

Abstract
Systemic lupus erythematosus (SLE) is an autoimmune disease that can attack many different body organs; the triggering event is unknown. SLE has been associated with more than 100 different autoantibody reactivities – anti-dsDNA is prominent. Nevertheless, autoantibodies to dsDNA occur in only two-thirds of SLE patients. We previously reported the use of an antigen microarray to characterize SLE serology. We now report the results of an expanded study of serology in SLE patients and scleroderma (SSc) patients compared with healthy controls. The analysis validated and extended previous findings: two-thirds of SLE patients reacted to a large spectrum of self-molecules that overlapped with their reactivity to dsDNA; moreover, some SLE patients manifested a deficiency of natural IgM autoantibodies. Most significant was the finding that many SLE patients who were negative for autoantibodies to dsDNA manifested abnormal antibody responses to Epstein–Barr virus (EBV): these subjects made IgG antibodies to EBV antigens to which healthy subjects did not respond or they failed to make antibodies to EBV antigens to which healthy subjects did respond. This observation suggests that SLE may be associated with a defective immune response to EBV. The SSc patients shared many of these serological abnormalities with SLE patients, but differed from them in increased IgG autoantibodies to topoisomerase and centromere B; 84% of SLE patients and 58% of SSc patients could be detected by their abnormal antibodies to EBV. Hence an aberrant immune response to a ubiquitous viral infection such as EBV might set the stage for an autoimmune disease.
Keywords: anti-dsDNA, autoantibodies, Epstein–Barr virus, scleroderma, systemic lupus erythematosus
Introduction
Systemic lupus erythematosus (SLE), a prototypic autoimmune disease, is associated with a large spectrum of autoantibodies. IgG antibodies to more than 100 different antigens including DNA, nucleosomes, histones, viral antigens and transcription factors have been reported in different SLE patients.1 Surprisingly, there is no serological diagnosis of SLE; the diagnosis is made when at least four of 11 criteria are fulfilled, and antibody abnormalities are only two of the criteria (anti-nuclear antibodies and anti-DNA and some others); hence a diagnosis of SLE cannot be made by serology alone, using current assays.2
Viral and bacterial infections are known to be associated with autoimmune diseases. SLE patients manifest high titres of antibodies to Epstein–Barr virus (EBV) and increased circulating EBV viral loads.3–4 Group A streptococcus infection causes the autoimmune diseases acute rheumatic fever and post-streptococcal glomerulonephritis.5 In a previous study,6 we found that SLE patients manifested increased IgG reactivity to hyaluronic acid (HA), a molecule that forms the capsule of the group A streptococcus.7
In the present study, we used antigen microarray technology and informatic analysis8 to study antibody repertoires of SLE patients compared with healthy controls, scleroderma (SSc) patients, and some pemphigus patients. Guided by the results from our previous antigen microarray study,6 we limited the chip array to the significant SLE antigens and added EBV antigens (EBVEA, EBV early antigen; EBNA1, EBV nuclear antigen 1; EBVp23 and EBVp18), along with several additional antigens associated with autoimmunity. Overall the chip contained 64 different antigens.
The aims of the study were: (i) to characterize serologically SLE patients who are negative as well as positive for anti-dsDNA; (ii) to examine antibody reactivities to defined antigen preparations of EBV; (iii) to determine whether the serological reactions of SLE patients differ from those of SSc patients, pemphigus patients and healthy controls; and (iv) to validate our previous findings on an expanded group of subjects.
Materials and methods
Human subjects
The study was approved by the Institutional Review Boards of each participating clinical unit; informed consent was obtained from all participants. Sera from 49 SLE patients, 24 SSc patients and 23 healthy controls were studied. An additional test set of 16 SLE patients, eight pemphigus patients and six healthy controls was tested separately for validation. The SLE patients fulfilled the American College of Rheumatology criteria for SLE.2 Blood samples and clinical data were collected from patients arriving at the Rheumatology and Nephrology Units at Rabin Medical Centre, PetachTikva, Israel; the Rheumatology Unit and the Haematology Department of the Sheba Medical Centre, Israel; the Department of Dermatology, Tel Aviv Sourasky Medical Centre; and the Dipartimento di Scienze Mediche e Chirurgiche, Sezione di Clinica Medica, Polo Didattico, Ancona, Italy.
Antigen microarrays and serum testing
Sixty-four antigens, some in several concentrations or in different solvents (110 different preparations overall), were spotted in triplicates on epoxy-activated glass substrates (ArrayIt SuperEpoxi microarray substrate slides, Sunnyvale, CA) using a 48-pin robot (Microgrid 600; Genomics Solutions, Ann Arbor, MI). These antigens included proteins, synthetic peptides from the sequences of selected proteins, nucleotides, phospholipids, and other self and non-self-molecules (see list of antigens in the Appendix). The microarrays were then blocked for 1 hr at 37° with 1% BSA. Test serum in 1% BSA blocking buffer (1 : 10 dilution) was incubated under a coverslip for 1 hr at 37°. The arrays were then washed and incubated for 1 hr at 37° with a 1 : 500 dilution of two detection antibodies, mixed together: a goat anti-human IgG Cy3-conjugated antibody, and a goat anti-human IgM Cy5-conjugated antibody (both purchased from Jackson ImmunoResearch Laboratories Inc., West Grove, PA). Image acquisition was performed by laser (Agilent Technologies, Santa Clara, CA) and the results were analysed using quantarray software (Packard BioChip Technologies, Billerica, MA). The quantitative range of signal intensity of binding to each antigen spot was 0–65 000; this range of detection made it possible to obtain reliable data at a 1 : 10 dilution of test samples.
