

Abstract
Background and Purpose: Chlormethiazole (CMZ), a clinical sedative/anxiolytic agent, did not reach clinical efficacy in stroke trials despite neuroprotection demonstrated in numerous animal models. Using CMZ as a lead compound, neuroprotective methiazole (MZ) analogues were developed, and neuroprotection and GABAA receptor dependence were studied.
Experimental Approach: Eight MZs were selected from a novel library, of which two were studied in detail. Neuroprotection, glutamate release, intracellular calcium and response to GABA blockade by picrotoxin were measured in rat primary cortical cultures using four cellular models of neurodegeneration. GABA potentiation was assayed in oocytes expressing the α1β2γ2 GABAA receptor.
Key Results: Neuroprotection against a range of insults was retained even with substantial chemical modification. Dependence on GABAA receptor activity was variable: at the extremes, neuroprotection by GN-28 was universally sensitive to picrotoxin, while GN-38 was largely insensitive. In parallel, effects on extracellular glutamate and intracellular calcium were associated with GABAA dependence. Consistent with these findings, GN-28 potentiated α1β2γ2 GABAA function, whereas GN-38 had a weak inhibitory effect. Neuroprotection against moderate dose oligomeric Aβ1–42 was also tolerant to structural changes.
Conclusions and Implications: The results support the concept that CMZ does not contain a single pharmacophore, rather that broad-spectrum neuroprotection results from a GABAA-dependent mechanism represented by GN-28, combined with a mechanism represented in GN-38 that shows the least dependence on GABAA receptors. These findings allow further refinement of the neuroprotective pharmacophore and investigation into secondary mechanisms that will assist in identifying MZ-based compounds of use in treating neurodegeneration.
Keywords: Alzheimer's disease, chlormethiazole, excitotoxicity, GABA, GABAA receptor, neuroprotection, neurodegeneration, stroke
Introduction
Selected thiazole derivatives are known to manifest sedative/hypnotic and anticonvulsant effects, which is indicative of GABA-mimetic activity (Lindberg, 1971a,b1971b). Chlormethiazole (CMZ) was selected as a potent anticonvulsant from limited structure-activity studies (Lechat et al., 1965a,b1965b,c1965c), and entered clinical use in the 1960s for treatment of epilepsy, alcohol withdrawal, and agitation, and is currently used for management of restlessness and insomnia in the elderly; all indications consistent with the CNS bioavailability of CMZ and its proposed mechanism of action as a potentiator of GABA activity at the GABAA receptor (Cross et al., 1989; Moody and Skolnick, 1989). Two decades ago, initial observations of CMZ's neuroprotection were repeated in several rodent models of cerebral ischaemia, which demonstrated that neuroprotection correlated with reduction in extracellular glutamate, an effect consistent with the hypothesis that CMZ was acting via GABAA receptors to reduce excitotoxicity in the stroke penumbra (Snape et al., 1993; Baldwin et al., 1994; Sydserff et al., 1995a,b1995b). Importantly, the observed neuroprotection also correlated with improvements in behavioural models of memory, and in further studies both neuroprotection and functional recovery were demonstrated in non-human primates after focal cerebral ischaemia (Liang et al., 1997; Marshall et al., 1999; 2000).
Based on the above animal model data and a history of tolerability during clinical use, CMZ was advanced into clinical trials for stroke (Green, 1998; Farooque et al., 1999; Marshall et al., 1999; Wahlgren et al., 1999). However, a large phase III clinical trial did not achieve the primary end point of improvement in the general population, although significant improvement was reported in a subset of the population with more extensive infarction (Mucke, 1999; Wahlgren et al., 1999; 2000). Speculation on the lack of success in human trials ranged from a relatively short reported half-life to an inability to reach the site of infarct, while some authors interpreted the failure in the broader context of the universal failure of neuroprotective drugs in stroke clinical trials (De Keyser et al., 1999; Muir and Grosset, 1999; Gladstone et al., 2002; Hankey, 2002; Wilby and Hutchinson, 2004). Such failures in the latter part of the 20th century have all but halted clinical studies on neuroprotection in diseases that may benefit from agents such as CMZ, including Alzheimer's disease (AD).
Neuroprotective agents derived from CMZ as a lead molecular scaffold and containing the 4-methylthiazole (MZ) pharmacophore are a potential source of novel therapeutics. However, pharmacological data on only a very limited number of CMZ derivatives have been published and CMZ has not been studied extensively in cellular models (Lechat et al., 1965b; Bengtsson and Lindberg, 1982; Colado et al., 2001; Green et al., 2001; Nelson et al., 2001). A library of CMZ analogues and MZ derivatives was synthesized (Qin et al., 2012), and selected members studied herein in rat primary cortical neuronal cultures subjected to four types of insult, providing models of ischaemia-reperfusion injury, excitotoxicity and AD: oxygen-glucose deprivation (OGD); application of glutamate; application of NMDA; and application of oligomeric amyloid-β1–42 (oAβ).
The involvement of GABAA signalling in neuroprotection was probed with picrotoxin (PTX) (Olsen, 1982), and based upon these data, GN-28 and GN-38 were selected for further study on GABA potentiation using the Xenopus oocyte model expressing α1β2γ2 GABAA receptors. These two MZ derivatives were further compared in vivo with CMZ itself, demonstrating that novel, brain-bioavailable MZ derivatives can be designed with enhanced neuroprotective efficacy. The results support the concept that CMZ contains more than one neuroprotective pharmacophore: GN-28 highlights the GABAA-dependent pharmacophore, whereas in GN-38 a neuroprotective pharmacophore is dominant with much less reliance on GABAA receptors. These two agents represent brain bioavailable chemical probes that may be used to understand and refine neuroprotective mechanisms for CMZ and non-sedative MZ derivatives for treatment of neurodegenerative disorders including AD and stroke.
Methods
Ethical animal handling and care
Experiments on rats and mice were performed at the Biologic Resources Laboratory (BRL) at University of Illinois at Chicago (UIC). The BRL ensures that the UIC program meets the Federal regulations, the requirements of the American Association for the Accreditation of Laboratory Animal Care, and currently accepted standards for providing adequate veterinary care and proper animal husbandry. Use of animals was approved by the Institutional Animal Care and Use Committee at the UIC (Chicago, IL, USA; protocol number 09-012). All experiments conformed to the Animal Welfare Act, Guide to Use and Care of Laboratory Animals, and the U.S. Government Principles on the Utilization and Care of Vertebrate Animals Used in Testing, Research and Training guidelines on the ethical use of animals. In addition, efforts were made to reduce the required number of animals, minimize suffering and employ alternative methods when possible. Pregnant female Sprague–Dawley rats (n = 10) and male C57Bl/6 mice (n = 20) originated from Charles River Laboratories (Wilmington, MA, USA). Xenopus laevis toads, used as the source of oocytes for engineered expression of α2β2γ2 GABAA receptors, were obtained from Xenopus One (Ann Arbor, MI, USA). All animal maintenance and surgical procedures on X. laevis conformed to UIC institutional policies (BRL protocol 13-125) and to the Statement for the Use of Animals in Ophthalmic and Vision Research adopted by the Association for Research in Vision and Ophthalmology.
All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Primary neuronal cultures
Cell cultures were prepared from the cortex and hippocampus of mixed sex E16-18 Sprague–Dawley rat embryos as previously described (Abdelhamid et al., 2011). Briefly, cells were harvested and plated in a medium of DMEM, 10% neonatal horse serum and 10% FBS (Invitrogen, Carlsbad, CA, USA) at a density of 1 × 105 cells per well in 96-well plates coated with poly-L-lysine (Sigma, St Louis, MO, USA). One day after plating, media was replaced with growth media consisting of Neurobasal media (Invitrogen), B27 supplement (Thermo Fisher Scientific, Waltham, MA, USA), 0.5 mM glutamine and 1% pen/strep, with further media changes every 3–4 days. This protocol reproducibly results in a neuronal culture of >99.5% purity. Three days before the start of any assay, B27 supplement was changed to B27 supplement minus antioxidants. In all experiments, final concentration of DMSO vehicle was consistent and kept <0.5%.
Oxygen-glucose deprivation
After 10-11 days in vitro (DIV) neuronal cultures were transferred to a sealed hypoxic chamber (Coy Lab, Grass Lake, MI, USA; dimensions 41 L × 23″ D × 23″ H) with an atmosphere of 5% CO2/95% N2 (oxygen tension was monitored with an electrode and kept <0.5%). Culture media was replaced with a solution containing the following (in mM): NaCl 116, CaCl2 1.8, MgSO4 0.8, KCl 5.4, NaH2PO4 1, NaHCO3 14.7, HEPES 10. All compounds were added at 50 μM at start of OGD period, and this concentration was kept constant through media changes. For GABAA receptor blockade, PTX (Sigma) was added at 100 μM 1 h before the start of the OGD, and this concentration was kept constant through media changes. After 2 h, cells were removed from hypoxic chamber and resupplied with growth media. After 24 h, supernatant was aspirated and preserved at −80°C for HPLC analysis, and cell survival was assayed via colorimetry by adding the organic dye, thiazolyl blue tetrazolium bromide (MTT), which is reduced to a formazan dye in the presence of active mitochondrial enzymes. Dye crystals were dissolved in acidified organic propanol and absorbance was read at λ = 570 nm, using λ = 630 nm as a reference wavelength, on a Dynex MRX ll micro-plate spectrophotometer.
