

Abstract
Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) and its receptor (TRAIL-R) play important roles in immune regulation and cancer cell death. Although TRAIL has been shown to induce chemokine release in various tumour cells, the function of TRAIL-R in the development of colitis and colitis-associated carcinogenesis has not been explored. In this study, we found that TRAIL-R-deficient mice exhibited a higher incidence of colitis and colitis-associated cancer than that of wild-type (WT) mice, and TRAIL-R expression was down-regulated in WT mice that were fed dextran sulphate sodium. Chemokines, including CCL2 and CXCL1, were highly expressed in the serum and inflammatory colon tissues of TRAIL-R−/− mice compared with WT mice, and TRAIL-R−/− mice showed a marked infiltration of immune cells during colitis. Hyperactivation of Janus kinase and nuclear factor-κB in colon epithelial cells was also observed, which correlated with the severity of colonic inflammation in TRAIL-R−/− mice. These data suggest that TRAIL-R plays a protective role in chemical-induced colon injury and negatively regulates mucosal immune responses.
Keywords: colitis, colitis-associated cancer, inflammation, tumour necrosis factor-related apoptosis-inducing ligand receptor
Introduction
Tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is a type-II membrane protein belonging to the TNF family that preferentially induces apoptotic cell death in a variety of tumour cell types, but not in most normal cells, both in vitro and in vivo.1 In humans, five TRAIL receptors (DR4, DR5, DcR1, DcR2 and OPG) have been characterized. As DR4 and DR5 contain an intracellular protein motif known as a death domain, which can transmit an apoptotic signal, these receptors are termed death receptors. In contrast, DcR1 (decoy receptor 1) and DcR2 (decoy receptor 2) lack a functional death domain, and OPG (osteoprotegerin) is a secreted receptor with a lower affinity for TRAIL under physiological conditions.2 DcR1, DcR2 and OPG are termed decoy receptors because they can bind to TRAIL but cannot induce cell death by apoptosis. A large number of in vitro and in vivo studies have shown that TRAIL and agonistic antibodies against DR4 or DR5, which are currently in clinical trials, are promising anti-cancer therapeutic strategies.3–4
In contrast to humans, mice express a single TRAIL-binding pro-apoptotic death receptor (TRAIL-R), which mimics human DR4 and DR5.5 The physiological role of TRAIL/TRAIL-R has been further elucidated with the development of TRAIL−/− and TRAIL-R−/− mice. Recent studies have demonstrated that the TRAIL/TRAIL-R pathway influences the regulation and homeostasis of the immune system. For example, in mice, the genetic ablation of TRAIL increased the severity of remitting and non-remitting disease in experimental autoimmune encephalomyelitis,6 and TRAIL deficiency promoted features of diabetes associated with pancreatic inflammation.7 It has also been reported that the TRAIL/TRAIL-R pathway was involved in the regulation of infection, the development of autoimmune disease and immune surveillance against tumour metastasis.8–9
We previously reported that TRAIL induced CXCL2, CCL4 and CCL20 secretion in a nuclear factor-κB (NF-κB) -dependent manner in both TRAIL-resistant and TRAIL-sensitive tumour cells.10 Furthermore, all of the cytokines induced by TRAIL are inflammatory chemokines, which serve to recruit leucocytes in response to physiological stress. TRAIL was shown to induce chemotactic migration in THP-1 human leukaemia cells, lipopolysaccharide (LPS) -primed primary human monocytes, and LPS-stimulated BALB/c mouse monocytes in vivo.11 A recent study from our group further demonstrated that adeno-associated virus-mediated soluble TRAIL expression in fibroblast-like synoviocytes exhibited an anti-arthritic effect in mice with collagen-induced arthritis.12 These results suggest that TRAIL may be a ligand with multiple functions depending on the cellular context.
Approximately 25% of TRAIL−/− mice develop lymphoid malignancies after 500 days of life, and in the context of the loss of at least one p53 allele, TRAIL was shown to suppress the initiation and development of tumours of both lymphoid and sarcoma origin. However, there was no critical role for TRAIL in Her2/neu oncogene-driven mammary epithelial cancer.13 Furthermore, TRAIL−/− mice were shown to be more sensitive to tumorigenesis induced by the chemical carcinogen methylcholanthrene.14 Interestingly, TRAIL-R−/− mice do not spontaneously develop tumours, and the loss of TRAIL-R was shown to have no effect on tumour generation or growth in p53−/− mice and adenomatous polyposis coli (APC) mutant mice.15 Hence, the observed differences in tumour incidence between TRAIL-deficient and TRAIL-R-deficient mice may a result of the distinct biological causes of the tumours evaluated.
Inflammation is considered a risk factor for many common malignancies, including lung, breast and colon cancers, and the clearest link between inflammation and colon cancer is observed in patients with inflammatory bowel disease (IBD).16 Novel insight into the immunological functions of TRAIL/TRAIL-R may help to determine whether they are also involved in the development of colitis and colitis-associated cancer. In the present study, we report that TRAIL-R−/− mice exhibited a higher incidence of dextran sulphate sodium (DSS) -induced colitis and colitis-associated carcinogenesis, and these results demonstrate a protective role for TRAIL-R in response to chemical-induced injury.
Materials and methods
Mice
TRAIL-R-deficient mice (TRAIL-R−/−) on the C57BL/6 background were kindly provided by Dr Tak Mak (University of Toronto). Heterozygote mice were inter-mated to generate TRAIL-R−/− and wild-type (WT) littermates. Seven- to eight-week-old male mice were used in DSS studies. All animals were housed in specific pathogen-free conditions. Animal experiments were performed in accordance with the institutional guidelines for animal care and were approved by the committee for the use and care of animals of the Chinese Academy of Medical Sciences.
Induction and assessment of colitis and colitis-associated cancer
Experimental colitis was induced by administering 2·5% (weight/volume) DSS (molecular weight 50 000, International Laboratory, South San Francisco, CA) in the drinking water ad libitum for 5 days. Colitis severity scores were recorded based on the daily observation of mouse stool consistency and the presence of blood for up to 6 days. Stool scores were expressed as follows: 0 = normal; 2 = loose stools; 4 = diarrhoea. Bleeding scores were described as follows: 0 = negative; 2 = positive; 4 = gross bleeding. Histological analysis of the severity of inflammation was carried out in the mouse colon after DSS administration. For the animal survival test, mice were continuously fed 3% DSS.