Image analysis and data processing
The foreground and background intensities of multiple spots of each antigen were averaged, and the difference between the foreground and the background was calculated. The resulting value was taken as the antigen reactivity of the antibodies binding to that spotted antigen. All antigens showed meaningful reactivity in a significant number of slides; hence no antigen was excluded.
Statistical analysis
We sought to identify antigens whose reactivity is higher or lower in a specific study subgroup compared with other subgroups. An antigen i was determined to characterize study subgroup A with respect to subgroup B, if at least 20% of the subjects in subgroup A manifested reactivity higher than a given threshold, which we set at a positive predictive value (PPV) of 90% – in other words, the rank order of reactivities to the particular antigen showed that 90% or more of the highest reactivities belonged to subgroup A relative to subgroup B subjects. Subjects who manifested reactivity higher than that threshold were termed ‘positive’ and antigen i was considered to be ‘increased’ in subgroup A. The same procedure was performed in the case that group A manifested lower reactivity than group B, namely at least 20% of the subjects in subgroup A showed reactivity lower than the threshold level set at a PPV of 90% – in other words, at least 90% of the lowest reactivities belonged to subgroup A compared with subgroup B. Subjects for which reactivity was lower than threshold were termed ‘positive’ and antigen i was declared as ‘decreased’ in subgroup A. The cut-off and positivity were determined specifically for each antigen and for a specific analysis, for example, SLE versus SSc, or SLE versus healthy controls.
Values of p were calculated via randomization and were subjected to multiple comparisons correction. All ‘decreased’ cases passed a false discovery rate (FDR) of up to 10%.9
Antigens that were ranked as ‘increased’ with a sensitivity score of at least 30% passed the FDR test. However, because of the over-representation of SLE specimens compared with SSc patients and healthy controls, some of the ‘increased’ antigens that manifested a sensitivity score below 30% did not pass the 10% FDR level. Nevertheless, these antigens were included in the data because ‘positive’ slides for such antigens overlapped with slides that were ‘positive’ for dsDNA (corresponding p-values were smaller than 8 × 10−3, for an FDR level of 5%).
Results
IgG and IgM reactivities in SLE patients compared with those of healthy controls and SSc patients
Table 1 shows antigen reactivities of the IgG and IgM isotypes that were either increased or decreased in the sera of the SLE patients compared with the reactivities of SSc patients and healthy controls. We found IgG reactivities to be increased for known SLE antigens such as DNA, Sm, and β2GP1, in addition to other antigens. Increased IgG reactivities to HA from both human and streptococcus were prominent in SLE patients. Reactivities to EBVEA and EBVp23 were found to be increased in SLE patients, compared with healthy controls, but not compared with SSc patients. Increased reactivities in SLE patients to HA and EBVEA were also found in our previous study.6
Table 1.
Sensitivity of antibody reactivities in systemic lupus erythematosus (SLE) patients compared to healthy controls and to scleroderma (SSc) patients
| Antigen | Sensitivity(%) for PPV ≥ 90% | ||
|---|---|---|---|
| SLE compared with controls | SLE compared with SSc | ||
| Increase in IgM | |||
| dsDNA | 47 | NS | |
| HA (human)1 | 48 | NS | |
| ssDNA | 40 | NS | |
| FOXp3-p221 | 39 | NS | |
| Sm1 | 29 | NS | |
| Buseralin1 | 27 | NS | |
| MOG1 | 24 | NS | |
| BMP41 | 20 | NS | |
| β2GP1 | NS | 22 | |
| Decrease in IgM | |||
| GST | 22 | 43 | |
| Increase in IgG | |||
| ssDNA | 69 | 55 | |
| dsDNA | 65 | 63 | |
| EBVEA | 55 | NS | |
| HA (streptococcal)2 | 55 | 43 | |
| FOXp3-p222 | 41 | 39 | |
| HA (human)2 | 40 | 33 | |
| MOG2 | 35 | 24 | |
| BMP42 | 34 | 30 | |
| EBVp23 | 31 | NS | |
| HSP60-peptide 262 | 29 | 29 | |
| p53 – peptide 102 | 20 | 20 | |
| p53p112 | 20 | 20 | |
| IGFBP12 | 20 | NS | |
| Sm | NS | 33 | |
| β2GP1 | NS | 29 | |
| HGF | NS | 30 | |
| Decrease in IgG | |||
| EBNA1 | 22 | NS | |
| EBVp18 | 20 | NS | |
NS, non-significant; PPV, positive predictive value.
IgM significant antigens – antigens other than DNA antigens that significantly characterize SLE patients.
IgG significant antigens – antigens other than DNA or Epstein–Barr virus antigens that significantly characterize SLE patients.
The IgG reactivities to EBVp18 and EBNA1 were found to be present in most healthy subjects; but unexpectedly, some of the SLE and SSc patients were found to have decreased reactivities to these EBV antigens (Table 1 and Fig. 1).
Figure 1.