Extracellular glutamate by HPLC
Aliquots of culture supernatant were deproteinized by rapid centrifugation (10 000× g for 20 min) at 4°C. All samples were analysed for glutamate using a binary gradient HPLC with fluorescence detection at 450 nm and pre-column derivatization with o-phthaldialdehyde (OPA; Pierce, Rockford, IL, USA); C-18 column (5 μm Hypersil BDS C18 column, 100 × 4.6 mm; Thermo Fisher Scientific) with guard column; flow rate 1.0 mL·min−1 at 35°C; mobile phase (A) 0.1 M sodium acetate, 5% methanol, and 2.5% tetrahydrofuran (THF) solution (pH 6.95) and (B) 97.5% methanol, 2.5% THF. Each experimental condition was assayed at least in triplicate and contained pooled supernatant obtained from six independent cultures in a 96-well plate. The concentration of glutamate in the supernatant of cultures not exposed to OGD was below the threshold of detection for this assay (∼25 nM per 105 cells), so the increase in extracellular glutamate attributable to OGD was substantial but not directly measurable with this approach.
Intracellular calcium
Cortical cultures were prepared as described above, with the exception that 29 mm culture dishes with 10 mm glass-bottomed wells (Invitrogen) were used to prepare cells. Intracellular Ca2+ was measured using fluo-4 AM dye in 1 mM DMSO stock solution (Invitrogen). Dye loading was done at final concentration of 5 μM fluo-4 over 45 min at 37°C in growth media described above followed by a triplicate wash using PBS and addition of compounds in fresh phenol red-free culture media. After 30 min, cells were visualized with an inverted fluorescence microscope (Olympus, Center Valley, CA, USA) and intracellular dye was excited at 488 nm and recorded at 515 nm at 1 Hz for 1 min with addition of 1 mM glutamate 2 s after beginning the recording. Results were analysed using ImageJ, with total fluorescence recorded using total integrated density value for each image normalized to starting fluorescence, and average peak fluorescence reported for each experimental condition. Representative images demonstrating change in intracellular calcium were prepared by subtracting peak fluorescence from starting fluorescence using image calculator and merged with bright field images taken prior to recording.
Glutamate and NMDA toxicity
At 10-11, DIV neuronal cultures were resupplied with fresh growth media. PTX blockade was performed as in OGD and added 1 h before excitotoxic insult. NMDA (100 μM; Sigma) or glutamate (1 mM; Sigma) was added to each culture as indicated in figures. One hour after addition of excitotoxic insult, each compound was supplied at a final concentration of 50 μM. After incubation for 24 h, final cell survival was assayed by MTT as above.
Electrophysiological measurement of GABAA potentiation
Experiments were conducted on X. laevis oocytes expressing α1β2γ2 GABAA receptors. Oocytes expressing α1β2γ2 receptors (rat α1, rat β2 and human γ2s) were prepared by cRNA injection and studied by two-electrode voltage-clamp recording (holding potential: −70 mV; amplifier: GeneClamp500B; Axon Instruments, Foster City, CA, USA) (Yue et al., 2012). Oocytes were superfused with Ringer solution (physiological saline) at a rate of ∼1 mL·min−1. The standard Ringer solution contained 100 mM NaCl, 2 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, and 10 mM glucose, at pH 7.4. Glass micropipettes for oocyte recording were prepared using a programmable puller (Sutter Instruments, Novato, CA, USA) and yielded a resistance of 1–10 MΩ when filled with 3 M KCl. The voltage-clamp procedure was controlled by a computer running Clampex 8.2 (Axon Instruments) interfaced with the apparatus. Electrophysiological data were obtained in response to the presentation, to the oocyte, of Ringer solution containing pharmacological agents. Test solutions were delivered via multiple channels from separate reservoirs by a gravity flow system (Automate Scientific, Berkeley, CA, USA) operated under computer command. Membrane current data were obtained using Clampex 8.2 and analysed using Clampfit 10.0 (Axon Instruments) and OriginPro7.5 (OriginLab Corporation, Northampton, MA, USA).
Aβ oligomer toxicity
Soluble oligomers of Aβ1–42 (oAβ) were prepared 24 h before their addition to cultures as previously described (Stine et al., 2003). Briefly, lyophilized full-length human-sequence peptide was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol and evaporated to leave a peptide film, which was then dissolved to 5 mM in DMSO and added to cold phenol-free F-12 cell culture media and kept at 4°C for 24 h. This procedure reliably develops soluble oligomers upon addition to culture media as confirmed by atomic force microscopy. At DIV 10–11, neuronal cultures were resupplied with fresh growth media. PTX blockade was performed as in OGD and added 1 h before oligomers. oAβ was added at a final concentration of 250 nM or 5 μM. One hour after addition of oAβ, each compound was supplied at a final concentration of 50 μM. After incubation for 4 days, final cell survival was assayed by MTT as above.
Brain bioavailability
C57BL/6 male mice 8–10 weeks old were administered compounds (4.45 μmol·kg−1 i.p.) in 10% DMSO solution. Twenty minutes after drug administration, mice were killed using CO2. Blood was rapidly collected from the dorsal aorta in Greiner Vacuette tubes (0.5 mL sterile plastic vials with K3EDTA) and kept on ice. After centrifugation (10 000× g for 10 min at 4°C), plasma supernatant was collected and kept at −80°C. Subsequent to blood draw, each animal was intracardially perfused with PBS buffer (pH 7.4) and then decapitated. Brains were collected, and cortices and hippocampi were rapidly dissected and flash frozen in liquid N2 to be stored at −80°C. Detailed sample preparation for LC-MS/MS is described in Supporting Information Methods. Briefly, quantitative analysis of drug concentrations in plasma and brain used internal standards spiked into plasma and brain homogenates before liquid extraction, and separation and measurement by LC-MS/MS tandem mass spectrometry.
Data analysis
Analysis of data was performed using ANOVA with either Dunnett's post-hoc test when comparing to vehicle control or Bonferroni's post-hoc test for multiple within group comparisons (as indicated in each figure legend) using GraphPad Prism v5 (San Diego, CA, USA). P-values of 0.05 or less (P < 0.05) were considered significant, and P-values of less than 0.01 or less than 0.001 are additionally reported.
All drug/molecular target nomenclature conforms to BJP's Concise Guide to PHARMACOLOGY (Alexander et al., 2013).
Results
Initial screen of novel MZs reveals neuroprotection with a variable sensitivity to PTX blockade
CMZ has been shown to be associated with a sedative effect at 100 μM in vivo, while in vitro studies show neuroprotection against OGD at doses as low as 10 μM (Green et al., 2000). Consistent with these findings, we have found that CMZ at 50 μM consistently demonstrates ∼125% cell viability over vehicle against OGD with no associated toxicity in primary cortical culture models (Supporting Information Figure S1A). Using this approach, a small, focused library of over 40 MZs that preserved the 4-methylthiazole ring of CMZ was designed and synthesized, and limited structure activity relationships were explored (Qin et al., 2012). For further investigation, eight compounds were selected from our novel library that sampled the pharmacophore space (Figure 1A) and were neuroprotective in the OGD model with equal or greater efficacy to CMZ (Figure 1B). Co-treatment with PTX (100 μM), a GABAA chloride channel blocker, revealed varied dependence on the GABAA receptor at a dose known to completely inhibit activity (Olsen, 1982); for example, MZs GN-38 and GN-46 maintained significant neuroprotection relative to control in the presence of PTX, while the neuroprotective action of GN-28 and GN-12 was almost completely abolished.
Figure 1.

MZs protect neurons from OGD with variable dependence on GABAA receptor. (A) Neuroprotection of MZs was demonstrated in primary cortical cultures against OGD. OGD was maintained for 2 h with (+) or without (−) 100 μM PTX added 1 h prior to start of OGD. MZs were added at initiation of OGD period (50 μM), and after 24 h survival were measured with MTT assay normalized to vehicle (0%) and CMZ (100%). (B) Extracellular glutamate was measured in above cortical cultures after 24 h of exposure to OGD by pre-column derivatization with OPA and fluorescent detection by HPLC, with results normalized to vehicle control (100%). All data show mean and SEM, with statistical significance determined by one-way anova and post hoc Dunnett's MCT comparing to vehicle control exposed to insult: *P < 0.05, **P < 0.01, ***P < 0.001. Data were obtained from at least six replicates from one plate for each experimental condition.