Colitis-associated cancer was established in 7-week-old mice by intraperitoneal injection with azoxymethane (AOM, Sigma Aldrich, St Louis, MO) in PBS at a dose of 12·5 mg/kg body weight on day 0. Then, mice were administered 2% DSS from day 5 to day 10, 1% DSS from day 31 to day 36, and drinking water only from day 0 to day 4, day 11 to day 30, and day 37 to day 60. Mice were killed on day 60. Mouse colon tissues were removed, flushed carefully with PBS, and then cut longitudinally. The number of colon tumour nodes was counted, and tumour sizes were measured with a calliper.
Isolation of colonic epithelial cells
Mouse colons were dissected, washed with PBS, and cut into sections 3 cm in length. Colon segments were incubated in Hanks’ balanced salt solution, supplemented with 5 mm EDTA and 15 mm HEPES (Sigma Aldrich), for 30 min at 37° with gentle shaking followed by vigorous shaking for 10 seconds. The solution was first passed through an 80-mesh stainless steel sieve, followed by a 200-mesh stainless steel sieve to remove the mucosal strips.17 The epithelial crypts remained on the upper side of the 200-mesh sieve, whereas other cells such as leucocytes passed through the sieve. Colonic epithelial cells were immediately washed with PBS and lysed with lysis buffer for use in the TRAIL-R expression assays.
Measurement of mRNA expression
Total RNA was extracted from colonic epithelial cells using the TRIzol reagent (Life Technologies, Grand Island, NY) according to the manufacturer’s instructions. The SYBR Green PCR Master Mix (Toyobo, Osaka, Japan) was used to analyse the expression of mRNA. The following primers were used for the amplification of TRAIL-R: 5′-AAAACGGCTTGGGCATCTTGGC-3′ and 5′-AGACGGTTCCAGGAGTCAAAGG-3′, β-actin: 5′-GACCTGACAGACTACCTC-3′ and 5′-AGACAGCTGTGTTGGC-3′. Quantification of mRNA by quantitative PCR was performed using an ABI7900 system. Reactions were performed in triplicate with β-actin as an internal control. Cycle threshold values were converted to relative gene expression levels with the 2−ΔΔCt method.
Western blotting
Colon epithelial cell or mesenteric lymph node cell lysates were subjected to SDS–PAGE. Proteins in the gel were transferred onto a PVDF membrane. The membrane was then probed with antibodies against DR5 (Abcam, Cambridge, MA), p-Janus kinase (JNK), JNK, p-IκBα (Cell Signaling Technology, Danvers, MA), IκBα (Santa Cruz Biotechnology Inc., Dallas, TX), and β-actin (Proteintech Group, Chicago, IL), followed by incubation with a horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology). Protein expression was visualized with an ECL system (Millipore, Billerica, MA) and was then exposed to X-ray film.
Isolation of lamina propria cells
Colon segments 3 cm in length were gently shaken in Hanks’ balanced salt solution, supplemented with EDTA and HEPES, for 20 min to remove epithelial cells. The colon segments were then washed, cut into small pieces, and suspended in 4% fetal bovine serum RPMI-1640 medium (Life Technologies). Then, the colon fragments were digested with 0·5 mg/ml of collagenase, 0·5 mg/ml of dispase and 40 μg/ml DNase (Roche Applied Science, Mannheim, Germany) for 1 hr at 37°. After filtration through a 200-mesh sieve, isolated lamina propria cells were washed with PBS and subjected to flow cytometry with an Accuri C6 Cytometer (BD, Franklin Lakes, NJ).
Histopathological assay
Intact colons were excised, washed and fixed in 10% neutral buffered formalin for 24 hr. Paraffin-embedded colon tissues were cut into 5-μm sections, which were then stained with haematoxylin & eosin (H&E). Histology scoring was described according to the following characteristics: (i) severity of inflammation (0–3, corresponding to none, mild, moderate or severe); (ii) crypt damage (0–4, corresponding to none, basal one-third damaged, basal two-thirds damaged, only surface epithelium intact, or entire crypt and epithelium lost); and (iii) sub-mucosal oedema [0–3, corresponding to no change, mild oedema (where the sub-mucosa accounts for < 30% of the diameter of the entire intestinal wall), moderate oedema (where the sub-mucosa accounts for 30–60% of the diameter of the entire intestinal wall), and profound oedema (where the sub-mucosa accounts for > 60% of the diameter of the entire intestinal wall)]. Each parameter was further multiplied by a factor reflecting the percentage of tissue involved (× 1, 0–25%; × 2, 26–50%; × 3, 51–75%; × 4, 76–100%). The sum of the scores from these three parameters was defined as the total histological injury score.
Immunohistochemical assay
Paraffin-embedded colon sections were de-paraffinized, rehydrated and pre-treated with 0·3% hydrogen peroxidase in PBS for 10 min. Sections were treated with Tris-EDTA buffer (pH 9) for heat-mediated antigen retrieval of CD3, β-catenin and DR5, and they were then pre-treated with proteinase K for F4/80 and Gr-1 retrieval. After blocking with goat serum, the sections were incubated with antibodies to CD3 (1 : 100, DakoCytomation, Glostrup, Denmark), F4/80 (1 : 50, Abdserotec, Oxford, UK), DR5 (1 : 500, Abcam), β-catenin (1 : 1000, Abcam) and Gr-1 (1 : 500, BD) for 1 hr at room temperature, followed by incubation with an horseradish peroxidase-conjugated secondary antibody for 30 min. Positive signals were visualized using a DAB kit, and the sections were counterstained with haematoxylin.
Determination of cytokine expression
Approximately 200 mg of distant colon tissue was washed in cold PBS, supplemented with penicillin and streptomycin, and cut into small pieces. The tissue was then cultured in 12-well, flat-bottom plates (Corning, Tewksbury, MA) with serum-free RPMI-1640 medium. A high concentration of penicillin (500 U/ml) and streptomycin (500 μg/ml) was supplemented to prevent bacteria growth. After incubation at 37° for 24 hr, the medium was collected and stored at −80° until use. The chemokine levels in whole-colon cultures and the serum from DSS-treated mice were determined using an ELISA kit (R&D Systems, Minnesota, MN) according to the manufacturer’s instructions.
Statistical analysis
All data are presented as the mean ± standard error of the mean. Statistical comparisons were performed using the two-tailed Student’s t-test. P-values < 0·05 were considered statistically significant.