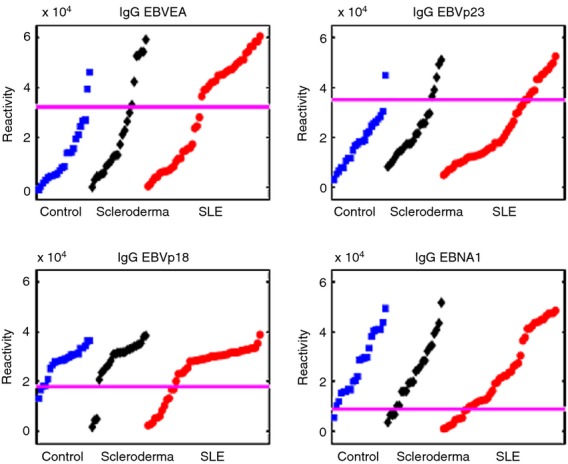
IgG reactivities to Epstein–Barr virus (EBV) antigens in healthy controls and in systemic lupus erythematosus (SLE) and scleroderma (SSc) patients. Note that subgroups of SLE patients show increased reactivities to EBV Early Antigen and EBVp23 or decreased reactivities to EBVp18 or EBV nuclear antigen 1. SLE patient reactivities to EBV antigens do not differ significantly from those of SSc patients. The horizontal lines mark the value that distinguished SLE patients from controls using a positive predictive value ≥ 90%. Each spot represents a single subject.
The IgM reactivities that characterized SLE patients compared with healthy controls did not differ significantly when compared with SSc patients; the IgG reactivities that distinguished between SLE and healthy controls also tended to discriminate between the SLE and SSc patients (Table 1). Increases in IgM reactivities to DNA and HA were most prominent. IgM and IgG reactivities to dsDNA overlapped; 18 of 23 (78%) SLE patients positive for IgM anti-dsDNA were also positive for IgG anti-dsDNA. In addition, the significant IgM and IgG reactivities were found to overlap: five of the six antigens significant for IgM reactivity were also significant for IgG reactivity (Table 1).
The IgM reactivities to glutathione S-transferase (GST) were relatively high in all the study groups, but a subgroup of SLE patients had decreased reactivities compared with controls and SSc patients (Fig. 2). Upon review of the data from our previous study,6 a similar decrease in IgM reactivities to GST was also found in some SLE patients.
Figure 2.
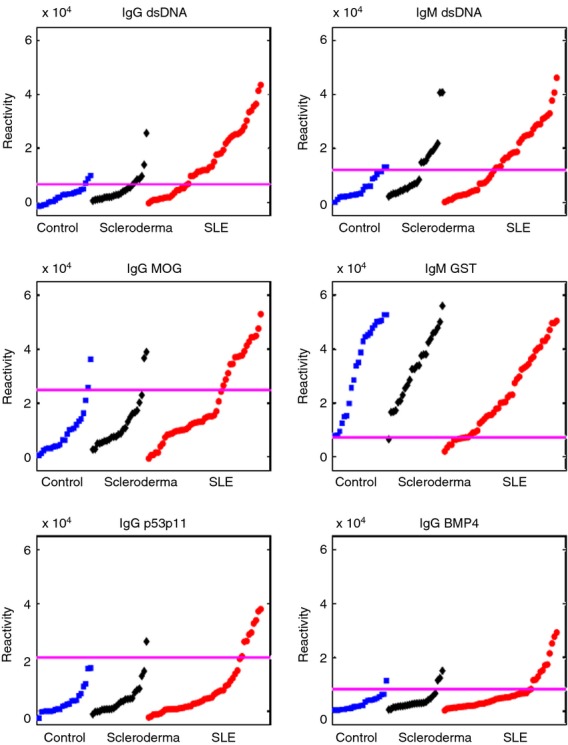
IgG and IgM reactivities to selected antigens in healthy controls and in systemic lupus erythematosus (SLE) and scleroderma patients. The horizontal lines mark the value that distinguished SLE patients from controls within a positive predictive value ≥90%. Each spot represents a single subject.
Similar to our previous study, we found decreases in IgM reactivities to CD99 and cardiolipin in SLE patients, although the threshold of PPV ≥ 90% was not passed (not shown). Decreases in IgM reactivities to myeloperoxidase, collagen III and insulin-like growth factor binding protein 1, which were described in our previous study,6 were not found in our current study.
In general, the different subgroups of SLE patients with increases or decreases in IgM and IgG reactivities partially overlapped each other; no reactivities or lack of reactivities were correlated in any subgroup. No clear correlation was found between the increases or decreases in IgM or IgG reactivities and the clinical manifestations of the disease. The SLE antibody profile overlapped that of the SSc patients with regard to EBV antigens but was significantly different with regard to the other antigens.
IgG and IgM reactivities in SSc patients
Table 2 shows the per cent sensitivities to antigens that were found to be increased in SSc patients compared with healthy controls and SLE patients. Only reactivities to topoisomerase and centromere B differed significantly in SSc patients compared with both healthy controls and SLE patients.
Table 2.
Sensitivity of antibody reactivities in scleroderma (SSc) patients compared with healthy controls and with systemic lupus erythematosus (SLE) patients
| Increased reactivities in SSc patients compared with healthy controls and SLE patients | |||
|---|---|---|---|
| Antigen | Sensitivity(%) for PPV ≥ 90% | ||
| SSc compared with healthy controls | SSc compared with SLE | ||
| Increased IgM | |||
| Centromere B | 42 | NS | |
| Increased IgG | |||
| Topoisomerase | 50 | 33 | |
| Centromere B | 33 | 25 | |
NS, non-significant; PPV, positive predictive value.
Similar to the SLE patients, increases in IgG reactivities to EBVEA and EBVp23 and decreases in IgG reactivities to EBVp18 and EBNA1 were found in SSc patients compared with controls. The apparent lack of significance can be attributed to the requirement of PPV ≥ 90% and the small number of SSc patients (Fig. 1).