Extracellular glutamate is recognized as a marker of excitotoxicity in tissues and cell cultures. Fluorescence detection of glutamate after separation with HPLC and pre-column derivatization with OPA was used to measure the concentration of extracellular glutamate in culture supernatant 24 h after initiation of OGD, normalizing to in-plate vehicle controls. The concentration of glutamate in cultures not exposed to OGD was below the threshold of quantification (∼50 nM per 2 × 105 cells); however, OGD caused significant elevation of extracellular glutamate level, which was reduced by CMZ and five MZs, with GN-28 showing the greatest reduction and GN-38 showing the least effect on extracellular glutamate (Figure 1C). On the basis of these results, two novel MZs, GN-28 and GN-38, were selected for more in-depth study as representatives of neuroprotective CMZ derivatives with greater and lesser dependence on GABAA receptors respectively. Toxicity assay showed that after 24 h, GN-28 and GN-38 had a negligible effect on cell survival at doses of 100 nM–100 μM (Supporting Information Figure S1C), results consistent with previous observations on CMZ; and furthermore, no artefactual interaction was found between these MZs and MTT (Supporting Information Figure S1B).
GN-28 and GN-38 showed neuroprotection in the OGD model of stroke, with opposite sensitivity to PTX blockade
GN-28 and GN-38 elicited survival of primary rat neuronal cell cultures 24 h after OGD when measured by MTT and normalized to CMZ (100% ± 7.7) and vehicle controls (0% ± 6.3): GN-28 provided increased survival over that of CMZ (160.8% ± 15.7; P < 0.01), attenuated by PTX (29.1% ± 22.3; P < 0.01); while GN-38 resulted in survival equal to that of CMZ (133.5% ± 15.2; P > 0.05), which was not significantly affected by PTX (96.8% ± 18.5; P > 0.05) (Figure 2A). These observations were extended to obtain concentration–response curves for GN-28 and GN-38 (100 nM–100 μM) for cellular protection after OGD, demonstrating a similar and approximately linear dependence of cell viability on concentration (Figure 2B).
Figure 2.
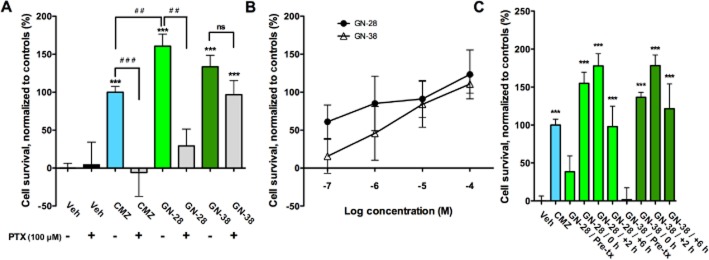
GN-28 and GN-38 protect neurons from OGD with variable dependence on GABAA receptor. (A) Neuroprotection of MZs was demonstrated in primary cortical cultures against OGD, with efficacy significantly attenuated by GABAA receptor blockade for CMZ and GN-28, while GN-38 remained neuroprotective without significant dependence on GABAA receptor. OGD was maintained for 2 h with (+) or without (−) 100 μM PTX added 1 h prior to start of OGD. MZs were added at initiation of the OGD period, and after 24 h survival was measured with MTT assay normalized to vehicle (0%) and CMZ (100%). (B) Concentration–response relationships for neuroprotection in OGD by GN-28 and GN-38, added at initiation of OGD period (0 h). (C) Effect of time of administration on neuroprotective effect in OGD for GN-28 and GN-38. No significant neuroprotective effect was seen after pretreatment of cell cultures with GN-28 or GN-38 (50 μM). However, neuroprotection was significant for both treatments when added any time from 0 to 6 h after start of OGD. All data show mean and SEM, with statistical significance determined in panels A and C by one-way anova and post hoc Bonferroni's MCT: *** P < 0.001 compared to first vehicle control; ## P < 0.01, ###P < 0.001 compared between marked groups; ns = not significant. Data for panels A–C were obtained from at least six replicates from one 96-well plate for each experimental condition.
OGD is a model of ischaemia-reperfusion injury. The mechanisms of cell death elicited during cellular hypoxia and ischaemia are not identical to those caused by reoxygenation and subsequent apoptotic and inflammatory mechanisms. Consequently, drug treatment during and after ischaemia can have varied outcomes. Pre-conditioning provides a further array of mechanisms for protection against subsequent insult. To study the role of drugs in each stage of OGD damage, GN-28 or GN-38 was added at four different timepoints: (i) 1 h pretreatment with removal of drug at commencement of OGD (pre-tx); (ii) immediately at the start of OGD (+0 h); (iii) immediately after the 2 h OGD period (+2 h); and (iv) 6 h after start of OGD (+6 h). OGD was transient for 2 h in each paradigm. Both GN-28 and GN-38 showed a similar protection profile, with no effect seen on pretreatment, but neuroprotection was seen for both GN-28 and GN-38 even up to 6 h after initiation of OGD and 4 h after oxygen/glucose reperfusion (Figure 2C).
Reductions in extracellular glutamate and intracellular calcium correlate with PTX sensitivity in OGD.
As introduced above, extracellular glutamate was measured in pooled supernatants after exposure to OGD. The attenuation of glutamate release, normalized to in-plate vehicle control (100% ± 3.4), was for CMZ and GN-28, 76.9% ± 5.2 and 47.5% ± 7.1, respectively, and was greater for GN-28 over CMZ (P < 0.01). Although GN-38 was neuroprotective against OGD, attenuation of glutamate release was not significant (91.9% ± 0.8; P > 0.05), compatible with the absence of significant blockade by PTX (Figure 3A,B). Increased intracellular calcium levels are associated with apoptosis after excitotoxic insult. Accordingly, we tested for an effect of these compounds on intracellular compounds using a fluorescent dye reporter in cortical cultures after exposure to 1 mM of glutamate. Paralleling the results on extracellular glutamate, CMZ and GN-28 showed significant reduction in intracellular calcium increase over baseline compared with vehicle control (113.4% ± 8.1, P < 0.05; and 106.3% ± 8.4, P < 0.01; vs. 136.2% ± 8.3), while reduction by GN-38 did not reach significance (119.4 ± 15.4, P > 0.05) (Figure 3C,D).
Figure 3.
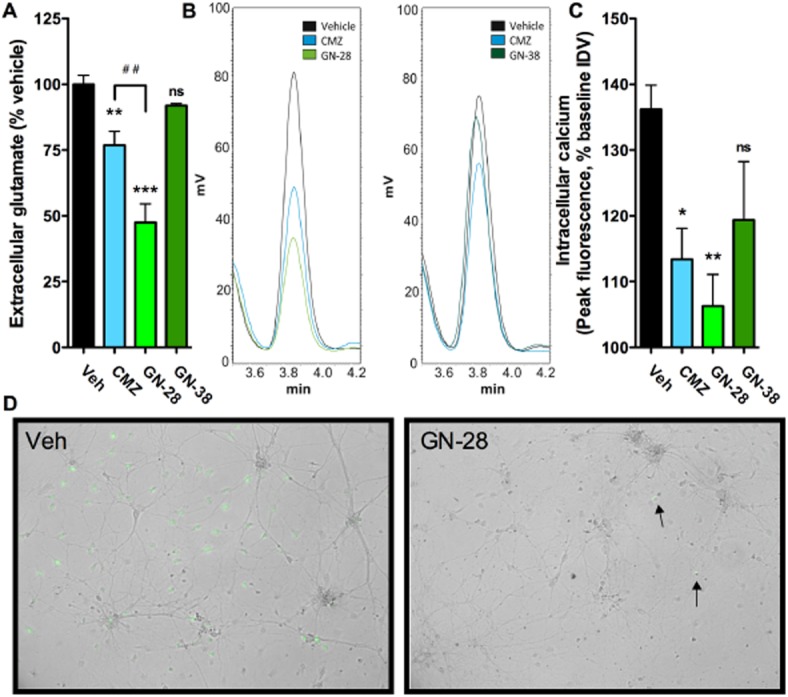
GN-28 significantly reduces extracellular glutamate release and intracellular calcium after excitotoxic insult, while GN-38 does not. (A,B) Extracellular glutamate concentration was significantly attenuated compared with vehicle control by CMZ and GN-28, but not by GN-38. GN-28 showed significantly more reduction than CMZ. Glutamate was measured from supernatants pooled from at least six replicates from one 96-well plate for each experimental group and assayed at least in triplicate by HPLC-UV after pre-column derivatization with OPA, which represents peaks shown in (B). (C,D) An increase in intracellular calcium was significantly reduced by CMZ and GN-28 after application of 1 mM glutamate, but not by GN-38. Intracellular calcium was measured using a fluo-4 dye reporter with fluorescence after glutamate addition normalized to baseline levels, and peak fluorescence reported. Measurements were taken in three independent cultures in 29 mm glass bottom dishes for each experimental group, with change in fluorescence shown in representative experiments after subtraction of baseline fluorescence and merged with bright field images in [D: compared with vehicle control, after treatment with GN-28, only a few cells showed substantial change in calcium-dependent fluorescence (arrows)]. All data show mean and SEM, with statistical significance determined in panels (A) and (C) by one-way anova and post hoc Bonferroni's MCT: * P < 0.05, ** P < 0.01, *** P < 0.001, compared to vehicle control; ## P < 0.01, compared between marked groups; ns = not significant.
