

Abstract
Structure is a key determinant of function, with the nervous system being no exception. For example, in the nervous system the physiological properties of different synapses may be understood by comparing their structures. However, it is not clear whether specific structural properties of some neurons might play a role in driving their selective removal during chronic neurodegeneration or whether the structural properties might underpin why particular types of synapses or other neuronal compartments are more susceptible to degeneration (i.e., become dysfunctional) in certain brain regions than in others. Our recent study of the ultrastructure of the hippocampus and the cerebellum revealed that early synaptic loss is not a ubiquitous event in a brain undergoing chronic neurodegeneration. The prominent structural differences in proximity of the synaptic environment that are brought about by a degree of synaptic ensheathment by glial cells may help explain why Purkinje cell synapses remain intact, while pyramidal cell synapses progressively degenerate. The intrinsic structural organization of the hippocampal neuropil could contribute to the susceptibility of synapses to extracellular protein misfolding by a relatively higher degree of synaptic exposure to the extracellular environment. We suggest that neuronal structure may determine more than function; it might also predict dysfunction.
Keywords: structure-function relationship, synaptic degeneration, prion disease, hippocampus, cerebellum, glial ensheathment
The morphology of a neuron, with its capacity for structural rearrangement, is an example of how structure and function work together to engineer cellular diversity and adaptability in living organisms. In humans, during nervous system development, different neuronal populations are formed and integrated into neuronal networks; later, during the natural aging process, individual neurons may become redundant and are eliminated. However, it is not clear what drives the selection process of neuronal structure removal during chronic neurodegeneration. In this context, it would be important to know whether particular structural properties of some neuronal populations might play a role in determining the onset and outcome of neurodegeneration. Furthermore, to determine the neuronal compartments in different brain regions that are more likely to be targeted by pathological agents (for example, neurotoxic peptide oligomers such as soluble amyloid-β) based on their structural organization might provide new insights into the mechanism underlying the development of certain types of neuronal dysfunction.
Structural and functional abnormalities of hippocampal synapses are a widely recognized hallmark of neurodegeneration in various animal models of pathological protein misfolding.1 In our recent study, we demonstrated that, within a brain undergoing chronic neurodegeneration, region-selective processes involve distinct neuronal compartments. Following a focal brain injection of the mouse-adapted 22L scrapie prion strain, the ultrastructure of the hippocampus and the cerebellum revealed that synaptic loss is not a ubiquitous outcome of chronic brain insult.2 In contrast to early synaptic changes in the stratum radiatum of the hippocampus, which are virtually identical to those detected with a different prion strain (ME7),3 dendritic rather than synaptic disintegration was observed in the cerebellum, even in late-stage disease. Similar levels of protease-resistant pathological prion protein have been found in the hippocampus and in the cerebellum of ME7 and 22L prion strains.2,4 In Creutzfeldt-Jakob disease, the most common prion disease in humans, cerebellar alterations are well documented;5 notably, flattening and reduction of dendritic arbors of Purkinje cells have been reported.6 In agreement with the studies above, dendrite degeneration was described as a feature of cerebellar pathology in a mouse model of another human prion disease Gerstmann-Sträussler-Scheinker syndrome.7 However, synaptic degeneration occurred in the cerebellum infected with a different prion strain (RML) in TG3 mice, a transgenic mouse line in which the prion protein was expressed exclusively in astrocytes.8 The hippocampus and the cerebellum are both structurally regular, with a well-defined synaptic circuitry; unmyelinated parallel axons innervating Purkinje neurons in the cerebellum and CA1 pyramidal neurons in the hippocampus are both glutamatergic, and both contact dendrites of the principal cells via dendritic spines.3,9 Using either of the prion strains, the glutamatergic, asymmetric type I synapses in the stratum radiatum of CA1 were selectively reduced in their numbers in comparison to age-matched controls.2,3 The synaptic terminals and their postsynaptic sides degenerated, with clear-cut abnormalities detected at 12 weeks postinjection, and without other obvious ultrastructural signs of degeneration in CA1 or CA3 neurons.10 The most conspicuous correlate of synaptic degeneration was a progressive change of synaptic geometry, with an aberrant curvature of spine postsynaptic density, which suggests a process of engulfment of the presynaptic element by the dendritic spine, rather than phagocytosis by activated microglia. However, dendritic pathology is absent in the hippocampus at an early stage, and only a few degenerating dendrites have been observed in late-stage disease.