Increased IgG reactivities to other antigens characterize the SLE patients scoring positive for IgG anti-dsDNA
We examined whether a combination of IgG reactivities to antigens other than EBV or dsDNA might increase the serological detection of SLE patients. We identified nine IgG reactivities that significantly characterized SLE patients compared with controls (termed IgG significant antigens; Table 1). An SLE patient was classified as positive for IgG significant antigens if he or she were positive for at least two of the nine antigens. Of the 49 SLE patients, 57% were detected by their IgG reactivities to IgG significant antigens; reactivity to dsDNA alone detected 65% of SLE patients. Indeed, the detection rate improved by only 6% by the addition of the IgG significant antigens to the dsDNA detection rate. Hence the information provided by the IgG significant antigens was mostly redundant to that provided by IgG anti-dsDNA (Fig. 3).
Figure 3.
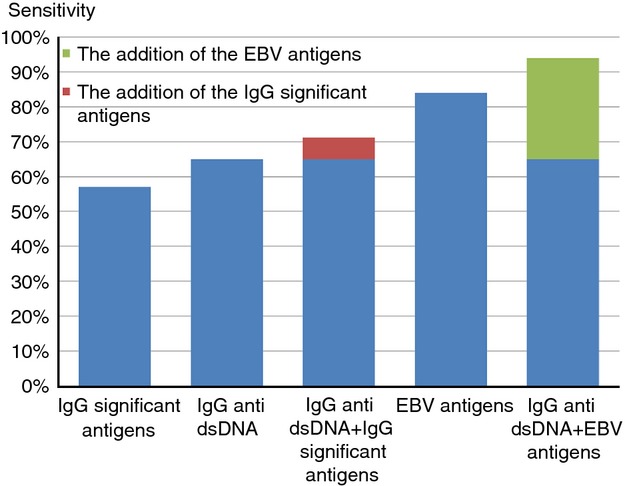
Detection rate of the IgG significant antigens, dsDNA and Epstein–Barr virus (EBV) antigens and their combinations. The systemic lupus erythematosus (SLE) patients detected by IgG significant antigens mostly overlapped with those detected by dsDNA and added little to the detection rate of IgG anti-dsDNA (red rectangle). In contrast, SLE patients detected by EBV antigens only partly overlapped with those detected by anti-dsDNA and significantly added to the detection rate of dsDNA (green rectangle).
A subgroup of anti-dsDNA negative SLE patients is characterized by IgG reactivities to EBV antigens
IgG antibodies to EBV antigens characterized 84% of SLE patients, and unlike the reactivities to the IgG significant antigens, 29% of the SLE patients positive for EBV antigens were not detected by their anti-dsDNA reactivity. Hence, combining dsDNA and EBV antigens increased the serological detection of SLE to 94% (Fig. 3). Reactivity to EBV antigens therefore contrasts with the reactivity to the 59 other antigens, which failed to provide significant information that was not already provided by anti-dsDNA reactivity. No significant clinical difference was found between these different serological subgroups of SLE patients. Similarly, the reactivity to EBV antigens detected SSc patients who were negative for dsDNA antibodies. Using the thresholds set by the SLE patients, 14 (58%) SSc patients were detected by the EBV antigens but only two of them were positive for dsDNA.
Increased IgM reactivities characterize the SLE patients scoring positive for IgM anti-dsDNA
We investigated whether SLE patients manifest an overlap between IgM anti-dsDNA and IgM reactivities to the six antigens found to characterize SLE patients (termed IgM significant antigens; Table 1). An SLE patient was classified as positive for IgM significant antigens if he or she were IgM positive for at least two out of the six antigens. Of the 49 SLE patients, 43% were detected by their reactivities to IgM significant antigens; IgM reactivity to dsDNA alone detected 47% of SLE patients. Indeed, the detection rate improved by only 8% by the addition of the IgM significant antigens to the IgM anti-dsDNA detection rate. Hence, similar to the overlap between IgG anti-dsDNA and the IgG significant antigens, the information provided by the reactivities to the IgM significant antigens was mostly redundant to that provided by IgM anti-dsDNA.
SLE patients can be distinguished serologically from SSc patients
To distinguish SLE patients from SSc patients, we first used IgG reactivities to dsDNA and IgM reactivities to GST to detect the SLE patients. Forty of the 49 SLE patients and three of the 24 SSc patients were detected in this way; however, two of the SSc patients and one SLE patient were positive for IgG to either topoisomerase or centromere B, and their diagnosis was changed to SSc; as a result we were left with one SSc patient false positive for SLE and 39 SLE patients who were true positives. Overall, the combination of these four reactivities yielded a sensitivity and specificity of 80% and 96%, respectively, for detecting SLE patients (PPV = 98%, negative predictive value = 70%).
The present study validates our earlier results
To validate the findings of our present study (‘learning set’), we examined two test sets, namely a new additional test set of 16 SLE patients, eight pemphigus patients and six controls, and a second test set composed of data from our previous study, which included 40 SLE patients and 16 healthy controls.6 In our previous study, the only EBV antigen we used was EBVEA; hence we tested whether the combination of EBVEA and dsDNA would yield similar results between these different studies when the SLE patients were compared with healthy controls and pemphigus patients. We set the threshold values for dsDNA and EBVEA based on the learning set. A sample was classified as ‘SLE’ if either reactivities for dsDNA or EBVEA were higher than the PPV ≥ 90% threshold as defined by the learning set. In our current study (the learning set), the IgG reactivities to EBVEA and dsDNA detected 82% of the SLE patients; 83% of the healthy controls were negative for both antigens. As SSc patients were positive for EBV antigens, they were not included in the analysis. In the new additional test set, 81% of the SLE patients were correctly classified and 86% of the healthy controls and the pemphigus patients were negative for both antigens. Using the samples of the previous study as the test set, we correctly classified 83% of the SLE patients, and 14 controls were negative for both antigens (88%). Overall, the three studies (the learning set and the two independent test sets) yielded similar results and show that over 80% of SLE patients are positive for at least one of these two antigens: EBVEA and dsDNA (Table 3).