GN-28 and GN-38 show protection against direct excitotoxins with similar sensitivity to PTX blockade as in OGD
As mentioned previously, the OGD model incorporates a relatively complex pathophysiology of insult, which includes reperfusion injury and disrupted energy homeostasis. To compare the results from OGD and test the direct ability of compounds to prevent cell death in a model with more limited oxidative and metabolic disruption but sustained excitotoxicity, cell survival was measured for each compound at 50 μM using the MTT assay 24 h after addition of either 1 mM glutamate or 100 μM NMDA, and results were normalized to vehicle controls either subjected to excitotoxic insult (0%) or no insult (100%). These concentrations were shown to result in reproducible and significant cell death after 24 h (Supporting Information Fig. S1d).
Against 1 mM of glutamate, GN-28 showed a comparable behaviour to CMZ, eliciting significant neuroprotection (53.4% ± 6.6, P < 0.001; 47.3% ± 7.1, P < 0.01 respectively) (Figure 4A). The neuroprotective effect of both agents was reduced by PTX blockade with no difference over vehicle-treated control (GN-28 −19.5% ± 4.6, P > 0.05; CMZ 4.5% ± 9.6, P > 0.05). GN-38, in contrast, was neuroprotective against glutamate toxicity (41.3% ± 9.6, P < 0.01), and even in the presence of PTX cell viability was greater than vehicle control (37.4% ± 12.2, P < 0.05). Concentration–response curves for GN-28 and GN-38 (100 nM–100 μM) showed an approximately linear response within the concentrations tested, with no difference between the two agents (Figure 4B): an IC50 of 2.8–3.5 μM was estimated from curve fitting. The pattern of results in response to NMDA neurotoxicity was qualitatively similar to that observed for glutamate toxicity (Figure 4C) with respect to the dependence of neuroprotection on the GABAA receptor. The concentration–response curves revealed attenuated efficacy of both neuroprotective agents and again no difference was observed between the two novel MZ derivatives (Figure 4D).
Figure 4.
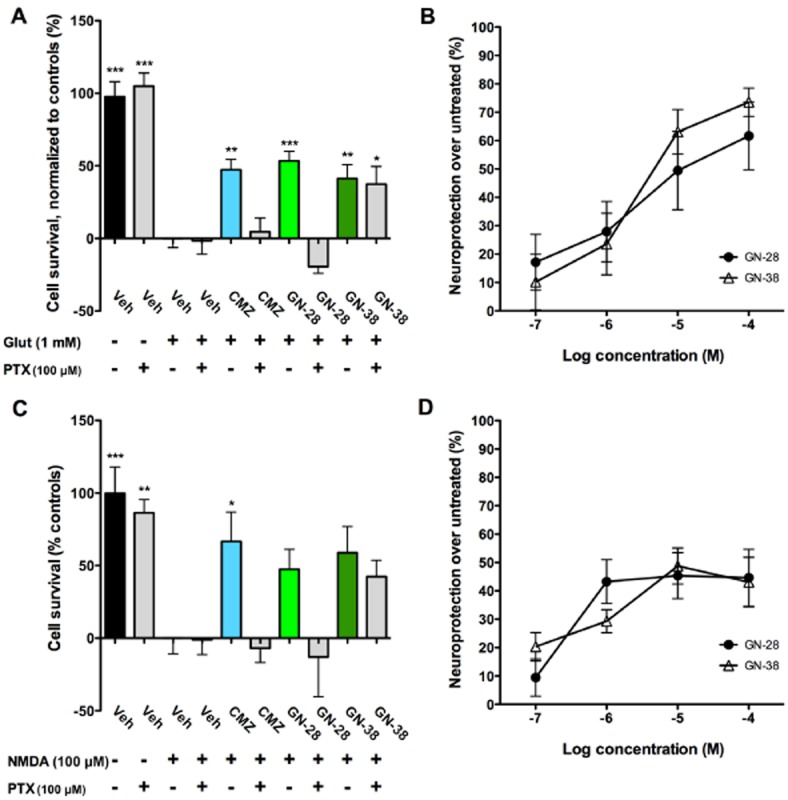
MZs protect neurons against excitotoxic insult with variable dependence on GABAA receptor. (A) Primary cortical cultures were subjected to glutamate-induced (1 mM) excitotoxicity with survival measured by MTT 24 h after start of insult and normalized to vehicle controls with (+, 0%) or without glutamate (−, 100%). Neuroprotection determined with CMZ and GN-28 (50 μM) was attenuated by addition of PTX (100 μM), but GABAA receptor blockade did not significantly decrease the efficacy of GN-38. (B) Concentration–response relationships for neuroprotection against 1 mM glutamate by GN-28 and GN-38, added 1 h after glutamate. (C) Primary cortical cultures were subjected to NMDA-induced (100 μM) excitotoxicity with survival measured by MTT 24 h after start of insult and normalized to vehicle controls with (+, 0%) or without glutamate (–, 100%). Neuroprotection was only significant for CMZ, but a trend towards a decreased effect was observed for CMZ and GN-28 (50 μM) after GABAA blockade with PTX (100 μM), no significant effect on neuroprotection was observed for GN-38. (D) Concentration–response relationships for neuroprotection against 100 μM NMDA by GN-28 and GN-38, added 1 h after NMDA. All data show mean and SEM, with statistical significance determined in panels (A) and (C) by one-way anova and post hoc Dunnett's MCT compared to vehicle control exposed to insult: * P < 0.05, ** P < 0.01, *** P < 0.001. Data for panels (A)–(D) were obtained from at least six replicates from one 96-well plate for each experimental condition.
GN-28 potentiates the α1β2γ2 GABAA receptor while GN-38 is weakly inhibitory
Application of GN-28 and GN-38 to α1β2γ2 GABAA–receptor expressing oocytes was assayed in the presence of GABA to confirm the ability of GN-28 to modulate GABAA receptor-mediated ion channels and to investigate any effect of GN-38 on this receptor isoform. GN-28 exhibited a dose-dependent potentiating effect on membrane currents elicited by 3 μM GABA (∼EC6; Figure 5A). In the absence of GABA, GN-28 did not elicit a measurable response, and in addition, GN-28-potentiated GABA responses were eliminated by PTX: 200 μM PTX inhibited >99% of the GABA response potentiated by 100 μM GN-28 (n = 7, not shown). In contrast, GN-38 did not exhibit any potentiation at this GABAA receptor (Figure 5B). On the contrary, normalized response amplitudes obtained with GN-38 and 3 μM GABA exhibited, on average, a modest decline with increasing GN-38 concentration (Figure 5C, open triangles). The concentration response obtained for potentiation of GABA response by GN-28 demonstrated that at 50 μM GN-28, a ∼2.5-fold increase of the response amplitude was elicited (Figure 5C, filled circles), compatible with the PTX-sensitive neuroprotection observed in primary neuronal cell cultures at the same concentration of GN-28.
Figure 5.
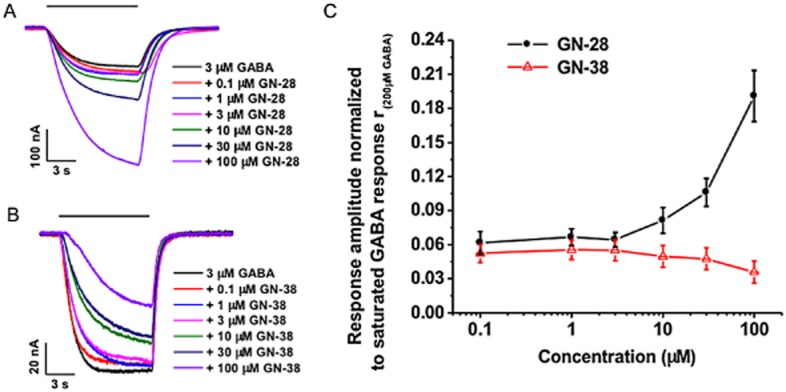
GN-28 shows potentiation at α1β2γ2 GABAA receptor expressing oocytes, while GN-38 is weakly inhibitory. (A) Representative waveforms of the responses to the application of 3 μM GABA supplemented with varying concentrations of GN-28. (B) Representative waveforms of the responses to the application of 3 μM GABA supplemented with varying concentrations of GN-38. (C) Dose-response curve for 3 μM GABA supplemented with increasing concentrations of GN-28 and GN-38. Data normalized to the saturated 200 μM GABA response. Data for GN-28 obtained from four oocytes. Data for GN-38 obtained from seven oocytes. All data are shown as mean ± SD.
Protection against oligomeric Aβ suggests different mechanisms of insult at two doses, with variable dependence on GABAA receptor blockade
To extend observations to an in vitro model of neuroprotection relevant to the amyloid-β hypothesis of AD aetiology, MZ derivatives were tested against neurotoxicity resulting from application of oligomeric human Aβ1–42 (oAβ). The exact nature of the insult by oAβ is not completely understood. However, neurotoxicity mediated through direct action at NMDA receptors has been hypothesized (De Felice et al., 2007). oAβ also increases glutamate release, disrupts calcium ion homeostasis, and leads to apoptosis, potentially via direct activation of a glutamate-mediated pathway leading to neuronal loss (Mattson et al., 1992; Brito-Moreira et al., 2011). Therefore, it was reasonable to hypothesize MZ derivatives exhibiting neuroprotection against OGD and direct excitotoxins might have the potential to protect neurons against an insult induced by oAβ. However, to observe neurotoxicity caused by oAβ1–42 in vitro, supraphysiological concentrations are routinely employed. Even in primary neuronal cultures, levels of cell death caused by oAβ1–42 are modest (Supporting Information Figure S1D). Some authors have presented more complex pictures of oAβ activity, where at moderate doses, excitotoxic effects at the network level predominate, whereas at higher doses direct effects on the cell are observed (Mucke and Selkoe, 2012). Therefore, two models were developed for use in rat primary neuronal cultures, both using application of soluble oligomeric Aβ1–42, widely held to be the primary neurotoxic form of Aβ (Lambert et al., 1998; Haass and Selkoe, 2007; Yu et al., 2010), using either a moderate (250 nM) or high (5 μM) concentration of oAβ.
