

Abstract
Prions are self-seeding alternate protein conformations. Most yeast prions contain glutamine/asparagine (Q/N)-rich domains that promote the formation of amyloid-like prion aggregates. Chaperones, including Hsp104 and Sis1, are required to continually break these aggregates into smaller “seeds.” Decreasing aggregate size and increasing the number of growing aggregate ends facilitates both aggregate transmission and growth. Our previous work showed that overexpression of 11 proteins with Q/N-rich domains facilitates the de novo aggregation of Sup35 into the [PSI+] prion, presumably by a cross-seeding mechanism. We now discuss our recent paper, in which we showed that overexpression of most of these same 11 Q/N-rich proteins, including Pin4C and Cyc8, destabilized pre-existing Q/N rich prions. Overexpression of both Pin4C and Cyc8 caused [PSI+] aggregates to enlarge. This is incompatible with a previously proposed “capping” model where the overexpressed Q/N-rich protein poisons, or “caps,” the growing aggregate ends. Rather the data match what is expected of a reduction in prion severing by chaperones. Indeed, while Pin4C overexpression does not alter chaperone levels, Pin4C aggregates sequester chaperones away from the prion aggregates. Cyc8 overexpression cures [PSI+] by inducing an increase in Hsp104 levels, as excess Hsp104 binds to [PSI+] aggregates in a way that blocks their shearing.
Keywords: prion, yeast, amyloid, chaperone, Hsp104, Sis1, [PSI+], Sup35, Pin4, Cyc8
Prions are transmissible self-propagating protein conformations. Most known prions are highly ordered protein aggregates that exploit the efficient self-templating capacity of amyloid. The de novo appearance of these prions starts from the formation of small seeds, composed of molecules of the same protein that undergo conformational conversion into amyloid. Prion propagation proceeds by the addition of more molecules of the same protein to the infectious ends of the amyloid seeds leading to aggregate growth. This is followed by fragmentation of the aggregate, which creates new seeds with infectious ends.1
Amyloid-based prions were first discovered as infectious proteins associated with neurodegenerative diseases. Their appearance was attributed to sporadic or mutation-driven misfolding of the PrP protein.2 It now appears that multiple proteins in various organisms maintain the ability to fold in prion or prion-like states, and that some of these prions can be beneficial.1,3
Most known prion-forming proteins contain conserved functional domain(s) responsible for the protein’s cellular activity, and a much less conserved prion domain that is responsible for aggregation and maintenance of the prion state. Several distinct structures have been described for prion domains of amyloid-based prions. All of them contain an ordered and extensive network of intermolecular bonds. Curiously there are many different self-seeding amyloid forms possible for most prion proteins. These different forms are called prion variants and often result in distinct phenotypic and biochemical properties.1,4
The most common type of prion domain is characterized by a very high proportion of polar uncharged glutamine and asparagine (Q/N) residues. Low complexity of primary structure and, especially, a low content of structure-promoting hydrophobic and charged amino acids, keeps these domains unstructured in the monomeric state, thus simplifying conformational switching. In the prion state, the Q and N residues engage in intermolecular interactions leading to the formation of monotonous β-rich aggregates.5,6 Genes encoding potentially prionogenic Q/N-rich domains constitute up to 5% of eukaryotic genomes,7 and, strikingly, most of these Q/N-rich sequences are indeed prone to drive the formation of prion-like aggregates in vivo and in vitro.8-10 Thus cells are likely to simultaneously harbor multiple prions carrying similar Q/N-rich prion domains. This underscores the importance of understanding how prions affect each other.