Table 3.
The results of three different studies using IgG reactivities to EBVEA and dsDNA. Systemic lupus erythematosus (SLE) patients were compared with healthy controls and with pemphigus patients. Note the similar results between each of the studies
| Current study (‘Learning Set’) | New test set | Previous study used as test set | |
|---|---|---|---|
| SLE-49, Controls-23 | SLE-16, Controls-6, pemphigus-8 | SLE-40, Controls-16 | |
| Sensitivity/Specificity (%) | 82/83 | 81/86 | 83/88 |
| PPV/NPV (%) | 91/68 | 87/80 | 94/67 |
NPV, negative predictive value; PPV, positive predictive value.
Discussion
This study has characterized antibody repertoires in SLE sera using sera from SSc patients and healthy control subjects as specificity controls. We used an antigen microarray device and informatic analysis that differ significantly from standard ELISAs in the concentration of sera of the test samples (1 : 10), the concentrations and amounts of the antigens, and the technology of the testing platform; moreover, we related only to antibody reactivities that manifested a very high PPV for SLE – a PPV ≥ 90% (Tables 3). Hence, this methodology provides a novel view of serum antibodies. This technology, in addition to detecting known autoimmune antibodies, enabled us to discover the following:
Approximately two-thirds of SLE patients manifested strong IgG reactivities to dsDNA. These reactivities overlapped with IgM anti-dsDNA antibodies and IgG and IgM reactivities to a broad spectrum of other molecules including mammalian proteins and peptides, and HA derived from both humans and bacteria.
The SLE patients also manifested antibodies to four defined EBV antigens; some patients manifested increased and others showed decreased IgG reactivities to several of these antigens. The antibodies to EBV, in contrast to the other antibodies, overlapped only partly with those binding to dsDNA; 29% of SLE patients were negative for anti-dsDNA but were positive for antibody changes to the EBV antigens.
Some SLE patients manifested decreased IgM autoantibodies to GST.
The clinical diagnosis of SLE is based on the fulfilment of four out of eleven criteria.2 Despite the high prevalence of autoantibodies in SLE, specific autoantibodies such as anti-dsDNA antibodies are only one of the 11 criteria; significant numbers of patients diagnosed with SLE are sero-negative to dsDNA. This study did not compare the microarray methodology with the results of standard tests used clinically; we are presently testing whether the microarray device and our array of antigens might contribute to the serological diagnosis of SLE (Fig. 3), particularly with regard to the patients negative for IgG antibodies to dsDNA.
The overlap of dsDNA autoantibody binding with a spectrum of IgM and IgG autoantibodies to other self-antigens suggests that SLE autoimmunity is not limited to B-cell clones specific to dsDNA; indeed, some two-thirds of SLE patients manifested coordinated reactivity to a whole set of self-antigens somehow connected to anti-dsDNA reactivity, both IgM and IgG (Fig. 3). Hence a common pathological process might mark this major subgroup of SLE patients. Indeed, clusters of IgM and IgG reactivities including reactivities to DNA and HA were found to characterize SLE patients.10
It is interesting that SLE-like conditions are associated with apparently non-antigen-specific perturbations of the immune systems such as genetic defects in Fas or Fas ligand molecules – the lpr and gld mutations11 – or in genetically susceptible NZB/W mice.12 Indeed, T-cell regulation of B cells is generally defective in SLE patients and leads to hyper-reactive B cells and over-production of many autoantibodies.13–14 What connects these various autoantibodies is not known. It has been suggested that SLE is associated with abnormal apoptosis and that SLE features autoantibodies to a number of molecules expressed in the apoptotic process.15 However, the list of significant autoantibodies includes more than those linked specifically to apoptosis (Table 1). Elevated IgG reactivities to HA extracted from both humans and streptococci suggest that infections might be linked to the induction of SLE in a subgroup of patients. It is intriguing that a significant number of SLE patients of the dsDNA-positive subgroup manifest both IgM and IgG antibodies that bind to a peptide epitope of FOXp3, a transcription factor for regulatory T cells.16 It is not yet known how such autoantibodies might affect regulatory T cells.
The IgG reactivities to EBV antigens are an exception to the self-antigen reactivities associated with SLE: IgG anti-EBV did not overlap with IgG reactivities to dsDNA in all SLE patients (Fig. 3). We found that 84% of the SLE patients showed at least one of the following: increased IgG reactivities to EBVEA or EBVp23, or decreased IgG reactivities to EBVp18 or EBNA1 (Fig. 3). Interestingly, 29% of the SLE patients were negative for dsDNA but positive for at least one of the EBV antigens. These four EBV reactivities were not confined to SLE patients, but appeared in SSc patients too, suggesting a possible role of an aberrant immune response to EBV in these autoimmune diseases.