Results were normalized to vehicle controls either subjected to insult (0%) or no insult (100%). At the high oAβ dose, only CMZ and GN-38 treatment elicited neuroprotection compared with vehicle (73.1% ± 14.1, P < 0.001; 71.3% ± 7.5, P < 0.001, respectively), while GN-28 showed no response (−8.5% ± 12.7, P > 0.05) (Figure 6A). PTX co-treatment blocked the effect of CMZ (26.6% ± 9.8, P > 0.05), but neuroprotection by GN-38 was insensitive (67.7% ± 28.9, P < 0.01). Response curves for neuroprotection by GN-28 and GN-38 (100 nM–100 μM) showed a concentration-dependence for both compounds. However, the effects of GN-28 against high-dose oAβ were only significant at the highest concentration studied (Figure 6B). Against the moderate dose of oAβ, all MZs were observed to provide significant neuroprotection relative to the vehicle control, and, unlike other models, each compound was sensitive to PTX co-treatment (Figure 6C). Overall, these results are consistent with a role for excitotoxicity in the presence of oligomeric Aβ, and the capacity of MZ derivatives to provide neuroprotection against this insult.
Figure 6.
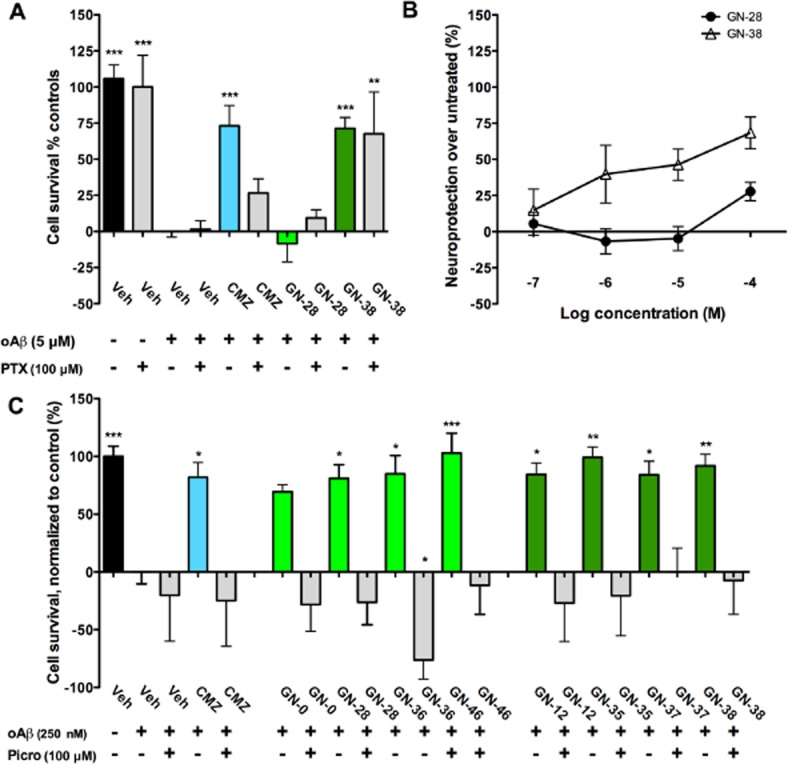
MZs protect neurons against 250 nM oligomeric Aβ with dependence on GABAA receptor, while protection against 5 μM oligomeric Aβ by CMZ and GN-38 requires GABAA-independent activity. (A) Primary cortical cultures were subjected to toxicity induced by oligomers of Aβ1–42 (5 μM) with survival measured by MTT 4 days after start of insult and normalized to vehicle controls with (+, 0%) or without oAβ (–, 100%). Neuroprotection seen with CMZ and GN-38 (50 μM) was attenuated by addition of 100 μM PTX (+), while GN-28 did not show significant neuroprotection at this dose. (B) Concentration–response relationships for neuroprotection against 5 μM oAβ by GN-28 and GN-38, added 1 h after oAβ. (C) Primary cortical cultures were subjected to toxicity induced by oAβ (250 μM) with survival measured by MTT 4 days after start of insult and normalized to vehicle controls with (+, 0%) or without oAβ (–, 100%). Neuroprotection demonstrated by all MZs (50 μM) was attenuated by addition of 100 μM PTX (+). All data show mean and SEM, with statistical significance determined in panels (A) and (C) by one-way anova and post hoc Dunnett's MCT compared to vehicle control exposed to insult: * P < 0.05, ** P < 0.01, *** P < 0.001. Data for panels (A)–(C) were obtained from at least six replicates from one 96-well plate for each experimental condition.
Both GN-28 and GN-38 show CNS bioavailability by LC-MS/MS
An important consideration in advancing a neuroprotective compound to in vivo studies is whether the compound crosses the blood–brain barrier to provide sufficient brain bioavailability. Therefore, for GN-28 and GN-38, CNS bioavailability was evaluated in male C57BL/6 mice at 8–10 weeks of age after i.p. administration, using solution-phase extraction from plasma and brain after perfusion and subsequent detection by LC-MS/MS. Compounds (4.45 μmol·kg−1) injected 20 min before collection of plasma and brain tissue were identified by LC-MS/MS and quantified using internal standards that were added prior to liquid extraction (see Supporting Information for details). Both MZ derivatives crossed the blood–brain barrier, with a superior brain/plasma ratio for GN-28, but with a higher concentration of free drug observed in the brain of animals treated with GN-38 ( Table 2011). Further metabolism and protein binding studies were not conducted at this stage; however, both MZ derivatives demonstrated brain bioavailability.
Table 1.
Plasma and brain concentration 20 min after administration
| Analyte | Plasma (ng·mL−1) | Brain (ng·mL−1) | [Brain]/[Plasma] |
|---|---|---|---|
| GN-28 | 18.4 ± 0.8 | 32.9 ± 3.7 | 1.8 |
| GN-38 | 559 ± 24 | 56.4 ± 5.5 | 0.1 |
Quantified using LC-MS/MS after i.p. administration of equimolar doses.
Data are expressed as the mean ± SEM (n = 4).
Discussion
Clinical agents are needed to treat a variety of intractable neurodegenerative disorders including AD and stroke. However, neuroprotective agents have yet to show sufficient clinical efficacy to support therapeutic application despite numerous promising mechanisms. One oft-proposed mechanism to block the resulting apoptotic neuronal death in the penumbra after ischaemic stroke is selective activation of inhibitory GABAA receptors to allow compensatory chloride ion influx causing a so-called voltage ‘clamp’ at lower resting potentials (Rudolph and Knoflach, 2011), which would inhibit both excessive neuronal firing and overactivation of NMDA receptors, leading to decreased glutamate release, reduced intracellular calcium and thus reduced apoptosis. For AD, the aetiology of the initial insult remains a matter of debate, as does the exact nature of the neurotoxicity caused by oligomeric Aβ1–42. However, excitotoxicity occurs in the human AD brain, evidenced strongly in the reduced threshold for seizure activity, and selective activation of inhibitory GABAA receptors has been proposed as a target for AD therapeutics (Vellas et al., 2011; Limon et al., 2012).
CMZ, shown extensively to be neuroprotective in animal models of ischaemic infarct (Marshall et al., 2000), continues to be suggested as a potential component of future combination therapies for neuronal injury (Hankey, 2002; Wilby and Hutchinson, 2004). CMZ is primarily understood to act as a GABAA receptor potentiator, independent of the benzodiazepine site (Usala et al., 2003), and potential applications for GABAergic compounds have been proposed in diseases as diverse as stroke, AD, schizophrenia, depression, and analgesia, complimenting their current utility as anticonvulsants, anxiolytics and sedative/hypnotics (Mohler, 2011; Rudolph and Knoflach, 2011). Intriguingly, CMZ has also been reported to inhibit pro-inflammatory pathways associated with TNF-α (Harmon et al., 2003; Clarkson et al., 2005) and to rescue mitochondrial function in brain tissues (Clarkson et al., 2007). These observations further stimulate interest in MZ derivatives, because recent evidence supports inhibition of TNF-α and restoration of mitochondrial function as therapeutic targets in AD (Alvarez et al., 2007; Strum et al., 2007; McAlpine et al., 2009; Santos et al., 2011; Shi et al., 2011).
