun TW, Davey RT, Jr., Engel D, Lane HC, Fauci AS. Re-emergence of HIV after stopping therapy. Nature. 1999;401:874–5. doi: 10.1038/44755. [DOI] [PubMed] [Google Scholar]
- 90.Dybul M, Hidalgo B, Chun TW, Belson M, Migueles SA, Justement JS, et al. Pilot study of the effects of intermittent interleukin-2 on human immunodeficiency virus (HIV)-specific immune responses in patients treated during recently acquired HIV infection. J Infect Dis. 2002;185:61–8. doi: 10.1086/338123. [DOI] [PubMed] [Google Scholar]
- 91.Stellbrink HJ, van Lunzen J, Westby M, O’Sullivan E, Schneider C, Adam A, et al. Effects of interleukin-2 plus highly active antiretroviral therapy on HIV-1 replication and proviral DNA (COSMIC trial) AIDS. 2002;16:1479–87. doi: 10.1097/00002030-200207260-00004. [DOI] [PubMed] [Google Scholar]
- 92.van Praag RM, Prins JM, Roos MT, Schellekens PT, Ten Berge IJ, Yong SL, et al. OKT3 and IL-2 treatment for purging of the latent HIV-1 reservoir in vivo results in selective long-lasting CD4+ T cell depletion. J Clin Immunol. 2001;21:218–26. doi: 10.1023/A:1011091300321. [DOI] [PubMed] [Google Scholar]
- 93.Levy Y, Lacabaratz C, Weiss L, Viard JP, Goujard C, Lelièvre JD, et al. Enhanced T cell recovery in HIV-1-infected adults through IL-7 treatment. J Clin Invest. 2009;119:997–1007. doi: 10.1172/JCI38052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sereti I, Dunham RM, Spritzler J, Aga E, Proschan MA, Medvik K, et al. ACTG 5214 Study Team IL-7 administration drives T cell-cycle entry and expansion in HIV-1 infection. Blood. 2009;113:6304–14. doi: 10.1182/blood-2008-10-186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Imamichi H, Degray G, Asmuth DM, Fischl MA, Landay AL, Lederman MM, et al. HIV-1 viruses detected during episodic blips following interleukin-7 administration are similar to the viruses present before and after interleukin-7 therapy. AIDS. 2011;25:159–64. doi: 10.1097/QAD.0b013e328340a270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bosque A, Famiglietti M, Weyrich AS, Goulston C, Planelles V. Homeostatic proliferation fails to efficiently reactivate HIV-1 latently infected central memory CD4+ T cells. PLoS Pathog. 2011;7:e1002288. doi: 10.1371/journal.ppat.1002288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lévy Y, Sereti I, Tambussi G, Routy JP, Lelièvre JD, Delfraissy JF, et al. Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: results of a phase I/IIa randomized, placebo-controlled, multicenter study. Clin Infect Dis. 2012;55:291–300. doi: 10.1093/cid/cis383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thibault S, Imbeault M, Tardif MR, Tremblay MJ. TLR5 stimulation is sufficient to trigger reactivation of latent HIV-1 provirus in T lymphoid cells and activate virus gene expression in central memory CD4+ T cells. Virology. 2009;389:20–5. doi: 10.1016/j.virol.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 99.Schlaepfer E, Audigé A, Joller H, Speck RF. TLR7/8 triggering exerts opposing effects in acute versus latent HIV infection. J Immunol. 2006;176:2888–95. doi: 10.4049/jimmunol.176.5.2888. [DOI] [PubMed] [Google Scholar]
- 100.Schlaepfer E, Speck RF. TLR8 activates HIV from latently infected cells of myeloid-monocytic origin directly via the MAPK pathway and from latently infected CD4+ T cells indirectly via TNF-α. J Immunol. 2011;186:4314–24. doi: 10.4049/jimmunol.1003174. [DOI] [PubMed] [Google Scholar]
- 101.Scheller C, Ullrich A, Lamla S, Dittmer U, Rethwilm A, Koutsilieri E. Dual activity of phosphorothioate CpG oligodeoxynucleotides on HIV: reactivation of latent provirus and inhibition of productive infection in human T cells. Ann.N.Y. Acad.Sci. 2006;1091:540–7. doi: 10.1196/annals.1378.095. [DOI] [PubMed] [Google Scholar]
- 102.Scheller C, Ullrich A, McPherson K, Hefele B, Knöferle J, Lamla S, et al. CpG oligodeoxynucleotides activate HIV replication in latently infected human T cells. J Biol Chem. 2004;279:21897–902. doi: 10.1074/jbc.M311609200. [DOI] [PubMed] [Google Scholar]
- 103.Søgaard OS, Lohse N, Harboe ZB, Offersen R, Bukh AR, Davis HL, et al. Improving the immunogenicity of pneumococcal conjugate vaccine in HIV-infected adults with a toll-like receptor 9 agonist adjuvant: a randomized, controlled trial. Clin Infect Dis. 2010;51:42–50. doi: 10.1086/653112. [DOI] [PubMed] [Google Scholar]