Results
TRAIL-R deficiency increases susceptibility to DSS-induced colitis
To explore the role of TRAIL-R in colitis, TRAIL-R−/− and WT mice were fed DSS to induce colitis. The severity of colitis was determined by characterizing bloody diarrhoea and ulcerations daily. As shown in Fig. 1(a), both the TRAIL-R−/− (n = 10) and WT (n = 9) groups of mice fed DSS for 5 days exhibited acute colitis. However, the disease activity index of TRAIL-R−/− mice, as determined by intestinal bleeding and stool consistency, was significantly higher than that of WT mice (P = 0·032). A high concentration of DSS in the drinking water resulted in the death of TRAIL-R−/− mice as early as day 5, whereas death did not occur in WT mice until day 6 (P < 0·001, Fig. 1b). The median survival times were 6·5 and 11 days for TRAIL-R−/− (n = 8) and WT (n = 10) mice, respectively (Fig. 1c). Body weights were recorded daily throughout the course of the study, and no significant difference in percentage weight loss was observed between TRAIL-R−/− and WT mice. However, because the TRAIL-R−/− mice had a shorter survival time, their body weights on the day they were killed were 78·1% of their initial weight, whereas the WT mice retained 66·4% of their initial weight (P < 0·001, Fig. 1d). H&E staining of colon sections from TRAIL-R−/− and WT mice did not show significant infiltration of inflammatory cells or notable tissue damage (Fig. 1e, d0 section), which is in agreement with previous reports showing that TRAIL-R−/− mice show no developmental abnormalities or histological changes.18 However, after feeding with DSS in the water for 5 days, increased damage to the epithelial layer was observed in TRAIL-R−/− mice compared with WT mice (Fig. 1e, d5 section). TRAIL-R−/− mice displayed severe colon inflammation, with near complete loss of colonic crypts and epithelium, obvious sub-mucosal oedema, and prominent inflammatory cell infiltration. In contrast, WT animals displayed partially preserved colonic crypt structure and only slight sub-mucosal oedema. Histological scoring revealed that TRAIL-R−/− mice fed DSS had significantly increased inflammation, crypt damage and sub-mucosal oedema compared with WT mice (Fig. 1f). Taken together, these data clearly demonstrate that TRAIL-R deficiency promoted the susceptibility of mice to DSS-induced colitis.
Figure 1.
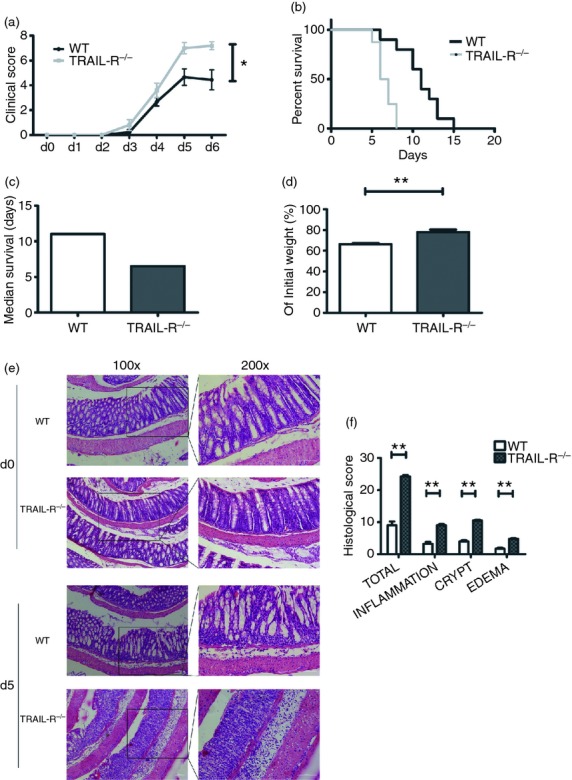
Tumour necrosis factor-related apoptosis-inducing ligand receptor deficiency (TRAIL-R−/−) enhances susceptibility to dextran sulphate sodium (DSS) -induced colitis. (a) Clinical scoring of wild-type (WT) (n = 9) and TRAIL-R−/− (n = 10) mice administered 2·5% DSS for 5 days. The clinical score is addition of stool and bleeding score. The maximum clinical score was 8 (P = 0·032). WT (n = 8) and TRAIL-R−/− (n = 10) mice were continuously administered 3% DSS in (b), (c) and (d). (b) Kaplan–Meier survival curve. The P-value was calculated according to the log-rank test. P < 0·001. (c) Median survival time. (d) Body weights on the day they were killed, shown as a percentage of the initial body weight. P < 0·001. (e) Representative haematoxylin & eosin staining of WT and TRAIL-R−/− mouse colons, from animals fed with or without 2·5% DSS for 5 days (left panels, magnification 100 ×; right panels, magnification 200 ×). Bar, 100 μm. (f) Histological injury score of DSS-induced colitis in WT (n = 4) and TRAIL-R−/− mice (n = 4). All mice were fed 2·5% DSS in water for 5 days. Histological analysis included the severity of inflammation, crypt damage and sub-mucosal oedema. The sum of the scores from these three parameters was defined as the total histological injury score. P-values of total histological score, inflammation, and crypt damage were < 0·001; oedema, P = 0·001.
TRAIL-R knockout facilitates colitis-associated tumorigenesis
Chronic colitis strongly contributes to colorectal tumorigenesis.16–19 Here, we investigated whether TRAIL-R deficiency has an impact on colitis-associated tumorigenesis. TRAIL-R−/− and WT mice (n = 10 for both groups) were injected with a single intraperitoneal injection of the carcinogen, AOM, on day 0, followed by feeding with 2% DSS from day 5 to day 10, water only from day 11 to day 30, 1% DSS from day 31 to day 36, and finally water only from day 37 to day 60 (Fig. 2a). Mice were killed on day 60, and their colons were dissected to examine tumour development. As shown in Fig. 2(a), 80% of TRAIL-R−/− mice and 100% of WT mice were alive on day 60. All of the TRAIL-R−/− mice developed colon tumours, whereas only half of the WT mice developed tumours (Fig. 2b). In both groups of animals, the colon tumours were exclusively located in the distal colon (Fig. 2c). The average tumour number per mouse was 9·5 ± 2·1 for TRAIL-R−/− mice, which was three times higher than that observed for WT mice (2·6 ± 0·6) (Fig. 2d). In both groups, tumour size ranged from < 2 mm to > 5 mm; however, tumours with a diameter > 5 mm were found only in TRAIL-R−/− mice (Fig. 2e). Furthermore, histological analysis by H&E staining demonstrated that these tumours were colonic adenomas with a high grade of dysplasia (Fig. 2f), and only a small proportion of nuclear expression of β-catenin was observed in the tumour tissue of TRAIL-R−/− mice (Fig. 2f), indicating that the Wnt pathway might not be involved in the colitis-associated tumorigenesis in TRAIL-R−/− mice. These results indicated that TRAIL-R deficiency facilitated the incidence and growth of colitis-associated colonic adenomas.