Positive prion-prion interactions were first uncovered in Saccharomyces cerevisiae upon the discovery of the [PIN+] prion (also known as [RNQ+]).11-13 [PIN+] was found to dramatically enhance the de novo formation of the [PSI+] prion when the Sup35 prion domain is overexpressed.11,12,14 Subsequent studies revealed that such positive prion-prion interactions are widespread among Q/N-rich proteins. Indeed, [PIN+] also facilitates the appearance of several other prions and promotes aggregation of constructs encompassing Q/N-rich prion domains or poly-Q stretches.8,15,16 In addition, a genetic screen in a strain lacking [PIN+], revealed that overexpression of 11 different proteins and protein fragments, all containing Q/N-rich sequences, could promote the induction of the de novo formation of [PSI+] by overexpressed Sup35 prion domain.12 Likewise, overexpression of long poly-Q stretches also enhances the appearance of [PSI+] in the absence of [PIN+].17
Negative prion-prion interactions have also been reported for the three most extensively studied yeast prions, [PSI+], [PIN+], and [URE3]. They include mutual inhibition and destabilization of co-existing prions, and inhibition of de novo prion appearance by a pre-existing prion.18-20 Also, [PSI+] and [URE3] can be cured by overexpression of Rnq1 deletion or point mutants in the presence of [PIN+], and overexpression of GFP-tagged Ure2 fragments capable of inducing the [URE3] prion can cure pre-existing [URE3].21-24
Two principally different mechanisms were proposed for interactions between heterologous prion proteins.12,14,16,18,19,23-25 The first postulates direct interaction of heterologous prion domains (Fig. 1A–C). If the pre-existing prion were used as a nidus for initial aggregation of the newly forming prion, this would lead to a positive prion-prion interaction, a phenomenon known as seeding or cross-seeding (Fig. 1A). However, a similar direct interaction between the overexpressed heterologous protein and the growing tips of the preexisting prion could result in “capping” of these growing tips, leading to a negative prion-prion interaction (Fig. 1B). Also, lateral association of heterologous prion aggregates could enlarge prion particles and interfere with their transmission to daughter cells (Fig. 1C).
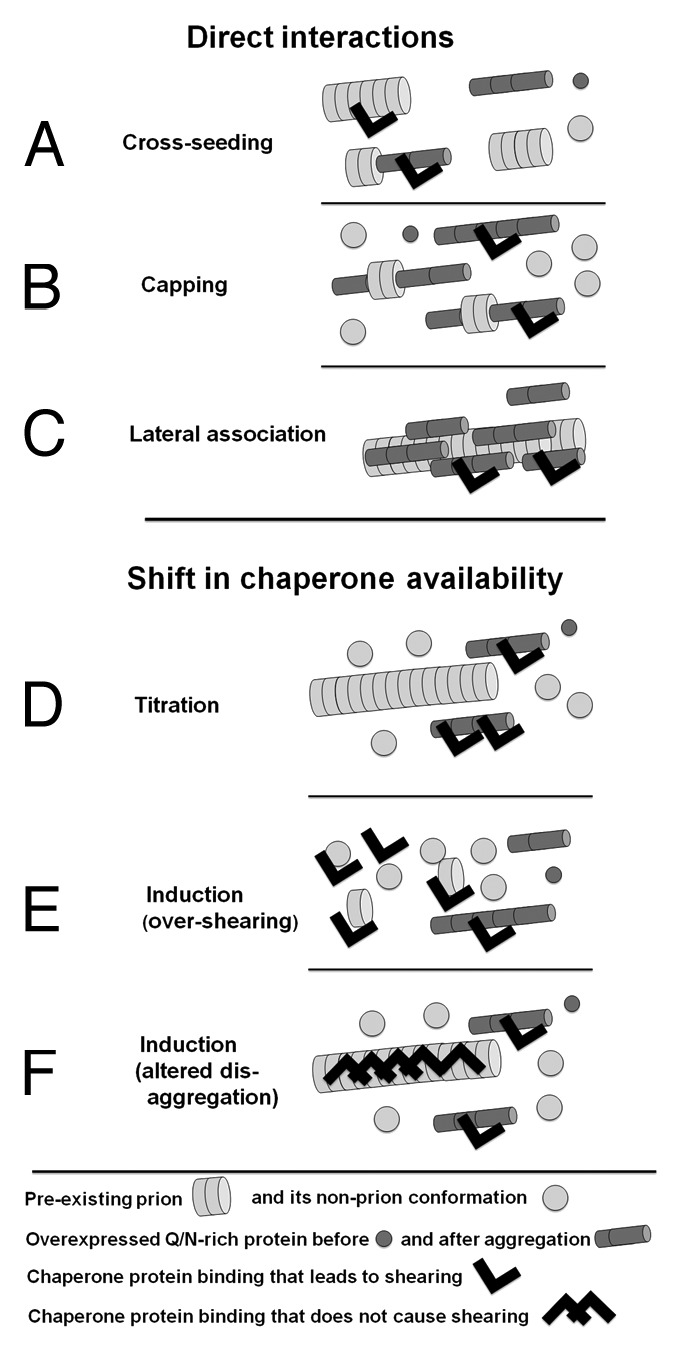
Figure 1. Models of interaction between different amyloids. See text for full description.