Epstein–Barr virus is a herpes virus that can cause a large spectrum of clinical manifestations, from a minor flu-like illness to infectious mononucleosis or even Burkitt’s lymphoma.17 The hallmark of the pathogenesis of EBV is the establishment of a latent infection in B cells. In the latent phase, the EBV genome can encode proteins such as latent membrane protein one, an EBV oncoprotein that can induce the B-cell activating factor BAFF, which, in turn, can activate self-reactive B cells and induce a lupus-like disease in transgenic mice.18 Recent work on murine gammaherpesvirus 68 (MHV68), a virus genetically and biologically related to EBV, has shown that the production of autoantibodies is associated with an expansion of T helper cells in response to the expression of the Orf73 viral gene.19 EBV-infected B cells appear to escape immune system surveillance and maintain a chronic infection; indeed, it was found that SLE patients have increased viral loads and a defective control of latent EBV infection.20 The increased IgG reactivities to EBV found in the present study might reflect a chronic state of EBV infection; in contrast, the decreased IgG reactivities may be linked to a defective immune reaction to the virus compared with the anti-EBV responses of healthy subjects.
It is interesting that we observed a significant decrease in IgM antibodies to GST and other self-molecules in some of the SLE patients. IgM autoantibodies, including antibodies to dsDNA, are prevalent in healthy persons and present in cord blood from birth.21 IgM autoantibodies have been suggested as possible protectors against the transition of natural autoimmunity to pathogenic autoimmune disease;22 and a relative lack of certain IgM reactivities may be a factor in SLE, and not only a serological marker. Until now, however, no link between GST and SLE has been reported. Note that healthy persons also manifest IgG antibodies that bind to self-antigens; healthy autoimmunity would appear to take part in the immunological homunculus – the image of body molecules encoded in immune repertoires.23 Indeed, Fig. 2 confirms that healthy subjects manifest measurable amounts of IgG autoantibody binding to key self-antigens;23 however, the difference is mainly quantitative: greater numbers of SLE patients have greater degrees of reactivity to these antigens.
Note that a number of SLE patients differed from SSc patients and healthy controls in manifesting increased IgG reactivity to peptides of p53 (Table 1); p53, like anti-DNA antibody, binds DNA, and we have reported that autoantibodies to p53 in SLE are idiotypically related to anti-DNA autoantibodies.24–25 Hence, anti-DNA autoantibodies in some SLE subjects might be related to heightened autoimmunity to p53, a molecule that functions in apoptosis26 and was found to be associated with disease activity in SLE.27
It is interesting that increased IgG and IgM reactivity to myelin oligodendrocyte glycoprotein (MOG) is present in many SLE subjects (Table 1 and Fig. 2); MOG is a myelin antigen targeted by autoreactive T cells 28 and associated with autoantibodies29 in subjects with multiple sclerosis. It remains to be seen whether anti-MOG autoimmunity is associated with nervous system manifestations of SLE.
In summary, the ability of the antigen microarray to measure quantitative reactivity to a relatively large set of antigens present in a relatively small volume of blood provides a novel view of complex diseases. Characterization of antibody repertoires in SLE, SSc and other autoimmune diseases may pave the way to serological diagnosis of disease and a better understanding of the complex pathological processes unique for SLE or shared with other conditions.29–30
Acknowledgments
This work was supported by the Open University of Israel grants (IG-0901 and IG-0902 to NS) and by a grant by the Laszlo N. Tauber Family Foundation to IRC and NS.
Disclosures
There is no conflict of interest to disclose.
Author contributions
IF designed the study, performed the experiments and wrote the article. NS designed the study, performed the statistical analysis and wrote the article. YM contributed to the recruitment of patients, sera and clinical data and wrote the article. AG contributed to the recruitment of patients; sera and clinical data and contributed to the writing of the article. EP-S, SO, AL, PL and RP contributed to the recruitment of patients, sera and clinical data. OS contributed to the design of the study, recruitment of patients, sera and clinical data. UG contributed to the recruitment of patients; sera and clinical data and contributed to the writing of the article. ED participated in the design of the study, performed the statistical analysis, and wrote the article. IRC designed the study, statistical analysis and wrote the article.