Figure 2.

Tumour necrosis factor-related apoptosis-inducing ligand receptor (TRAIL-R) deficiency promotes colitis-associated cancer (CAC) development. (a) Kaplan–Meier survival curve of azoxymethane (AOM)/dextran sulphate sodium (DSS) -induced colitis-associated cancer model of wild-type (WT) (n = 10) and TRAIL-R−/− mice (n = 10). (b) Tumour incidence. (c) Representative macroscopic tumorigenesis. Colon tissues were cut longitudinally, and the arrows indicate colonic tumours. Bar, 1 cm. (d) Total colonic polyps formed in the colons of WT and TRAIL-R−/− mice. P = 0·014 (e) Size distribution of colonic tumours formed in WT and TRAIL-R−/− mice (P = 0·008 for the < 2 mm group; P = 0·128 for the 2–5 mm group). (f) Representative haematoxylin & eosin staining of WT and TRAIL-R−/− mice; magnification, 100 ×. Bar, 50 μm. Left image is immunohistochemical staining of TRAIL-R−/− tumour tissue, where brown colour represents β-catenin-positive cells. Arrows indicate the positive nuclear staining; magnification, 400 ×.
DSS administration suppresses TRAIL-R expression in mouse epithelial cells
To investigate whether TRAIL-R expression is altered in the colon following DSS administration, mice were administered 2·5% DSS in their drinking water for 5 days to elicit colitis. Immunohistochemical analysis of mouse colonic tissue sections revealed that TRAIL-R was expressed at a high level in untreated WT mice. Both images of longitudinal and transverse sections showed that TRAIL-R was exclusively expressed on epithelial cells (Fig. 3a). However, TRAIL-R expression was significantly suppressed in DSS-fed mice (Fig. 3a). Real-time PCR analysis showed that TRAIL-R expression was down-regulated in the colon epithelial cells of DSS-administered mice on days 0 and 5, suggesting that TRAIL-R expression was regulated at the mRNA level (Fig. 3b). Western blot analysis of colon epithelial cell lysates from DSS-administered mice on days 0, 2 and 5 confirmed that TRAIL-R expression began to decrease at day 2 and continued to decline through day 5 (Fig. 3c). These data suggest that DSS administration suppresses TRAIL-R expression in mouse epithelial cells, which is consistent with the previous report by Brost et al. that significantly lower TRAIL, TRAIL-R1, and TRAIL-R2 expression in the epithelium of patients with IBD.20 Taken together, TRAIL-R may play an important role in colonic colitis formation and the epithelial barrier immune response.
Figure 3.
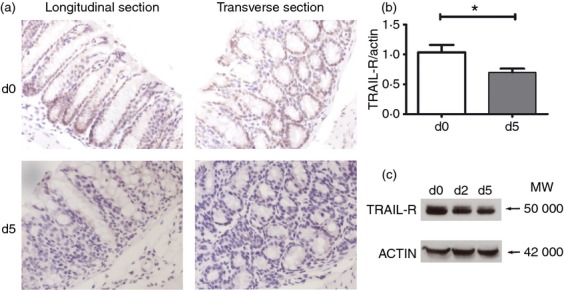
Tumour necrosis factor-related apoptosis-inducing ligand receptor (TRAIL-R) expression is reduced in wild-type (WT) mice fed dextran sulphate sodium (DSS). Eight-week-old WT mice were administered 2·5% DSS or vehicle in their drinking water for 5 days. (a) Immunohistochemical staining of colon tissues (brown, TRAIL-R; blue, haematoxylin stain). (b) Real-time PCR analysis for the expression of TRAIL-R in colon epithelial cells (CEC) of the mice fed with DSS for 0 and 5 days. (c) Western blot analysis of the expression of TRAIL-R in CEC. Cell lysates were pooled from two mice that were fed with DSS for 0 days, 2 days and 5 days.
TRAIL-R deficiency increases the expression of CCL2 and CXCL1
The cytokine network plays a key role in the initiation and progression of IBD.21 To determine the effect of TRAIL-R deficiency on the expression of pro-inflammatory cytokines and chemokines during DSS-triggered colitis, we analysed the expression of TNF-α, interferon-γ (IFN-γ), interleukin-6 (IL-6), IL-10, CCL2/MCP-1 and CXCL1/KC in the serum and colonic tissues of DSS-fed mice. Interestingly, no significant differences in the expression of TNF-α, IFN-γ, IL-6 and IL-10 were observed between TRAIL-R−/− and WT mice (n = 3), according to the results of the Cytometric Bead Array assay (data not shown). However, the expression of CCL2 (P = 0·034, Fig. 4a) and CXCL1 (P = 0·003, Fig. 4b) in the serum of TRAIL-R−/− mice was markedly higher than that observed in WT mice (n = 6). Furthermore, CCL2 expression was significantly higher in DSS-fed TRAIL-R−/− mouse colonic tissue compared with WT colonic tissue (P = 0·015, Fig. 4c). However, CXCL1 expression was almost identical between the two groups of colon tissues (P = 0·871, Fig. 4d). These data indicate that TRAIL-R deficiency increased the expression of CCL2 and CXCL1 in the serum of TRAIL-R−/− mice, which may have initiated the infiltration of leucocytes, macrophages and neutrophils into the inflammatory colon tissue.
Figure 4.
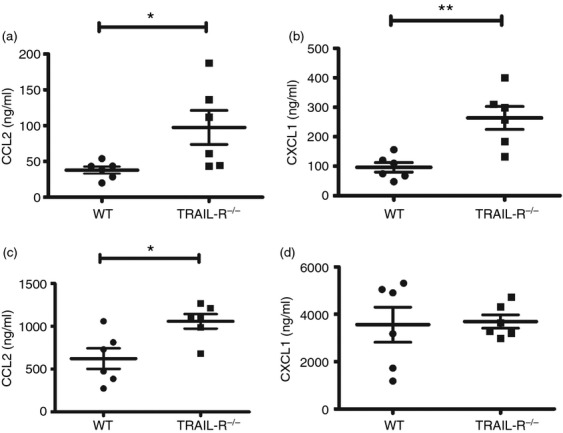
Chemokine expression is up-regulated in the serum and colon tissue of tumour necrosis factor-related apoptosis-inducing ligand receptor deficient (TRAIL-R−/−) mice. Wild-type (WT) (n = 6) and TRAIL-R−/− (n = 6) mice were fed 2·5% dextran sulphate sodium (DSS) for 5 days. Secreted chemokines in the serum were measured by ELISA. (a) CCL2/MCP-1 production in the serum; P = 0·034. (b) CXCL1/KC production in the serum; P = 0·003. (c) and (d) Colon tissues (200 ± 5 mg) from WT and TRAIL-R−/− mice were cut into small pieces and cultivated in serum-free RPMI-1640 medium for 24 hr. CCL2/MCP-1 (c, P = 0·015) and CXCL1/KC (d, P = 0·871) production in the colon tissue cultures was detected by ELISA.