- 104.Berg RK, Melchjorsen J, Rintahaka J, Diget E, Søby S, Horan KA, et al. Genomic HIV RNA induces innate immune responses through RIG-I-dependent sensing of secondary-structured RNA. PLoS One. 2012;7:e29291. doi: 10.1371/journal.pone.0029291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ramsdell JS, Pettit GR, Tashjian AH., Jr. Three activators of protein kinase C, bryostatins, dioleins, and phorbol esters, show differing specificities of action on GH4 pituitary cells. J Biol Chem. 1986;261:17073–80. [PubMed] [Google Scholar]
- 106.Mehla R, Bivalkar-Mehla S, Zhang R, Handy I, Albrecht H, Giri S, et al. Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS One. 2010;5:e11160. doi: 10.1371/journal.pone.0011160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kovochich M, Marsden MD, Zack JA. Activation of latent HIV using drug-loaded nanoparticles. PLoS One. 2011;6:e18270. doi: 10.1371/journal.pone.0018270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.DeChristopher BA, Loy BA, Marsden MD, Schrier AJ, Zack JA, Wender PA. Designed, synthetically accessible bryostatin analogues potently induce activation of latent HIV reservoirs in vitro. Nat Chem. 2012;4:705–10. doi: 10.1038/nchem.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rullas J, Bermejo M, García-Pérez J, Beltán M, González N, Hezareh M, et al. Prostratin induces HIV activation and downregulates HIV receptors in peripheral blood lymphocytes. Antivir Ther. 2004;9:545–54. [PubMed] [Google Scholar]
- 110.Pérez M, de Vinuesa AG, Sanchez-Duffhues G, Marquez N, Bellido ML, Muñoz-Fernandez MA, et al. Bryostatin-1 synergizes with histone deacetylase inhibitors to reactivate HIV-1 from latency. Curr HIV Res. 2010;8:418–29. doi: 10.2174/157016210793499312. [DOI] [PubMed] [Google Scholar]
- 111.Yang HC, Xing S, Shan L, O’Connell K, Dinoso J, Shen A, et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J Clin Invest. 2009;119:3473–86. doi: 10.1172/JCI39199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Doyon G, Zerbato J, Mellors JW, Sluis-Cremer N. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog (PTEN) AIDS. 2012 doi: 10.1097/QAD.0b013e3283570620. [DOI] [PubMed] [Google Scholar]
- 113.Spivak A, Andrade A, Hoh H, Bacchetti P, Eisele E, Buckheit R, et al. Safety and Feasibility of Using Disulfiram to Enhance HIV Transcription among Long-term ARV-treated Adults: Preliminary Results from a Pilot Study. 19th Conference on Retroviruses and Opportunistic Infections Seattle, 2012. [Google Scholar]
- 114.Imai K, Togami H, Okamoto T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J Biol Chem. 2010;285:16538–45. doi: 10.1074/jbc.M110.103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bouchat S, Gatot JS, Kabeya K, Cardona C, Colin L, Herbein G, et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4(+) T cells from HIV-1-infected HAART-treated patients. AIDS. 2012;26:1473–82. doi: 10.1097/QAD.0b013e32835535f5. [DOI] [PubMed] [Google Scholar]
- 116.Andreeff M, Stone R, Michaeli J, Young CW, Tong WP, Sogoloff H, et al. Hexamethylene bisacetamide in myelodysplastic syndrome and acute myelogenous leukemia: a phase II clinical trial with a differentiation-inducing agent. Blood. 1992;80:2604–9. [PubMed] [Google Scholar]
- 117.Klichko V, Archin N, Kaur R, Lehrman G, Margolis D. Hexamethylbisacetamide remodels the human immunodeficiency virus type 1 (HIV-1) promoter and induces Tat-independent HIV-1 expression but blunts cell activation. J Virol. 2006;80:4570–9. doi: 10.1128/JVI.80.9.4570-4579.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Choudhary SK, Archin NM, Margolis DM. Hexamethylbisacetamide and disruption of human immunodeficiency virus type 1 latency in CD4(+) T cells. J Infect Dis. 2008;197:1162–70. doi: 10.1086/529525. [DOI] [PubMed] [Google Scholar]
- 119.Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, et al. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature. 2011;477:95–8. doi: 10.1038/nature10347. [DOI] [PubMed] [Google Scholar]
- 120.Hersperger AR, Pereyra F, Nason M, Demers K, Sheth P, Shin LY, et al. Perforin expression directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control. PLoS Pathog. 2010;6:e1000917. doi: 10.1371/journal.ppat.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]