Appendix 1. List of antigens
| Actin – Actin from bovine muscle, A3563, Sigma. | EBNA1 (EBV nuclear antigen 1) – recombinant, EBV-276, Prospec. | Hyaluronic acid from rooster comb – H5388, Sigma. | p53 p10 – amino acids 14–33, KTCPVQLWVSATPPAGSRVR, UniProtKB: A5JTV6. |
| beta2GP1 – A2299-77E, US Biological. | EBV p18-recombinant, EBV-273, Prospec. | Hyaluronic acid sodium salt, from Streptococcus equi – 53747, Sigma. | p53p11 – amino acids 29–48 – GSRVRAMAIYKKSQHMTEVV, UniProtKB: A5JTV6 . |
| BMP4 (Bone morphogenic protein 4) – human recombinant, CYT- 36, Prospec. | EBV p23 – recombinant, EBV-278, Prospec. | Hyaluronic acid human – H1504, Sigma. | p53 amino acids 253–272 – DSSGNLLGRDSFEVRVCACP,UniProtKB: P02340. |
| Buserelin – HOR-255, Prospec. | EBVEA (Epstein–Barr virus early antigen) – recombinant, EBV-272, Prospec. | IGFBP1 (insulin growth factor binding protein 1) – recombinant, CRI232B, Cell Sciences. | p53 amino acids 53–72 – LPQDVEEFFEGPSEALRVSG. UniProtKB: P02340. |
| Cardiolipin – C0563, Sigma. | FABP 3 (fatty acid binding protein 3) – recombinant, PRO-340, Prospec. | IgG – I2511, Sigma. | PCNA (proliferating cell nuclear antigen) – recombinant, PRO-303, Prospec. |
| CD99 – human recombinant, PRO-294, Prospec. | Fibrinogen – F4753, Sigma. | IgM – I8260, Sigma. | PDGF receptor (platelet-derived growth factor receptor) – recombinant, D0946, Sigma. |
| Centromere A – human recombinant, PRO-389, Prospec. | FOX (Forkhead box) Protein 3 – p290–304 – TKASSVASSQGPVVP, UniProtKB: B7ZLG11 | La – recombinant, PRO-327, Prospec. | PDI (Protein disulphide Isomerase) – recombinant, ENZ-262, Prospec |
| Centromere B – human recombinant, PRO-390, Prospec. | GLP1 (glucagon-like peptide-1) – recombinant, HOR-236, Prospec. | Lipopolysaccharides from Pseudomonas aeruginosa – L9143, Sigma. | Pneumococcal capsular polysaccharide type 4 – was purchased from ATCC (Manassas, VA). |
| CMV (cytomegalovirus) Pp150 – recombinant, CMV-216, Prospec. | GROa (growth-regulated protein α) – recombinant, CHM-329, Prospec. | Lipopolysaccharides from Salmonella enterica – L5886, Sigma. | PR3 (Proteinase 3) – CSI14825A, Cell Sciences. |
| Collagen III – C4407, Sigma. | GST (glutathione-S-transferase) – G8642, Sigma. | Lysosomal membrane protein 2 – recombinant, H00003920, Abnova. | RO60 – recombinant, PRO-329, Prospec. |
| Collagen IV – C7521, Sigma. | HGF (hepatocyte growth factor) – recombinant, CYT-244, Prospec. | Methyl-CpG-binding domain protein 2 – recombinant, ab40707, Abcam. | SAP90 (Disks large homologue 4) amino acids 63–82 – VDVREVTHSAAVEALKEAGS. UniProtKB: K7EKU8. |
| DNA (cytosine-5) methyltransferase 1 – recombinant, ab91367, abcam. | Horseradish peroxidase – P6782, Sigma. | Methyl-CpG-binding domain protein 4 – recombinant, H00008930, Abnova. | Sm (Smith antigen) – , Cell Sciences. |
| dsDNA – D1501, Sigma. | HSP60 amino acids 21–41, QSIVPALEIANAHRKPLVIIA. UniProtKB: Q53QD5. | MOG (myelin oligodendrocyte glycoprotein) – p35–55 – PRO-371, Prospec. | SYPH (synaptophysin; rat) amino acids 81–100 – CVKGGTTKIFLVGDYSSSAE, UniProtKB: P07825. |
| ssDNA – D8899, Sigma | hsp60 amino acids 240–259, QDAYVLLSEKKOSSVQSIVP. UniProtKB: P108092 | MPO (myeloperoxidase) – ENZ-074, Prospec. | Thyroglobulin – T1001, Sigma. |
| DSG (desmoglein) 1 – human recombinant, H00001828-P01, Abnova. | HSP60p26- amino acids 376-395, EQLDITTSEYEKEKLNERLA, UniProtKB: P63038. | NRMJ amino acids 206-234- LGCSSRGVCVDGQCICDSE, UniProtKB: F1LQ63. | Topoisomerase 1 – recombinant, ENZ-306, Prospec. |
| DSG (desmoglein)3 – ab87441, abcam. | HSP 60- amino acids 436-455- IVLGGGCALLRCIPALDSLK, UniProtKB:P63038. | P278 (HSP60 amino acids 458–474) – NEDQKIGIEIIKRALKI UniProtKB: P63038 | U1RNP(U1 ribonucleoprotein complex) – recombinant, PRO-445, Prospec. |
EBV, Epstein–Barr virus.
The overlap between the sequence TKASSVASSQGPVVP that was tested, was 60% with the matching human sequence TKASSVASSDKGSCC.
The overlap between the sequence QDAYVLLSEKKOSSVQSIVP that was tested, was 90% with the matching human sequence QDAYVLLSEKKISSIQSIVP.
References
- Sherer Y, Gorstein A, Fritzler MJ, et al. Autoantibody explosion in systemic lupus erythematosus: more than 100 different antibodies found in SLE patients. Semin Arthritis Rheum. 2004;34:501–37. doi: 10.1016/j.semarthrit.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Criteria published by EM Tan et al. 1982. Arthritis Rheum; 25:1271, updated by MC Hochberg, Arthritis Rheum 1997; 40:1725.