In contrast to the hippocampus, our findings in the cerebellum indicated quite the opposite. Degenerative changes associated with the Purkinje cells were dominated by dendritic malformations, while the major synaptic inputs to the cerebellum were preserved. Electron microscopy confirmed that the spiny branchlets on Purkinje neurons and the parallel and climbing fiber terminals were both intact, whereas the Purkinje dendrites degenerated progressively.2 We concluded that neuronal vulnerability to pathological protein misfolding is strongly dependent on the structure and function of the target neurons. Similar findings were observed in a methylmercury model of neurotoxicity. Ultrastructural investigation revealed degeneration of granule cells and swelling of the parallel fiber synaptic boutons; however, the dendritic spines of Purkinje cells (i.e., the postsynaptic sides of parallel fibers) showed no obvious changes at any stage.11
Neuronal function including synaptic transmission emerges only from the coordinated activities of neurons and glia. One approach to understanding the physiological differences between synapses has been to compare their anatomical structures. This approach might be used to understand why synapses or other neuronal compartments are more susceptible to degeneration in certain brain regions than in others depending on their structural organization, including glial arrangement. A distinct structural feature of type I Purkinje synapses in the molecular layer of the cerebellum is a degree of glial ensheathment, a property that is not shared by all neurons. In fact, Purkinje cells are the only neurons in the cerebellar cortex that are fully ensheathed.12 Their somata and dendrites, and even the synapses with dendritic spines, are completely surrounded by astrocytic processes of Bergmann glial cells. This structural arrangement differs from that of the hippocampus, where only about 50% of synapses have astrocytic processes surrounding a part of their perimeters, in contrast to the cerebellum, where nearly all of the synapses are completely ensheathed13,14 (Fig. 1). Astroglia are a source of metabolic and physical support for neurons and it is clear that they are involved in direct neuronal signaling, also locally at the synapses.15,16 Astrocytes integrate, process, and control synaptic information and by being in direct physical contact with neurons astrocytes are able to shape the synaptic environment. Astrocytic processes contain glutamate transporters; therefore, the degree of synaptic ensheathment as a structural measure is crucial to understanding their contribution to synaptic activity. The glutamate receptors on the glial processes are indispensable for proper structural and functional relationships between Bergmann glia and glutamatergic synapses of Purkinje neurons.17 A study showed that conversion of Ca2+-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors into Ca2+-impermeable receptors in the Bergmann glia triggered morphological changes in the fine glial processes enveloping Purkinje cell synapses. Moreover, it prolonged the kinetics of glutamatergic synaptic transmission and caused multiple innervations of Purkinje cells by climbing fibers.18 These findings reinforce the view that synaptic function and the morphology of glial processes enveloping the synapses are interdependent. In addition, it has been suggested that Bergmann glia cells create independent compartments that are capable of autonomous interactions tuned to the particular need of the synapses they ensheath.19 These relatively self-sufficient, compartmentalized microdomains could provide a certain degree of synaptic protection, in contrast to the hippocampal synapses, which do not share this structural arrangement. Devoid of similar support mechanisms, the protective capacity of pyramidal neurons in the hippocampus might be also compromised intrinsically because their synapses are exposed to a higher degree to their direct extracellular environment. This is particularly relevant in the context of soluble amyloid-β protein oligomers. These oligomers, extracted directly from the cerebral cortex of individuals with Alzheimer disease and injected to a healthy rodent hippocampus, strongly impair synaptic structure and function.20
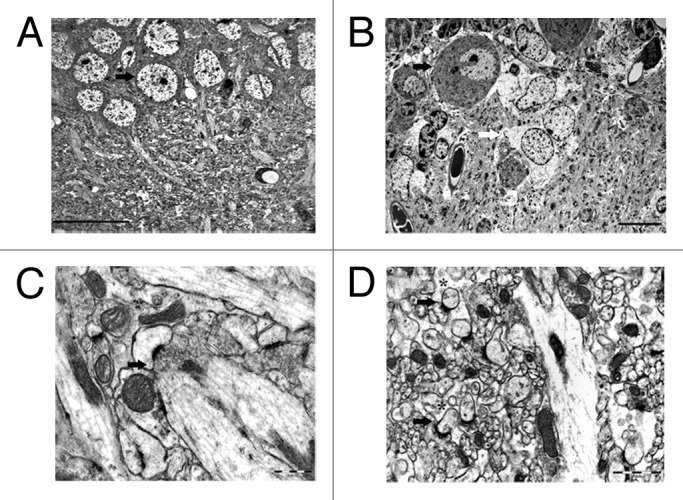
Figure 1. Electron micrographs of CA1 pyramidal cells (arrow) in the hippocampus (A) and Purkinje cells (black arrow) tightly enveloped by Bergmann glial cells (white arrow) in the cerebellum (B) illustrating typical structural organizations of the brain regions. A representative image of a type I synapse (arrow) in stratum radiatum of the hippocampus (C) and cerebellar molecular layer (D) illustrating prominent differences in the degree of synaptic ensheathment by glia cells (asterisks). Scale bars: 20 μm (A), 10 μm (B), 0.5 μm (C), 1 μm (D).
The molecular mechanisms corresponding to some of the structural changes within the hippocampus undergoing chronic neurodegeneration in prion disease have been discussed previously.10 However, within the cerebellum, fewer studies have been conducted and the exact molecular mechanisms underlying neurodegeneration are unknown. While it is not disputed that the identification of early molecular biomarkers is indispensable for the development of diagnostics and therapy, the structural changes are often neglected, but appear to be a robust correlate of ongoing neurodegeneration, providing a reliable readout of neuronal pathophysiology and behavioral abnormalities.