The second mechanism explains both positive and negative prion-prion interactions by a shift in the availability of factors, e.g., chaperones, involved in prion appearance orpropagation (Fig. 1D–F). Titration models (Fig. 1D) propose that prions can bind such factors pulling them away from the cytoplasm. This could lead to a positive or negative prion-prion interaction depending on whether the titrated factor promotes or inhibits the appearance or propagation of the other prion. Induction models (Fig. 1E and F) propose that prions could modulate each other’s formation and stability by inducing specific chaperone proteins. Because prions vary in their dependence on particular chaperone proteins, the same shift in chaperone concentrations can stabilize some prions and destabilize others. While all the above mechanisms are possible, until now, experimental evidence only supported a model for positive prion-prion interactions arising from direct cross-seeding interactions.10,17,26
In our recently published study,27 we explore negative prion-prion interactions. We find that negative interactions leading to destabilization of pre-existing prions upon overexpression of proteins with Q/N-rich prion domains are widespread. We also provide evidence supporting both chaperone titration and chaperone induction mechanisms for these interactions.
Negative Prion-Prion Interactions are Widespread but are Prion-Specific and Prion Variant-Specific
Our study27 began with an unbiased genetic screen for cellular factors that affect the stability of the yeast prion [PSI+]. [PSI+] is a prion form of a translation termination factor Sup35 (eRF3). Thus, in the presence of [PSI+], translation termination is inefficient leading to nonsense suppression. For the screen we used a collection of ~15 000 [psi-][pin-] colonies transformed with multicopy vectors from a yeast genomic library.28 These transformants were crossed to a [PSI+] strain and the expression of the library genes in the diploid was boosted by plasmid amplification. Diploids with a reduced level of nonsense-suppression, indicative of [PSI+] loss, were selected for further analysis. Several screen hits encoded proteins with Q/N-rich domains, and the strongest candidate encoded a Q/N-rich part of the Cyc8 protein, which forms the [OCT+] prion.29 Strikingly, a Q/N-rich Cyc8 fragment was previously selected in another genetic screen, where its overexpression promoted the de novo formation of the [PSI+] prion in a [pin-] strain.12
To see if other Q/N-rich proteins engage in both positive and negative prion-prion interactions, we used a candidate approach. Candidates included the proteins and protein fragments identified in the Derkatch et al.12 screen as being able, when overexpressed, to promote the de novo formation of [PSI+]: Ste18, Yck1, Pin2, Pin3, Ure2, Nup116, Lsm4, New1, the C-terminal parts of Pin4 (Pin4C), and Cyc8 (Cyc8C), and the N-terminal part of Swi1 (Swi1N). We now tested these hits from the positive prion-prion interaction screen for the ability to cause the loss of pre-existing [PSI+], and almost half of them did: Pin4C, Cyc8C, Ste18, Yck1, and Pin2. Interestingly, the proteins engaging in the negative interaction with [PSI+] were among the strongest inducers of [PSI+]. Furthermore, the destabilizing effect of the Q/N-rich proteins was not limited to [PSI+]. [URE3] was destabilized by the overexpression of Pin4C, Ste18, Pin3, and New1, and [PIN+] was lost in Pin4C-overexpressing cells (in this case only Pin4C was tested).