CMZ, an effective therapeutic currently used in the clinic, represents a lead compound for anti-neurodegenerative drug discovery, targeting a combination of mechanisms proposed to combat multifactorial neuronal insults. In the present work, CMZ provided significant neuroprotection in four in vitro models of neurodegeneration at doses that are pharmacologically relevant in vivo (Cross et al., 1995; Green et al., 2000). In initial screening for neuroprotection, a library of CMZ analogues and MZ derivatives was observed to show subtle relationships between structure and activity: for example, the 2-pyridyl isomer of GN-12 was significantly less active than GN-12 (Qin et al., 2012). A number of MZ derivatives were observed to have significantly greater efficacy than CMZ as neuroprotectants, and one objective of the present work was to examine the dependence on the GABAA receptor for CMZ and selected MZ neuroprotective agents in greater detail. GN-28 was found to equal or exceed the activity of CMZ in all models in which excitotoxicity was expected to provide the major contribution, and, in these models, the actions of both GN-28 and CMZ were sensitive to GABAA blockade. Nevertheless, GN-38 provided comparable levels of neuroprotection to GN-28, despite insignificant effects on glutamate release and intracellular calcium, with an effect largely independent of the GABAA receptor. A possible explanation for these results would be changes in binding affinity at the GABAA receptor, with GN-28 showing enhanced binding over CMZ, and GN-38 either losing binding affinity or losing potentiating activity at the receptor itself, but formal binding studies and other pharmacokinetic work have yet to be completed for all receptor subtypes, so this hypothesis remains speculative.
Measurement of ion currents through isolated GABAA receptors provides a definitive measure of GABAA activity. In accord with observations using PTX, GN-28 was confirmed to be an α1β2γ2 GABAA receptor potentiator, amplifying the GABA response ∼2.5-fold at the 50 μM concentration used in neuronal cell cultures. GN-28, like CMZ, was not found to be a direct agonist at α1β2γ2 GABAA at the concentrations studied. These data strongly support the hypothesis that GN-28 and CMZ are achieving neuroprotection through at GABAA receptor potentiation. Conversely, again in accord with observations using PTX, GN-38 showed neither agonist nor GABA potentiating activity at the α1β2γ2 GABAA receptor, supporting the hypothesis of substantially reduced activity at least at this receptor subtype, making an alternative mechanism for its activity much more likely in excitotoxic models.
CMZ was neuroprotective in primary cultures treated with both high and moderate doses of oAβ, and, at the moderate dose of oAβ (250 nM), both GN-28 and GN-38 provided almost complete neuroprotection. Importantly, GN-38 provided neuroprotection against high doses of oAβ even in the presence of GABAA receptor blockade, while GN-28 failed to show substantial neuroprotection except at the 100 μM dose, suggesting the GABAA independent mechanism is of more importance at higher doses of oAβ. Against more moderate doses of oAβ that result in only a small amount of cell death (Supporting Information Figure S1d), PTX blockade abolishes activity for all compounds studied, thus GABAA activity is necessary and sufficient for neuroprotection against this type of insult, which may more closely correlate to levels of oAβ found early in the AD brain. Interestingly, this experiment provides the sole evidence that GN-38 may still retain some activity at the GABAA receptor, while also showing that the unknown GABAA-independent activity is not sufficient for protection against this insult. Future electrophysiological studies in other receptor subtypes may help elucidate the GABAA receptor profile of these novel compounds, and planned screening against known drug targets will help determine the nature of the GABA-independent activity demonstrated most strongly by GN-38.
Disorders in which neuronal loss is a major pathophysiological event are likely to gain benefit from treatment with neuroprotective agents. Increased activity at the GABAA receptor is recognized as an unharnessed neuroprotective mechanism in stroke; and in AD, excitotoxicity, increased seizure activity and Aβ-induced GABAA receptor dysfunction commend non-benzodiazepine GABAA receptor potentiators as neuroprotective therapeutic agents (Rissman and Mobley, 2011; Vellas et al., 2011). Herein, novel, neuroprotective MZ derivatives, based upon the clinical sedative, anxiolytic agent, CMZ, are reported to provide neuroprotection in four different in vitro models, relevant to neurodegenerative disorders, including stroke and AD. Our observations support the concept of dual mechanisms of action for CMZ that are represented differentially in GN-28 and GN-38. Whereas the GABAA-dependent activity proves successful against a wide range of excitotoxic insults, contribution from the GABAA-independent mechanism would seem to be essential to provide protection against cell death induced by high-dose oAβ. The GABAA-dependent activity is increased in GN-28, while the GABAA-independent mechanism appears to predominate in the structure of GN-38. Finally, development as therapeutics is promising as GN-28 and GN-38 were both observed to remain protective even when administered 6 h after OGD insult, to cross the blood–brain barrier, and to have significantly attenuated sedative activity in vivo compared with CMZ (Supporting Information Figures S5 and S6). Further studies, including binding assays and a screen against known drug targets, of these GABAA-dependent and GABAA-independent neuroprotectants would expand our understanding of the mechanism and facilitate their development as therapeutics.
Acknowledgments
The present work was funded by NIH grants AG031294, EY016094 and EY001792; by the Arnold and Mabel Beckman Initiative for Macular Research (Los Angeles, CA); by Research to Prevent Blindness, Inc. (New York, NY); and by award UL1RR029879 from the University of Illinois at Chicago Center for Clinical and Translational Sciences (CCTS).
Glossary
- AD
Alzheimer's Disease
- CMZ
chlormethiazole (also clomethiazole)
- DIV
days in vitro
- MTT
thiazolyl blue tetrazolium bromide
- MZ
methiazole (4-methylthiazole)
- oAβ
oligomeric amyloid-β
- OGD
oxygen-glucose deprivation
- OPA
o-phthalaldehyde
- PTX
picrotoxin
- THF
tetrahydrofuran
Conflict of interest
G. R. T. is a scientific adviser for sGC Pharma, Incorporated, that holds a license for the compounds described.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12454
Figure S1 (A) CMZ showed substantial, reproducible neuroprotection compared with untreated vehicle controls in OGD. Data shown represent mean and SEM of over 30 independent experiments, with analysis by two-tailed Student's t-test. (B) No effect was seen when compounds were incubated for 24 h with MTT in growth media without cells, suggesting no artefactual reducing interaction between drugs and MTT. (C) Incubation of compounds (50 μM) in primary cortical cultures for 24 h did not result in significant protection or toxicity at any dose studied. (D) After treatment with various insults using protocols described in the text, the amount of cell death measured by MTT and normalized to untreated vehicle controls was reproducible and significant. All data from panels B.D show mean and SEM, with statistical significance determined in panels B and D by use of one-way ANOVA and post hoc Dunnett's MCT compared to vehicle control: *P < 0.05, ***P < 0.001; ns, not significant. Data for panels B.D were obtained from at least six independent replicates for each experimental condition.
Figure S2 Calibration curves were constructed for GN-28 and GN-38 in plasma spiked with standard solutions and internal standard (GN-27 and F-12 respectively). Final concentrations of analyte ranged from 1 to 100 ng EmL.1. The equation for the regression line was used to quantify analytes in plasma samples.
Figure S3 Total ion chromatograms (TIC) of GN-28 in mouse plasma 20 min after i.p. injection [GN-27 was used as an internal standard (IS)]; and of GN-38 in mouse plasma 20 min after i.p. injection [F-12 was used as an internal standard (IS)]. LC-MS/MS chromatograms showing fragmentation used for MRM quantification of analytes relative to internal standards: Transition MRM m/z 220→189 and m/z 238→207 were used to detect GN-28 and IS, respectively; transition MRM m/z 288→147 and m/z 352→147 were used to detect GN-38 and IS respectively.
Figure S4 TIC and MRM chromatograms of GN-28 and GN-38 in mouse brain 20 min after i.p. injection and spiking with internal standards.
Figure S5 CMZ was applied (i.p.) at 30, 40, 50 mg·kg-1. Mean latency to fall was determined at 30 and 60 min post injections. The data represent the average time (s) animals stayed on the rod before and after treatments. CMZ decreased the latency to fall in a dose-response fashion. CMZ (50 mg·kg-1) caused significant sedation, which translated to a great loss in animal balance when compared with pretreatment mean latency (***P < 0.001, n = 5). This effect faded away after 60 min. Vehicle group was injected with the same vehicle used to prepare i.p. injections. Solvent was composed of: water 90% (v v-1), DMSO 10% (v v-1). Vehicle did not show any significant effect on the mean latency to fall.
Figure S6 The accelerated rotarod performance task was performed with: CMZ (45 mg·kg-1, i.p.); GN-28 (equimolar dose, 59 mg·kg-1, i.p.); or GN-38 (equimolar dose, 64 mg·kg-1, i.p.). Mean latency to fall was determined at 10, 30 and 60 min post injections. The data represent average time (s) animals stayed on the rod after drug treatments. Vehicle, composed of water 90% (v v-1), DMSO 10% (v v-1), did not show any significant effect on the mean latency to fall. Bars represent mean and SEM, analysed by two-way repeated measures ANOVA with Bonferroni's post-test comparing with vehicle control: **P < 0.01, *** P < 0.001 (n = 5 for each group).
Table S1 Retention times, coefficients of determination (R2) of calibration curves and extraction recoveries for the different compounds.