TRAIL-R deficiency facilitates the infiltration of macrophage and T lymphocytes into colon tissue
To confirm whether TRAIL-R deficiency enhances the infiltration of inflammatory cells into colon tissue, lamina propria cells were isolated from mouse colon tissue and analysed by flow cytometry. As shown in Fig. 5(a), two populations, consisting of lymphocytes (Gate P1) and mononuclear cells (Gate P2), were observed in the isolated lamina propria cells from WT and TRAIL-R knockout mice. Cell counting revealed that there were 10-fold more lymphocytes and mononuclear cells in the colonic lamina propria of TRAIL-R knockout mice than that of WT mice (Fig. 5b). Histochemical analysis showed that there were more infiltrated inflammatory cells, including F4/80+ macrophages and CD3+ T lymphocytes, in the mucosa and sub-mucosal layer of the colon tissue from TRAIL-R−/− mice administered DSS for 5 days (Fig. 5c and 5d). Gr-1+ neutrophils were not detectable in the colon sections from TRAIL-R−/− and WT animals, compared with positive control staining from inflamed lung tissue (Fig. 5c). Furthermore, epithelial cell apoptosis, as evaluated by TUNEL staining, was equivalent in both groups of mice fed DSS for 5 days (data not shown).
Figure 5.
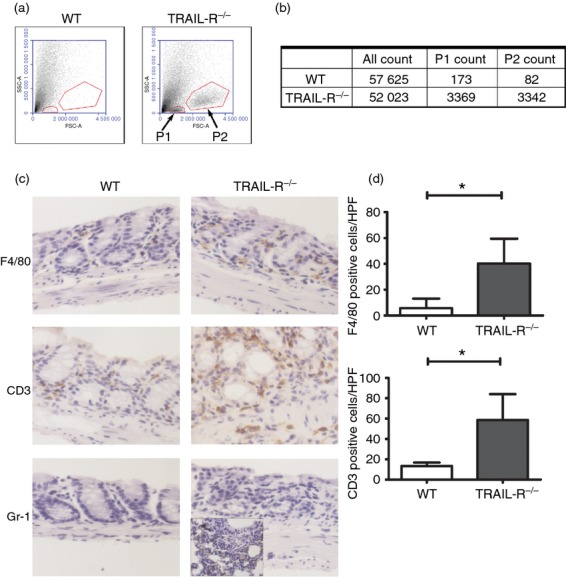
More extensive inflammatory cell infiltration in the colon tissues of tumour necrosis factor-related apoptosis-inducing ligand receptor deficient (TRAIL-R−/−) mice compared with wild-type (WT) mice after dextran sulphate sodium (DSS) administration. (a) Isolated colon lamina propria cells were analysed by flow cytometry. The cells from each group of mice were pooled from four individual mice. Target cells were gated based on forward light scatter (FSC) and side light scatter (SSC). Gate P1 represents lymphocytes, and Gate P2 represents mononuclear cells. (b) Counts of gated cells. (c) Immunohistochemical evaluation of colon tissue, where the brown colour represents F4/80-, CD3- and Gr-1-positive cells in WT and TRAIL-R−/− mice fed with 2·5% DSS for 5 days. For Gr-1 staining, inflamed lung tissue was used as a positive control. (d) Average numbers of F4/80+ cells (P = 0·033) and CD3+ cells (P = 0·030) in colonic tissue sections. Data represent the mean ± SEM of four mice per group, and three high-power fields (HPF) were counted per mouse.
Ikeda et al. reported that TRAIL might promote the increase of regulatory T cells and then suppress autoimmunity.22 To find out whether regulatory T cells were involved in our model, the number of regulatory T cells in the blood after DSS-treatment was analysed by flow cytometry. However, there was no difference between WT and TRAIL-R KO mice (data not shown).
These data indicate that TRAIL-R deficiency facilitated the infiltration of macrophages and T lymphocytes into mouse colonic tissue, which further demonstrates that TRAIL-R plays a critical role in DSS-induced colitis and colitis-associated carcinogenesis, as well as colonic epithelial homeostasis.
Hyperactivation of JNK and NF-κB in colon epithelial cells contributes to the increased severity of colitis in TRAIL-R−/− mice
To further examine the molecular mechanisms underlying the altered homeostasis associated with DSS-induced colitis, colon epithelial cells (CEC) and mesenteric lymph node cells (MLN) from WT and TRAIL-R−/− mice were isolated, and the activation status of JNK and NF-κB, known key regulators of inflammatory responses, was evaluated. As shown in Fig. 6(a), after DSS administration for 5 days, the phosphorylation of JNK and IκB was increased in CEC and MLN cells; however, elevated levels of phosphorylated JNK and IκB were observed in CEC, but not in MLN cells, from TRAIL-R−/− mice compared with their WT counterparts.
Figure 6.
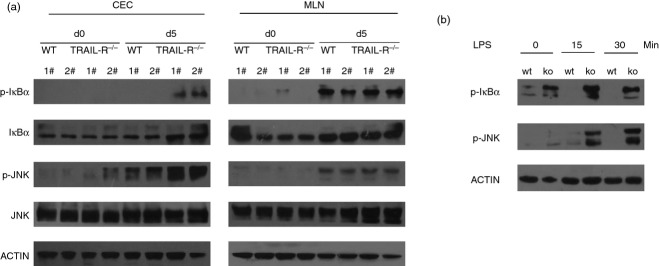
(a) The nuclear factor-κB (NF-κB) and Janus kinase (JNK) pathways are highly activated in tumour necrosis factor-related apoptosis-inducing ligand receptor deficient (TRAIL-R−/−) colon epithelial cells (CEC) but not in mesenteric lymph node (MLN) cells. Protein lysates were prepared from CEC or MLN cells from WT or TRAIL-R−/− mice on day 0 or day 5 after dextran sulphate sodium (DSS) administration. Western blotting was performed with antibodies against IκBα, phospho-IκBα, JNK, phospho-JNK and actin. Each bar represents cell lysates from two individual mice. (b) CEC from TRAIL-R−/− mice were hyper-responsive to lipopolysaccharide (LPS) stimulation. CEC from WT or TRAIL-R−/− mice were untreated or stimulated with LPS (20 μg/ml) for 15 or 30 min. Then, the cells were lysed, and the cell lysates were subjected to SDS–PAGE. Immunoblotting was performed to detect Toll-like receptor signalling components using anti-phospho-IκBα and anti-phospho-JNK antibodies.