While obviously widespread, such negative interactions between pre-existing prions and overexpressed proteins with Q/N-rich domains are specific. Some proteins destabilized only one of the prions tested. For example, Cyc8C efficiently cured [PSI+] but had no effect on [URE3], and Pin3 cured [URE3] but not [PSI+]. Also, as with previously described negative prion-prion interactions between [PIN+] and [PSI+],19 the curing effect was prion variant-specific: Cyc8C cured medium and very high [PIN+] variants, but not high [PIN+]. This specificity could reflect the existence of different mechanisms of prion destabilization. Alternatively the same curing mechanisms could have different effects on different prions or prion variants.
Curing of [PSI+] by Pin4C Involves Enlargement of [PSI+] Aggregates Leading to Reduced Transmission to Daughter Cells
To gain an insight into the mechanism of negative prion-prion interactions, we focused on the curing of [PSI+] by overexpressed Pin4C (see Fig. 2B). To be able to examine [PSI+] aggregates by microscopy in cells overexpressing Pin4C, we used a strain, in which the chromosomal SUP35 ORF was replaced by a fully functional SUP35-GFP fusion.30 This strain can stably maintain strong [PSI+] variants that are visualized as tiny fluorescent foci. PIN4C was introduced on a multicopy plasmid under the control of a tightly regulated GAL promoter. Overnight overexpression of Pin4C caused the Sup35-GFP aggregates to become brighter and bigger in 80% of cells (Fig. 2C). As yeast prion aggregates have been shown to be composed of SDS-resistant oligomers,31 we also analyzed the size of [PSI+] oligomers resistant to 2% SDS at room temperature in Pin4C overexpressing cells. Indeed, the enlargement of visible Sup35-GFP aggregates caused by overexpression of Pin4C, coincided with an increase of the size of SDS-resistant Sup35-GFP oligomers (Fig. 2C). However, these changes, while dramatic, were reversible if Pin4C overexpression was turned-off. [PSI+] was lost irreversibly only if Pin4C overexpression continued for another 4 days, at which time most Sup35-GFP foci were gone and fluorescence was evenly distributed in the cytoplasm.
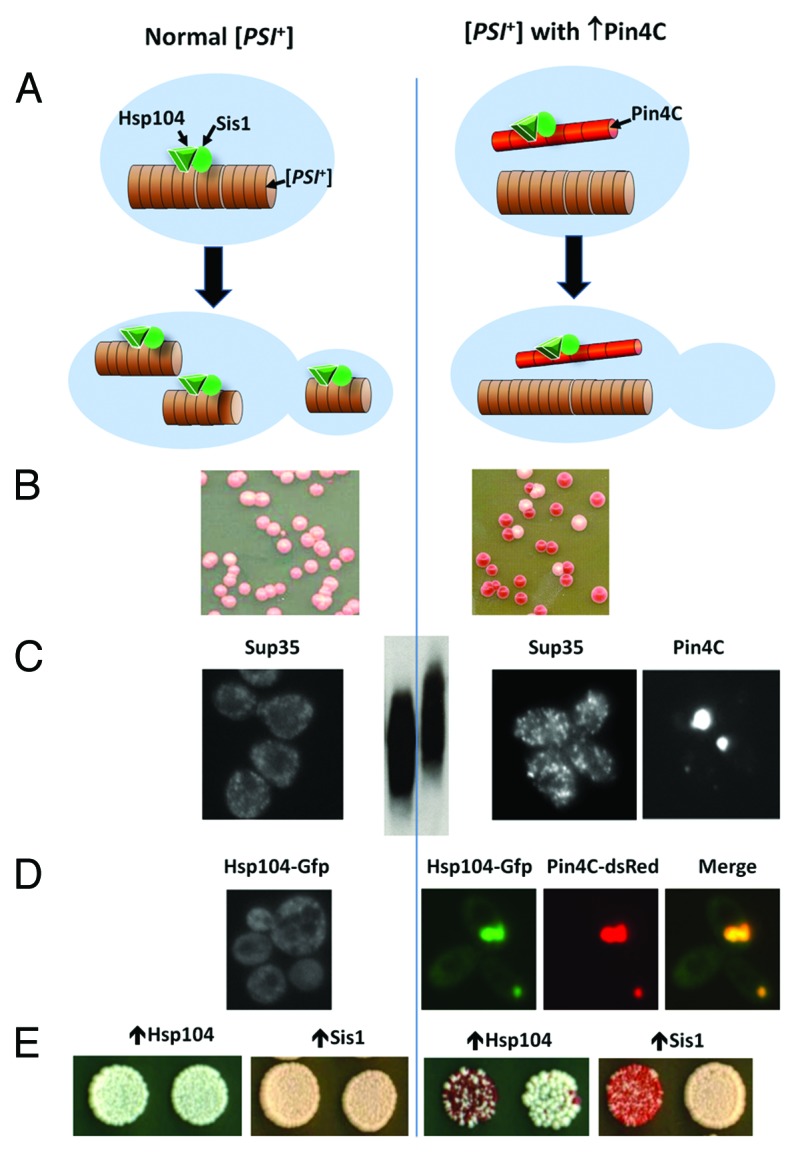
Figure 2. Curing of the [PSI+] prion as a result of titration of chaperone proteins by Pin4C aggregates. (A) Cartoon showing normal propagation of [PSI+] in the absence of overexpressed Pin4C (left), and rapid loss of [PSI+] when Pin4C is overexpressed (right). Normally Hsp104 and Sis1 bind to [PSI+] aggregates, and Hsp104 shears the aggregates producing small [PSI+] seeds that are easily transmitted to daughter cells. When Pin4C is overexpressed, it forms aggregates that titrate Hsp104 and Sis1 away from the [PSI+] aggregates preventing their shearing. The resulting large [PSI+] aggregates remain in the mother cell, so the daughter cells do not inherit the prion. (B) Nonsense suppression color test showing rapid loss of [PSI+] when Pin4C is overexpressed. The nonsense reporter is the chromosomal ade1–14 nonsense mutation, which causes cells to be Ade-, and colonies to be red on YPD medium. In [PSI+] cells, the inactivation of the translational release factor causes nonsense suppression and, depending upon the level of the restoration of the Ade+ phenotype, the colonies become white or pink. [PSI+] propagates stably, as shown by pink (Ade+) [PSI+] colonies (left). Upon Pin4C overexpression, red (Ade-) [psi-] colonies frequently appear (right). (C) Enlargement of visible [PSI+] aggregates upon Pin4C overexpression. Sup35 coalesces to small dots in [PSI+] cells expressing GFP-tagged Sup35 under the control of the native SUP35 promoter (left). These dots enlarge upon overexpression of Pin4C (right; Sup35). Appearance of enlarged Sup35 foci coincides with the formation of large Pin4C dots, which do not co-localize with Sup35 aggregates (right; Pin4C; the same cell cluster is shown for Sup35 and Pin4C panels). Also, SDS resistant oligomers formed by Sup35 in [PSI+] cells (middle, left lane) enlarge upon overexpression of Pin4C (middle, right lane). (D) A shift of Hsp104 into Pin4C aggregates. Hsp104-GFP is diffuse or in tiny particles in the cytoplasm in [PSI+] cells (left). Upon Pin4C-dsRED overexpression, Hsp104-GFP co-aggregates with Pin4C-dsRED leaving less Hsp104-GFP in the cytoplasm (right). (E) Overexpression of Hsp104 or Sis1 rescues the curing caused by overexpression of Pin4C. The Hsp104T160M allele used for this experiment is functional but, unlike wild-type Hsp104, it does not cure [PSI+] when overexpressed.35 While overexpression of this Hsp104 allele did not alter the aggregation of Pin4C, overexpression of Sis1 prevented Pin4C from forming large aggregates (data not shown). Parts B-E of this figure were adapted with permission from figures in Yang et al.27
Analyses of micro-colonies originating from single [PSI+] cells overexpressing Pin4C indicated that the enlarged [PSI+] aggregates are not transmitted efficiently from mothers to daughters. Cells devoid of Sup35-GFP foci were found on the outer edges of micro-colonies, usually right off the clusters of cells with enlarged Sup35-GFP. Furthermore, [PSI+] curing was shown to depend upon cell division: when cell division was inhibited with α-factor, curing of [PSI+] was reduced. Finally, the explanation that [PSI+] loss is due to reduced transmission of prion aggregates from mother to daughter cells was also supported by FRAP analyses of Sup35-GFP mobility in dividing cells. Compared with regular [PSI+] cells, fluorescence recovery in completely photobleached daughters was usually reduced in Pin4C-overexpressing [PSI+] cells with enlarged Sup35-GFP foci. However, in one cell Sup35-GFP mobility was sharply increased, suggesting that it had already become [psi-].