References
- 1.Abdelhamid R, Luo J, VandeVrede L, Kundu I, Michalsen B, Litosh VA, et al. Benzothiophene selective estrogen receptor modulators provide neuroprotection by a novel GPR30-dependent mechanism. ACS Chem Neurosci. 2011;2:256–268. doi: 10.1021/cn100106a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander SPH, et al. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013;170:1449–1867. doi: 10.1111/bph.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alvarez A, Cacabelos R, Sanpedro C, Garcia-Fantini M, Aleixandre M. Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol Aging. 2007;28:533–536. doi: 10.1016/j.neurobiolaging.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 4.Baldwin HA, Williams JL, Snares M, Ferreira T, Cross AJ, Green AR. Attenuation by chlormethiazole administration of the rise in extracellular amino acids following focal ischaemia in the cerebral cortex of the rat. Br J Pharmacol. 1994;112:188–194. doi: 10.1111/j.1476-5381.1994.tb13050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bengtsson S, Lindberg UH. Compounds related to clomethiazole. VI. Synthesis of some reference compounds in connection with biotransformation studies. Acta Pharm Suec. 1982;19:37–42. [Google Scholar]
- 6.Brito-Moreira J, Paula-Lima AC, Bomfim TR, Oliveira FB, Sepulveda FJ, De Mello FG, et al. Abeta oligomers induce glutamate release from hippocampal neurons. Curr Alzheimer Res. 2011;8:552–562. doi: 10.2174/156720511796391917. [DOI] [PubMed] [Google Scholar]
- 7.Clarkson AN, Clarkson J, Jackson DM, Sammut IA. Mitochondrial involvement in transhemispheric diaschisis following hypoxia-ischemia: clomethiazole-mediated amelioration. Neuroscience. 2007;144:547–561. doi: 10.1016/j.neuroscience.2006.09.040. [DOI] [PubMed] [Google Scholar]
- 8.Clarkson AN, Liu H, Rahman R, Jackson DM, Appleton I, Kerr DS. Clomethiazole: mechanisms underlying lasting neuroprotection following hypoxia-ischemia. FASEB J. 2005;19:1036–1038. doi: 10.1096/fj.04-3367fje. [DOI] [PubMed] [Google Scholar]
- 9.Colado MI, O'Shea E, Esteban B, Green AR. Studies on the neuroprotective effect of the enantiomers of AR-A008055, a compound structurally related to clomethiazole, on MDMA (‘ecstasy’)-induced neurodegeneration in rat brain. Psychopharmacology (Berl) 2001;157:82–88. doi: 10.1007/s002130100762. [DOI] [PubMed] [Google Scholar]
- 10.Cross AJ, Jones JA, Snares M, Jostell KG, Bredberg U, Green AR. The protective action of chlormethiazole against ischaemia-induced neurodegeneration in gerbils when infused at doses having little sedative or anticonvulsant activity. Br J Pharmacol. 1995;114:1625–1630. doi: 10.1111/j.1476-5381.1995.tb14949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cross AJ, Stirling JM, Robinson TN, Bowen DM, Francis PT, Green AR. The modulation by chlormethiazole of the GABAA-receptor complex in rat brain. Br J Pharmacol. 1989;98:284–290. doi: 10.1111/j.1476-5381.1989.tb16893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, et al. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- 13.De Keyser J, Sulter G, Luiten PG. Clinical trials with neuroprotective drugs in acute ischaemic stroke: are we doing the right thing? Trends Neurosci. 1999;22:535–540. doi: 10.1016/s0166-2236(99)01463-0. [DOI] [PubMed] [Google Scholar]
- 14.Farooque M, Isaksson J, Jackson DM, Olsson Y. Clomethiazole (ZENDRA, CMZ) improves hind limb motor function and reduces neuronal damage after severe spinal cord injury in rat. Acta Neuropathol (Berl) 1999;98:22–30. doi: 10.1007/s004010051047. [DOI] [PubMed] [Google Scholar]
- 15.Gladstone DJ, Black SE, Hakim AM. Toward wisdom from failure: lessons from neuroprotective stroke trials and new therapeutic directions. Stroke. 2002;33:2123–2136. doi: 10.1161/01.str.0000025518.34157.51. [DOI] [PubMed] [Google Scholar]
- 16.Green AR. Clomethiazole (Zendra) in acute ischemic stroke: basic pharmacology and biochemistry and clinical efficacy. Pharmacol Ther. 1998;80:123–147. doi: 10.1016/s0163-7258(98)00024-2. [DOI] [PubMed] [Google Scholar]
- 17.Green AR, Hainsworth AH, Jackson DM. GABA potentiation: a logical pharmacological approach for the treatment of acute ischaemic stroke. Neuropharmacology. 2000;39:1483–1494. doi: 10.1016/s0028-3908(99)00233-6. [DOI] [PubMed] [Google Scholar]
- 18.Green AR, Hainsworth AH, Misra A, Debens TA, Jackson DM, Murray TK, et al. The interaction of AR-A008055 and its enantiomers with the GABA(A) receptor complex and their sedative, muscle relaxant and anticonvulsant activity. Neuropharmacology. 2001;41:167–174. doi: 10.1016/s0028-3908(01)00053-3. [DOI] [PubMed] [Google Scholar]
- 19.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 20.Hankey GJ. Clomethiazole: an unsuccessful bachelor, but perhaps a prosperous married man? Stroke. 2002;33:128–129. [PubMed] [Google Scholar]
- 21.Harmon D, Coleman E, Marshall C, Lan W, Shorten G. The effect of clomethiazole on plasma concentrations of interleukin-6, -8, -1beta, tumor necrosis factor-alpha, and neutrophil adhesion molecule expression during experimental extracorporeal circulation. Anesth Analg. 2003;97:13–18. doi: 10.1213/01.ane.0000063821.60233.7d. [DOI] [PubMed] [Google Scholar]
- 22.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lechat P, Streichenberger G, Boime A, Lemeignan M. Relation between chemical structure and physiological activity of certain thiazole derivatives. I. 4-Methyl-5-(b-hydroxyethyl)thiazole. Ann Pharm Franc. 1965a;23:179–186. [PubMed] [Google Scholar]
- 24.Lechat P, Streichenberger G, Boime A, Lemeignan M. Relations between the chemical structure and physiological activity of certain thiazole derivatives. II. Effect of the length of the alkyl chain in 4-methyl-5-(w-chloroalkyl)thiazoles. Ann Pharm Franc. 1965b;23:369–576. [PubMed] [Google Scholar]
- 25.Lechat P, Van Den Driessche J, Lemeignan M, Deleau D. Pharmacological investigation of a quaternary ammonium compound with ganglion-exciting properties. Arch Int Pharmacodyn Ther. 1965c;155:262–272. [PubMed] [Google Scholar]
- 26.Liang SP, Kanthan R, Shuaib A, Wishart T. Effects of clomethiazole on radial-arm maze performance following global forebrain ischemia in gerbils. Brain Res. 1997;751:189–195. doi: 10.1016/s0006-8993(96)01292-9. [DOI] [PubMed] [Google Scholar]
- 27.Limon A, Reyes-Ruiz JM, Miledi R. Loss of functional GABA(A) receptors in the Alzheimer diseased brain. Proc Natl Acad Sci U S A. 2012;109:10071–10076. doi: 10.1073/pnas.1204606109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindberg UH. Compounds related to clomethiazole. IV. 4-Methylthiazoles and oxazoles with polar side-chains and some other analogs of clomethiazole. Acta Pharm Suec. 1971a;8:39–48. [PubMed] [Google Scholar]
- 29.Lindberg UH. Hypnotic and anticonvulsant agents related to the thiazole part of thiamine. Acta Pharm Suec. 1971b;8:647–660. [PubMed] [Google Scholar]
- 30.Marshall JW, Cross AJ, Jackson DM, Green AR, Baker HF, Ridley RM. Clomethiazole protects against hemineglect in a primate model of stroke. Brain Res Bull. 2000;52:21–29. doi: 10.1016/s0361-9230(99)00275-0. [DOI] [PubMed] [Google Scholar]
- 31.Marshall JW, Cross AJ, Ridley RM. Functional benefit from clomethiazole treatment after focal cerebral ischemia in a nonhuman primate species. Exp Neurol. 1999;156:121–129. doi: 10.1006/exnr.1998.6994. [DOI] [PubMed] [Google Scholar]
- 32.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. Beta-amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McAlpine FE, Lee JK, Harms AS, Ruhn KA, Blurton-Jones M, Hong J, et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer's disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis. 2009;34:163–177. doi: 10.1016/j.nbd.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohler H. The rise of a new GABA pharmacology. Neuropharmacology. 2011;60:1042–1049. doi: 10.1016/j.neuropharm.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 35.Moody EJ, Skolnick P. Chlormethiazole: neurochemical actions at the gamma-aminobutyric acid receptor complex. Eur J Pharmacol. 1989;164:153–158. doi: 10.1016/0014-2999(89)90242-2. [DOI] [PubMed] [Google Scholar]
- 36.Mucke H. Clomethiazole (Astra Arcus AB) Idrugs. 1999;2:184–193. [PubMed] [Google Scholar]
- 37.Mucke L, Selkoe DJ. Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2:a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muir KW, Grosset DG. Neuroprotection for acute stroke: making clinical trials work. Stroke. 1999;30:180–182. doi: 10.1161/01.str.30.1.180. [DOI] [PubMed] [Google Scholar]