To further investigate the TRAIL-R-mediated downstream pathway leading to sensitivity to DSS-induced colitis, CEC were treated with LPS for various times, and the activation of JNK and IκB was analysed by Western blotting. Upon LPS stimulation, TRAIL-R−/− CEC exhibited increased activation of JNK and IκB compared with WT cells (Fig. 6b), indicating that loss of TRAIL-R in epithelial cells, but not in leucocytes, sensitizes mice to DSS-induced colitis.
Discussion
In the present study, we found that TRAIL-R−/− mice were more susceptible to DSS-induced colitis and colitis-associated carcinogenesis. Furthermore, TRAIL-R expression in the epithelial barrier was reduced following DSS administration, and the expression of chemokines, including CCL2 and CXCL1, was largely increased in the serum and inflammatory colon tissues of TRAIL-R−/− mice than that of WT mice. The inflamed colons of TRAIL-R−/− mice enhanced the infiltration of inflammatory cells, including CD3+ T lymphocytes and F4/80+ macrophages, and we found that JNK and IκB were highly activated in TRAIL-R−/− colonic epithelial cells. Hence, we propose that TRAIL-R plays a critical role in DSS-induced colitis and colitis-associated carcinogenesis, as well as in the regulation of mucosal immune responses.
Upon stimulation, TRAIL is expressed in a variety of cells of the innate and adaptive immune systems. Our data indicated that TRAIL-R expression in epithelial cells was reduced during DSS-induced colitis. Previously, in human intestinal epithelial (Caco-2) cells, we observed that TRAIL-R2 (DR5) could be internalized upon TRAIL stimulation (data not shown), suggesting that TRAIL-R internalization may explain its reduced expression. Several studies have also proposed that TRAIL/TRAIL-R is involved in fighting infection and modulating the immune response. Zheng et al. reported that Listeria infection in the livers and spleens of normal mice was much more severe than that observed in TRAIL−/− mice, and reduced cell death was observed in lymphoid and myeloid cells.23 Furthermore, neutrophil-derived TRAIL was shown to induce apoptosis of DR5-expressing macrophages, thereby promoting early bacterial killing in pneumococcal pneumonia.24 TRAIL-R−/− mice have also been shown to exhibit increased clearance of murine cytomegalovirus and increased expression levels of IL-2, IFN-α and IFN-γ.25 These results suggest that TRAIL-R−/− mice possess an enhanced immune response, which may protect against infection or lead to uncontrolled inflammation and immunopathology, as observed in DSS-induced colitis.
Many colon cancer cell lines, such as Colo205, HCT15, HCT116, HCC-2998, SW620 and HT29, are sensitive to TRAIL cytotoxicity.26 Normally, the colonic crypt epithelium is completely resistant to TRAIL-induced apoptosis in vitro.27 However, cytomegalovirus infection was shown to significantly enhance TRAIL-induced apoptosis in human intestinal epithelial (Caco-2) cells.27 TRAIL, TRAIL-R1 (DR4) and TRAIL-R2 (DR5) are co-expressed in the human luminal surface epithelium, and Begue et al. reported that TRAIL was markedly up-regulated during intestinal inflammation. Hence, increased TRAIL expression in the intestine suggests that TRAIL may serve as a pro-inflammatory mediator.28 Brost et al. reported significantly lower TRAIL, TRAIL-R1 and TRAIL-R2 expression in the epithelium of IBD patients,20 and TRAIL-R1 and TRAIL-R2 staining was shown to be stronger in neoplastic cells from colorectal adenomas and carcinomas compared with normal mucosal cells.29 Furthermore, alterations in the TRAIL/TRAIL-R expression pattern in IBD may depend on the stage of the disease. Taken together, these reports indicate an important role for TRAIL/TRAIL-R in intestinal epithelial cell homeostasis.
DSS-induced colitis is characterized by body weight loss, bloody diarrhoea, intestinal ulceration and granulocyte infiltration.30–31 DSS is thought to be directly toxic to gut epithelial cells of basal crypts, thereby compromising intestinal epithelial integrity. T and B lymphocytes are generally not required for the development of DSS-induced colonic inflammation, this murine model is therefore particularly useful for investigating the contribution of innate immunity to colitis.32 In the present study, the median survival time of TRAIL-R−/− mice continually administered DSS was only 6·5 days, and the rate of body weight loss was not significantly different from that of WT mice. However, TRAIL-R−/− mice began to die as early as day 5 of DSS administration, which may have been a result of uncontrolled release of cytokines or chemokines, as we observed high expression levels of CXCL1 and CCL2 in the serum and of CCL2 in the inflammatory colon tissue of TRAIL-R−/− mice. However, CXCL1 expression in the inflammatory colon tissue was virtually identical between TRAIL-R−/− and WT mice, possibly because CXCL1 expression reached saturation in colon tissues. There was no difference between CCL2 and CXCL1 expression between the untreated WT and TRAIL-R−/− mice (data not shown). The results suggest that the higher level of CCL2 and CXCL1 expression was due to the DSS treatment. The body weights of TRAIL-R−/− mice administered 1% DSS for 7 days, followed by water only, began to increase at day 11, suggesting that DSS did not permanently damage the colon epithelial barrier and that TRAIL-R deficiency had no effect on the efficiency of tissue repair.