- James JA, Kaufman KM, Farris AD, et al. An increased prevalence of Epstein–Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J Clin Invest. 1997;100:3019–26. doi: 10.1172/JCI119856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James JA, Harley JB, Scofield RH. Epstein–Barr virus and systemic lupus erythematosus. Curr Opin Rheumatol. 2006;18:462–7. doi: 10.1097/01.bor.0000240355.37927.94. [DOI] [PubMed] [Google Scholar]
- Carapetis JR, Steer AC, Mulholland EK, et al. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–94. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- Fattal I, Shental N, Mevorach D, et al. An antibody profile of systemic lupus erythematosus detected by antigen microarray. Immunology. 2010;130:337–43. doi: 10.1111/j.1365-2567.2010.03245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stollerman GH, Dale JB. The importance of the group a streptococcus capsule in the pathogenesis of human infections: a historical perspective. Clin Infect Dis. 2008;46:1038–45. doi: 10.1086/529194. [DOI] [PubMed] [Google Scholar]
- Quintana FJ, Merbl Y, Sahar E, et al. Antigen-chip technology for accessing global information about the state of the body. Lupus. 2006;15:428–30. doi: 10.1191/0961203306lu2328oa. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. JR Stat Soc B. 1995;57:289–300. [Google Scholar]
- Quan LZ, Chun X, Tianfu W, et al. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J Clin Invest. 2005;115:3428–39. doi: 10.1172/JCI23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Tanaka M, Brannan CI, et al. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76:969–76. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- Howie JB, Helyer BJ. The immunology and pathology of NZB mice. Adv Immunol. 1968;9:215–66. doi: 10.1016/s0065-2776(08)60444-7. [DOI] [PubMed] [Google Scholar]
- Kyttaris VC, Juang Y, Tsokos GC. Immune cells and cytokines in systemic lupus erythematosus: an update. Curr Opin Rheumatol. 2005;17:518–22. doi: 10.1097/01.bor.0000170479.01451.ab. [DOI] [PubMed] [Google Scholar]
- Linker-Israeli M, Quismorio FP, Jr, Horwitz DA. CD8+ lymphocytes from patients with systemic lupus erythematosus sustain, rather than suppress, spontaneous polyclonal IgG production and synergize with CD4+ cells to support autoantibody synthesis. Arthritis Rheum. 1990;33:1216–25. doi: 10.1002/art.1780330823. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Ichino M, Mihara S, et al. Inhibition of Fas/Fas ligand-mediated apoptotic cell death of lymphocytes in vitro by circulating anti-Fas ligand autoantibodies in patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41:344–53. doi: 10.1002/1529-0131(199802)41:2<344::AID-ART19>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Buckner JH. Mechanisms of impaired regulation by CD4+ CD25+ FOXP3+ regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10:849–59. doi: 10.1038/nri2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawa K. Epstein–Barr virus-associated diseases in humans. Int J Hematol. 2000;71:108–17. [PubMed] [Google Scholar]
- Niller HH, Wolf H, Minarovits J. Regulation and dysregulation of Epstein–Barr virus latency: implications for the development of autoimmune diseases. Autoimmunity. 2008;41:298–328. doi: 10.1080/08916930802024772. [DOI] [PubMed] [Google Scholar]
- Gauld SB, De Santis JL, Kulinski JM, McGraw JA, Leonardo SM, Ruder EA, Maier W, Tarakanova VL. Modulation of B-cell tolerance by murine gammaherpesvirus 68 infection: requirement for Orf73 viral gene expression and follicular helper T cells. Immunology. 2013;139:197–204. doi: 10.1111/imm.12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang I, Quan T, Nolasco H, et al. Defective control of latent Epstein–Barr virus infection in systemic lupus erythematosus. J Immunol. 2004;172:1287–94. doi: 10.4049/jimmunol.172.2.1287. [DOI] [PubMed] [Google Scholar]
- Merbl Y, Zucker-Toledano M, Quintana FJ, Cohen IR. Newborn humans manifest autoantibodies to defined self molecules detected by antigen microarray informatics. J Clin Invest. 2007;117:712–8. doi: 10.1172/JCI29943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen IR, Cooke A. Natural autoantibodies might prevent autoimmune disease. Immunol Today. 1986;7:363–4. doi: 10.1016/0167-5699(86)90026-5. [DOI] [PubMed] [Google Scholar]
- Madi A, Bransburg-Zabary S, Kenett DY, et al. The natural autoantibody repertoire in newborns and adults: a current overview. Adv Exp Med Biol. 2012;750:198–212. doi: 10.1007/978-1-4614-3461-0_15. [DOI] [PubMed] [Google Scholar]
- Herkel J, Mimran A, Erez N, et al. Autoimmunity to the p53 protein is a feature of systemic lupus erythematosus (SLE) related to anti-DNA antibodies. J Autoimmun. 2001;17:63–9. doi: 10.1006/jaut.2001.0518. [DOI] [PubMed] [Google Scholar]
- Herkel J, Kam N, Erez N, et al. Monoclonal antibody to a DNA-binding domain of p53 mimics charge structure of DNA: anti-idiotypes to the anti-p53 antibody are anti-DNA. Eur J Immunol. 2004;34:3623–32. doi: 10.1002/eji.200425371. [DOI] [PubMed] [Google Scholar]
- Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012;28:128–36. doi: 10.1016/j.tig.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miret C, Molina R, Filella X, et al. Font J. Relationship of p53 with other oncogenes, cytokines and systemic lupus erythematosus activity. Tumour Biol. 2003;24:185–8. doi: 10.1159/000074428. [DOI] [PubMed] [Google Scholar]
- de Rosbo NK, Ben-Nun A. T-cell responses to myelin antigens in multiple sclerosis; relevance of the predominant autoimmune reactivity to myelin oligodendrocyte glycoprotein. Autoimmun. 1998;11:287–99. doi: 10.1006/jaut.1998.0202. [DOI] [PubMed] [Google Scholar]
- Quintana FJ, Farez MF, Viglietta V, et al. Antigen microarray identify unique serum autoantibody signatures in clinical and pathologic subtypes of multiple sclerosis. Proc Natl Acad Sci USA. 2008;105:18889–94. doi: 10.1073/pnas.0806310105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hueber W, Kidd BA, Tomooka BH, et al. Antigen microarray profiling of autoantibodies in rheumatoid arthritis. Arthritis Rheum. 2005;52:2645–55. doi: 10.1002/art.21269. [DOI] [PubMed] [Google Scholar]