- 39.Nelson RM, Hainsworth AH, Lambert DG, Jones JA, Murray TK, Richards DA, et al. Neuroprotective efficacy of AR-A008055, a clomethiazole analogue, in a global model of acute ischaemic stroke and its effect on ischaemia-induced glutamate and GABA efflux in vitro. Neuropharmacology. 2001;41:159–166. doi: 10.1016/s0028-3908(01)00052-1. [DOI] [PubMed] [Google Scholar]
- 40.Olsen RW. Drug interactions at the GABA receptor-ionophore complex. Annu Rev Pharmacol Toxicol. 1982;22:245–277. doi: 10.1146/annurev.pa.22.040182.001333. [DOI] [PubMed] [Google Scholar]
- 41.Qin Z, Luo J, VandeVrede L, Tavassoli E, Fa M, Teich AF, et al. Design and synthesis of neuroprotective methylthiazoles and modification as NO-chimeras for neurodegenerative therapy. J Med Chem. 2012;55:6784–6801. doi: 10.1021/jm300353r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rissman RA, Mobley WC. Implications for treatment: GABAA receptors in aging, down syndrome and Alzheimer's disease. J Neurochem. 2011;117:613–622. doi: 10.1111/j.1471-4159.2011.07237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rudolph U, Knoflach F. Beyond classical benzodiazepines: novel therapeutic potential of GABA(A) receptor subtypes. Nat Rev Drug Discov. 2011;10:685–697. doi: 10.1038/nrd3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santos RX, Correia SC, Carvalho C, Cardoso S, Santos MS, Moreira PI. Mitophagy in neurodegeneration: an opportunity for therapy? Curr Drug Targets. 2011;12:790–799. doi: 10.2174/138945011795528813. [DOI] [PubMed] [Google Scholar]
- 45.Shi JQ, Shen W, Chen J, Wang BR, Zhong LL, Zhu YW, et al. Anti-TNF-alpha reduces amyloid plaques and tau phosphorylation and induces CD11c-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res. 2011;1368:239–247. doi: 10.1016/j.brainres.2010.10.053. [DOI] [PubMed] [Google Scholar]
- 46.Snape MF, Baldwin HA, Cross AJ, Green AR. The effects of chlormethiazole and nimodipine on cortical infarct area after focal cerebral ischaemia in the rat. Neuroscience. 1993;53:837–844. doi: 10.1016/0306-4522(93)90628-s. [DOI] [PubMed] [Google Scholar]
- 47.Stine WB, Jr, Dahlgren KN, Krafft GA, Ladu MJ. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 48.Strum JC, Shehee R, Virley D, Richardson J, Mattie M, Selley P, et al. Rosiglitazone induces mitochondrial biogenesis in mouse brain. J Alzheimers Dis. 2007;11:45–51. doi: 10.3233/jad-2007-11108. [DOI] [PubMed] [Google Scholar]
- 49.Sydserff SG, Cross AJ, Green AR. The neuroprotective effect of chlormethiazole on ischaemic neuronal damage following permanent middle cerebral artery ischaemia in the rat. Neurodegeneration. 1995a;4:323–328. doi: 10.1016/1055-8330(95)90022-5. [DOI] [PubMed] [Google Scholar]
- 50.Sydserff SG, Cross AJ, West KJ, Green AR. The effect of chlormethiazole on neuronal damage in a model of transient focal ischaemia. Br J Pharmacol. 1995b;114:1631–1635. doi: 10.1111/j.1476-5381.1995.tb14950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Usala M, Thompson SA, Whiting PJ, Wafford KA. Activity of chlormethiazole at human recombinant GABA(A) and NMDA receptors. Br J Pharmacol. 2003;140:1045–1050. doi: 10.1038/sj.bjp.0705540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vellas B, Sol O, Snyder PJ, Ousset PJ, Haddad R, Maurin M, et al. EHT0202 in Alzheimer's disease: a 3-month, randomized, placebo-controlled, double-blind study. Curr Alzheimer Res. 2011;8:203–212. doi: 10.2174/156720511795256053. [DOI] [PubMed] [Google Scholar]
- 53.Wahlgren NG, Diez-Tejedor E, Teitelbaum J, Arboix A, Leys D, Ashwood T, et al. Results in 95 hemorrhagic stroke patients included in CLASS, a controlled trial of clomethiazole versus placebo in acute stroke patients. Stroke. 2000;31:82–85. doi: 10.1161/01.str.31.1.82. [DOI] [PubMed] [Google Scholar]
- 54.Wahlgren NG, Ranasinha KW, Rosolacci T, Franke CL, Van Erven PM, Ashwood T, et al. Clomethiazole acute stroke study (CLASS): results of a randomized, controlled trial of clomethiazole versus placebo in 1360 acute stroke patients. Stroke. 1999;30:21–28. doi: 10.1161/01.str.30.1.21. [DOI] [PubMed] [Google Scholar]
- 55.Wilby MJ, Hutchinson PJ. The pharmacology of chlormethiazole: a potential neuroprotective agent? CNS Drug Rev. 2004;10:281–294. doi: 10.1111/j.1527-3458.2004.tb00028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu C, Nwabuisi-Heath E, Laxton K, Ladu MJ. Endocytic pathways mediating oligomeric Abeta42 neurotoxicity. Mol Neurodegener. 2010;5:19. doi: 10.1186/1750-1326-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yue L, Pawlowski M, Dellal SS, Xie A, Feng F, Otis TS, et al. Robust photoregulation of GABA(A) receptors by allosteric modulation with a propofol analogue. Nat Commun. 2012;3:1095. doi: 10.1038/ncomms2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A) CMZ showed substantial, reproducible neuroprotection compared with untreated vehicle controls in OGD. Data shown represent mean and SEM of over 30 independent experiments, with analysis by two-tailed Student's t-test. (B) No effect was seen when compounds were incubated for 24 h with MTT in growth media without cells, suggesting no artefactual reducing interaction between drugs and MTT. (C) Incubation of compounds (50 μM) in primary cortical cultures for 24 h did not result in significant protection or toxicity at any dose studied. (D) After treatment with various insults using protocols described in the text, the amount of cell death measured by MTT and normalized to untreated vehicle controls was reproducible and significant. All data from panels B.D show mean and SEM, with statistical significance determined in panels B and D by use of one-way ANOVA and post hoc Dunnett's MCT compared to vehicle control: *P < 0.05, ***P < 0.001; ns, not significant. Data for panels B.D were obtained from at least six independent replicates for each experimental condition.
Figure S2 Calibration curves were constructed for GN-28 and GN-38 in plasma spiked with standard solutions and internal standard (GN-27 and F-12 respectively). Final concentrations of analyte ranged from 1 to 100 ng EmL.1. The equation for the regression line was used to quantify analytes in plasma samples.
Figure S3 Total ion chromatograms (TIC) of GN-28 in mouse plasma 20 min after i.p. injection [GN-27 was used as an internal standard (IS)]; and of GN-38 in mouse plasma 20 min after i.p. injection [F-12 was used as an internal standard (IS)]. LC-MS/MS chromatograms showing fragmentation used for MRM quantification of analytes relative to internal standards: Transition MRM m/z 220→189 and m/z 238→207 were used to detect GN-28 and IS, respectively; transition MRM m/z 288→147 and m/z 352→147 were used to detect GN-38 and IS respectively.
Figure S4 TIC and MRM chromatograms of GN-28 and GN-38 in mouse brain 20 min after i.p. injection and spiking with internal standards.
Figure S5 CMZ was applied (i.p.) at 30, 40, 50 mg·kg-1. Mean latency to fall was determined at 30 and 60 min post injections. The data represent the average time (s) animals stayed on the rod before and after treatments. CMZ decreased the latency to fall in a dose-response fashion. CMZ (50 mg·kg-1) caused significant sedation, which translated to a great loss in animal balance when compared with pretreatment mean latency (***P < 0.001, n = 5). This effect faded away after 60 min. Vehicle group was injected with the same vehicle used to prepare i.p. injections. Solvent was composed of: water 90% (v v-1), DMSO 10% (v v-1). Vehicle did not show any significant effect on the mean latency to fall.
Figure S6 The accelerated rotarod performance task was performed with: CMZ (45 mg·kg-1, i.p.); GN-28 (equimolar dose, 59 mg·kg-1, i.p.); or GN-38 (equimolar dose, 64 mg·kg-1, i.p.). Mean latency to fall was determined at 10, 30 and 60 min post injections. The data represent average time (s) animals stayed on the rod after drug treatments. Vehicle, composed of water 90% (v v-1), DMSO 10% (v v-1), did not show any significant effect on the mean latency to fall. Bars represent mean and SEM, analysed by two-way repeated measures ANOVA with Bonferroni's post-test comparing with vehicle control: **P < 0.01, *** P < 0.001 (n = 5 for each group).
Table S1 Retention times, coefficients of determination (R2) of calibration curves and extraction recoveries for the different compounds.