In conclusion, the intrinsic structural properties of the hippocampal synaptic environment could contribute to the relative susceptibility of synapses to a pathological protein misfolding or other similar types of brain insults. In this context, neuronal structure may influence more than function; it might, in fact, determine the onset and development of certain types of neuronal dysfunction.
Acknowledgments
I am grateful to Jr-Prof Dr Stefan Remy, Dr Martin Fuhrmann, Dr Erdem Tamgüney (DZNE, Bonn, Germany) and Dr Marie-Ève Tremblay (Université Laval, Québec, Canada) for their comments and helpful discussions.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/prion/article/26019
References
- 1.Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–5. doi: 10.1126/science.1067122. [Review] [DOI] [PubMed] [Google Scholar]
- 2.Šišková Z, Reynolds RA, O’Connor V, Perry VH. Brain region specific pre-synaptic and post-synaptic degeneration are early components of neuropathology in prion disease. PLoS One. 2013;8:e55004. doi: 10.1371/journal.pone.0055004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sisková Z, Page A, O’Connor V, Perry VH. Degenerating synaptic boutons in prion disease: microglia activation without synaptic stripping. Am J Pathol. 2009;175:1610–21. doi: 10.2353/ajpath.2009.090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cunningham C, Deacon RM, Chan K, Boche D, Rawlins JN, Perry VH. Neuropathologically distinct prion strains give rise to similar temporal profiles of behavioral deficits. Neurobiol Dis. 2005;18:258–69. doi: 10.1016/j.nbd.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 5.Ironside JW. Prion diseases: update on Creutzfeldt-Jakob disease. Neuropathol Appl Neurobiol. 1996;22:446. doi: 10.1111/j.1365-2990.1996.tb00919.x. [DOI] [PubMed] [Google Scholar]
- 6.Ferrer I, Kulisevski J, Vazquez J, Gonzalez G, Pineda M. Purkinje cells in degenerative diseases of the cerebellum and its connections: a Golgi study. Clin Neuropathol. 1988;7:22–8. [PubMed] [Google Scholar]
- 7.Jeffrey M, Goodsir C, McGovern G, Barmada SJ, Medrano AZ, Harris DA. Prion protein with an insertional mutation accumulates on axonal and dendritic plasmalemma and is associated with distinctive ultrastructural changes. Am J Pathol. 2009;175:1208–17. doi: 10.2353/ajpath.2009.090125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeffrey M, Goodsir CM, Race RE, Chesebro B. Scrapie-specific neuronal lesions are independent of neuronal PrP expression. Ann Neurol. 2004;55:781–92. doi: 10.1002/ana.20093. [DOI] [PubMed] [Google Scholar]
- 9.Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev. 2001;81:1143–95. doi: 10.1152/physrev.2001.81.3.1143. [Review] [DOI] [PubMed] [Google Scholar]
- 10.Gray BC, Siskova Z, Perry VH, O’Connor V. Selective presynaptic degeneration in the synaptopathy associated with ME7-induced hippocampal pathology. Neurobiol Dis. 2009;35:63–74. doi: 10.1016/j.nbd.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 11.Su M, Kakita A, Yamada M, Takahashi H, Ikuta F. Degeneration of the synaptic boutons of parallel fibers in rats treated with methylmercury: chronological observations. Neuropathology. 1996;16:172–7. doi: 10.1111/j.1440-1789.1996.tb00178.x. [DOI] [Google Scholar]
- 12.Palay SL, Chan-Palay V. Cerebellar Cortex. Cytology and Organization. New York: Springer, 1974. [Google Scholar]
- 13.Ventura R, Harris KM. Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci. 1999;19:6897–906. doi: 10.1523/JNEUROSCI.19-16-06897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spacek J. Three-dimensional analysis of dendritic spines. III. Glial sheath. Anat Embryol (Berl) 1985;171:245–52. doi: 10.1007/BF00341419. [DOI] [PubMed] [Google Scholar]
- 15.Bergles DE, Dzubay JA, Jahr CE. Glutamate transporter currents in bergmann glial cells follow the time course of extrasynaptic glutamate. Proc Natl Acad Sci U S A. 1997;94:14821–5. doi: 10.1073/pnas.94.26.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen NJ, Barres BA. Signaling between glia and neurons: focus on synaptic plasticity. Curr Opin Neurobiol. 2005;15:542–8. doi: 10.1016/j.conb.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009;32:421–31. doi: 10.1016/j.tins.2009.05.001. [Review] [DOI] [PubMed] [Google Scholar]
- 18.Iino M, Goto K, Kakegawa W, Okado H, Sudo M, Ishiuchi S, Miwa A, Takayasu Y, Saito I, Tsuzuki K, et al. Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science. 2001;292:926–9. doi: 10.1126/science.1058827. [DOI] [PubMed] [Google Scholar]
- 19.Grosche J, Matyash V, Möller T, Verkhratsky A, Reichenbach A, Kettenmann H. Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nat Neurosci. 1999;2:139–43. doi: 10.1038/5692. [DOI] [PubMed] [Google Scholar]
- 20.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]