The severity of inflammation is known to correlate with the incidence of colorectal cancer,16–19 and several pieces of evidence have linked inflammation with tumorigenesis. First, inflammatory cells and cytokines are present in the microenvironment of the epithelial barrier and inflammatory colon tissue. Second, key pro-inflammatory signalling pathways (such as JAK/signal transducer and activator of transcription and NF-κB) may be involved in the incidence of cancer.33 Third, non-steroidal anti-inflammatory drugs reduce the development of colon cancer.34 Moreover, the AOM/DSS animal model has been used in the literature to demonstrate the importance of colitis in the development of colon cancer, although a single injection of AOM does not lead to carcinogenesis.35 We found that both tumour incidence and tumour node number in TRAIL-R−/− mice were higher than in WT mice. In another animal model of gastrointestinal tumours, i.e. APCmin/+ mice, no difference in the number of intestinal polyps was found between TRAIL-R−/− and WT mice.15 APCmin/+ mice are heterozygous for a nonsense mutation at codon 850 of APC, and these heterozygous animals develop normally but are prone to the development of intestinal polyps, which is accelerated by a high-fat diet. However, spontaneously developing tumours may not typically induce a vigorous immune response. AOM-induced tumorigenesis can be significantly enhanced by chronic colitis, as demonstrated in the present study, which found that TRAIL-R-deficient mice exhibited a higher incidence of colitis-associated tumours and enhanced tumour growth.
Since the numbers of apoptotic epithelial cells were similar in DSS-treated TRAIL-R−/− mice and their WT counterparts (data not shown),the higher incidence of tumour in TRAIL-R-deficient mice may not be related to the role of TRAIL in apoptosis. However, due to the loss of TRAIL-R, tumour cells might have been less likely to die and to have grown faster. More dedicated analysis is required to reveal the role of TRAIL-R in tumour growth. Our finding suggests that the excessive immune response in TRAIL-R−/− mice may be the main cause of high colitis-associated cancer incidence.
In the face of constant immunological stimulation, there is a requirement for a homeostatic balance between tolerance and immunity. The tolerance of the intestinal epithelial layer to microbes is not only important for controlling inflammation but also for preventing tumorigenesis in the colon. Hence, deregulation of the balance between tolerance and immunity may contribute to IBD. Toll-like receptors (TLRs) and NOD-like receptors (NLRs) serve to alert the host immune system to the presence of bacteria in extracellular and intracellular spaces.36 In the gut, multiple tolerance mechanisms ensure reduced TLR activation by commensal microorganisms; for example, inhibitory molecules of TLRs, such as Toll-interacting protein, single immunoglobulin IL-1-related receptor, and peroxisome proliferator-activated receptor γ,37–38 may inhibit strong immune responses to microbes. Xiao et al. demonstrated that epithelium-derived single immunoglobulin IL-1-related receptor was critical for controlling homeostasis and the innate immune response of the colon to enteric microflora.39 TRAIL-R primarily affects late signalling events downstream of the TLRs, although TRAIL-R deficiency does not affect the initial activation of NF-κB. At 4 and 8 hr post-stimulation, IκB-α protein was shown to be re-expressed in WT cells but could not be detected in TRAIL-R−/− cells.25 We found that the JNK and NF-κB pathways were highly activated in CEC during the course of colitis. These data suggest that lack of IκB-α feedback inhibition and subsequent NF-κB activity may explain the increased cytokine production in TRAIL-R−/− cells.
In summary, these results reveal a novel pathophysiological role for TRAIL-R in IBD and colorectal cancer, whereby TRAIL-R may negatively regulate the mucosal immune response and tissue homeostasis in response to external stress or injury.
Acknowledgments
JZ and DZ designed the study and wrote the paper, JZ and LC performed the experiments, JS gave technical support, SL and YL gave conceptual advice. This work was partially supported by the State Key Basic Research Programme of China (Grant No. 2013CB530805) and the Natural Science Foundation of China (Grant No. 91029735 and 81372200).
Glossary
- APC
adenomatous polyposis coli
- AOM
azoxymethane
- CEC
colon epithelial cells
- DSS
dextran sulphate sodium
- H&E
haematoxylin & eosin
- IBD
inflammatory bowel disease
- IFN
interferon
- IL
interleukin
- JNK
Janus kinase
- LPS
lipopolysaccharide
- MLN
mesenteric lymph node
- NF-κB
nuclear factor-κB
- TLR
toll-like receptor
- TRAIL
tumour necrosis factor (TNF)-related apoptosis-inducing ligand
- WT
wild-type
Disclosures
The authors declare having no competing interest.
References
- Wiley SR, Schooley K, Smolak PJ, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–82. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- Emery JG, McDonnell P, Burke MB, et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem. 1998;273:14363–7. doi: 10.1074/jbc.273.23.14363. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Herbst RS. To kill a tumor cell: the potential of proapoptotic receptor agonists. J Clin Invest. 2008;118:1979–90. doi: 10.1172/JCI34359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox NL, Humphreys R, Luster TA, Klein J, Gallant G. Tumor Necrosis Factor-related apoptosis-inducing ligand (TRAIL) Receptor-1 and Receptor-2 agonists for cancer therapy. Expert Opin Biol Ther. 2010;10:1–18. doi: 10.1517/14712590903319656. [DOI] [PubMed] [Google Scholar]
- Wu GS, Burns TF, Zhan Y, Alnemri ES, El-Deiry WS. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res. 1999;59:2770–5. [PubMed] [Google Scholar]
- Cretney E, McQualter JL, Kayagaki N, Yagita H, Bernard CC, Grewal IS, Ashkenazi A, Smyth MJ. TNF-related apoptosis-inducing ligand (TRAIL)/Apo2L suppresses experimental autoimmune encephalomyelitis in mice. Immunol Cell Biol. 2005;83:511–19. doi: 10.1111/j.1440-1711.2005.01358.x. [DOI] [PubMed] [Google Scholar]
- Di Bartolo BA, Chan J, Bennett MR, Cartland S, Bao S, Tuch BE, Kavurma MM. TNF-related apoptosis-inducing ligand (TRAIL) protects against diabetes and atherosclerosis in Apoe/mice. Diabetologia. 2011;54:3157–67. doi: 10.1007/s00125-011-2308-0. [DOI] [PubMed] [Google Scholar]
- Schaefer U, Voloshanenko O, Willen D, Walczak H. TRAIL: a multifunctional cytokine. Front Biosci. 2007;12:3813–24. doi: 10.2741/2354. [DOI] [PubMed] [Google Scholar]
- Falschlehner C, Schaefer U, Walczak H. Following TRAIL’s path in the immune system. Immunology. 2009;127:145–54. doi: 10.1111/j.1365-2567.2009.03058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Wang W, Zhang Y, Liu S, Liu Y, Zheng D. Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced chemokine release in both TRAIL-resistant and TRAIL-sensitive cells via nuclear factor κB. FEBS J. 2009;276:581–93. doi: 10.1111/j.1742-4658.2008.06809.x. [DOI] [PubMed] [Google Scholar]
- Wei W, Wang D, Shi J, Xiang Y, Zhang Y, Liu S, Liu Y, Zheng D. Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) induces chemotactic migration of monocytes via a death receptor 4-mediated RhoGTPase pathway. Mol Immunol. 2010;47:2475–84. doi: 10.1016/j.molimm.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Shi J, Diao Z, Zhou J, Zhu J, Yuan H, You X, Liu Y, Zheng D. Epirubicin potentiates recombinant adeno-associated virus type 2/5-mediated TRAIL expression in fibroblast-like synoviocytes and augments the antiarthritic effects of rAAV2/5-TRAIL. Arthritis Rheum. 2012;64:1345–54. doi: 10.1002/art.33492. [DOI] [PubMed] [Google Scholar]
- Zerafa N, Westwood JA, Cretney E, Mitchell S, Waring P, Iezzi M, Smyth MJ. Cutting edge: TRAIL deficiency accelerates hematological malignancies. J Immunol. 2005;175:5586–90. doi: 10.4049/jimmunol.175.9.5586. [DOI] [PubMed] [Google Scholar]
- Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol. 2002;168:1356–61. doi: 10.4049/jimmunol.168.3.1356. [DOI] [PubMed] [Google Scholar]
- Yue HH, Diehl GE, Winoto A. Loss of TRAIL-R does not affect thymic or intestinal tumor development in p53 and adenomatous polyposis coli mutant mice. Cell Death Differ. 2005;12:94–7. doi: 10.1038/sj.cdd.4401523. [DOI] [PubMed] [Google Scholar]
- Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol. 2004;287:G7–17. doi: 10.1152/ajpgi.00079.2004. [DOI] [PubMed] [Google Scholar]
- Grossmann J, Walther K, Artinger M, et al. Progress on isolation and short-term ex-vivo culture of highly purified non-apoptotic human intestinal epithelial cells (IEC) Eur J Cell Biol. 2003;82:262–70. doi: 10.1078/0171-9335-00312. [DOI] [PubMed] [Google Scholar]
- Finnberg N, Gruber JJ, Fei P, et al. DR5 knockout mice are compromised in radiation-induced apoptosis. Mol Cell Biol. 2005;25:2000–13. doi: 10.1128/MCB.25.5.2000-2013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter M, Saunders B, Wilkinson K, et al. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology. 2004;126:451–9. doi: 10.1053/j.gastro.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Brost S, Koschny R, Sykora J, Stremmel W, Lasitschka F, Walczak H, Ganten TM. Differential expression of the TRAIL/TRAIL-receptor system in patients with inflammatory bowel disease. Pathol Res Pract. 2010;206:43–50. doi: 10.1016/j.prp.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Alex P, Zachos NC, Nguyen T, Gonzales L, Chen T-E, Conklin LS, Centola M, Li X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–52. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T, Hirata S, Fukushima S, Matsunaga Y, Ito T, Uchino M, Nishimura Y, Senju S. Dual effects of TRAIL in suppression of autoimmunity: the inhibition of Th1 cells and the promotion of regulatory T cells. J Immunol. 2010;185:5259–67. doi: 10.4049/jimmunol.0902797. [DOI] [PubMed] [Google Scholar]
- Zheng SJ, Jiang J, Shen H, Chen YH. Reduced apoptosis and ameliorated listeriosis in TRAIL-null mice. J Immunol. 2004;173:5652–8. doi: 10.4049/jimmunol.173.9.5652. [DOI] [PubMed] [Google Scholar]
- Steinwede K, Henken S, Bohling J, et al. TNF-related apoptosis-inducing ligand (TRAIL) exerts therapeutic efficacy for the treatment of pneumococcal pneumonia in mice. J Exp Med. 2012;209:1937–52. doi: 10.1084/jem.20120983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl GE, Yue HH, Hsieh K, et al. TRAIL-R as a negative regulator of innate immune cell responses. Immunity. 2004;21:877–89. doi: 10.1016/j.immuni.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Pai RC, Fong S, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–62. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strater J, Walczak H, Pukrop T, Von Muller L, Hasel C, Kornmann M, Mertens T, Moller P. TRAIL and its receptors in the colonic epithelium: a putative role in the defense of viral infections. Gastroenterology. 2002;122:659–66. doi: 10.1053/gast.2002.31889. [DOI] [PubMed] [Google Scholar]
- Begue B, Wajant H, Bambou JC, et al. Implication of TNF-related apoptosis-inducing ligand in inflammatory intestinal epithelial lesions. Gastroenterology. 2006;130:1962–74. doi: 10.1053/j.gastro.2006.03.022. [DOI] [PubMed] [Google Scholar]
- Koornstra JJ, Kleibeuker JH, van Geelen CM, Rijcken FE, Hollema H, de Vries EG, de Jong S. Expression of TRAIL (TNF-related apoptosis-inducing ligand) and its receptors in normal colonic mucosa, adenomas, and carcinomas. J Pathol. 2003;200:327–35. doi: 10.1002/path.1364. [DOI] [PubMed] [Google Scholar]
- Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–6. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107:1643–52. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- Eckmann L, Jung HC, Schurer-Maly C, Panja A, Morzycka-Wroblewska E, Kagnoff MF. Differential cytokine expression by human intestinal epithelial cell lines: regulated expression of interleukin 8. Gastroenterology. 1993;105:1689–97. doi: 10.1016/0016-5085(93)91064-o. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003;94:965–73. doi: 10.1111/j.1349-7006.2003.tb01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Tschopp J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol. 2005;26:447–54. doi: 10.1016/j.it.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Steenholdt C, Andresen L, Pedersen G, Hansen A, Brynskov J. Expression and function of toll-like receptor 8 and Tollip in colonic epithelial cells from patients with inflammatory bowel disease. Scand J Gastroenterol. 2009;44:195–204. doi: 10.1080/00365520802495529. [DOI] [PubMed] [Google Scholar]
- Dubuquoy L, Jansson EA, Deeb S, et al. Impaired expression of peroxisome proliferator-activated receptor γ in ulcerative colitis. Gastroenterology. 2003;124:1265–76. doi: 10.1016/s0016-5085(03)00271-3. [DOI] [PubMed] [Google Scholar]
- Xiao H, Gulen MF, Qin J, et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity. 2007;26:461–75. doi: 10.1016/j.immuni.2007.02.012. [DOI] [PubMed] [Google Scholar]