Out of the models postulating direct interaction of the prion-forming proteins, the finding of enlarged Sup35-GFP aggregates is obviously inconsistent with the capping model, where [PSI+] is proposed to be cured by the incorporation of Pin4C at the Sup35-GFP fiber ends, thereby preventing Sup35-GFP aggregate growth. Indeed, capping would lead to a diminution of Sup35-GFP foci, not their enlargement. A model that proposes the formation of mixed amyloid structures via lateral association of heterologous aggregates is consistent with the finding that Pin4C overexpression leads to the enlargement and reduced mobility of [PSI+] aggregates However, this model predicts co-localization of Pin4C with Sup35-GFP aggregates, which was not found. When we expressed a PIN4C-dsRED fusion controlled by a GAL promoter in a [PSI+] SUP35-GFP strain, we saw multiple bright cytoplasmic Pin4C-dsRED foci that became larger and less numerous in a few hours and by 24 h usually formed one large focus per cell. These large Pin4C-dsRED foci were accompanied by the appearance of enlarged Sup35-GFP foci, but co-localization of Pin4C-dsRED and Sup35-GFP foci was never observed (Fig. 2C). Furthermore, prion aggregate enlargement due to increased lateral association of prion aggregates should occur in the absence of the continuous synthesis of the prion-forming protein. However, we found that in the absence of newly synthesized Sup35-GFP, the foci remained small and dim. Thus we found no support for models implying direct interactions of prion-forming proteins.
Curing of [PSI+] in Pin4C-Overexpressing Cells is Caused by the Titration of the Hsp104 and Sis1 Chaperones by Pin4C Aggregates
We then asked if our results could be explained by shifts in the availability of chaperones involved in prion propagation, focusing on Hsp70/40 and Hsp104. Current models suggest that Hsp104 pulls out individual protein molecules from linear prion aggregates to thread these molecules through its central pore, just as it does while disassembling stress-induced amorphous protein aggregates. The key consequence, however, is not the unfolding of the threaded molecule, but the breaking of the prion aggregate at the site, where the molecule was removed. The Ssa Hsp70s recognize the prion aggregate, and attract Sis1 Hsp40, which facilitates the binding of Hsp104 to the prion, while Sse1 and Fes1 act as nucleotide exchange factors for the Ssa’s.32 Thus, depletion of Hsp104, Sis1, or Hsp70 is expected to lead to the enlargement of prion oligomers, their poor transmission to daughter cells and, as a result, prion loss.33
Western analyses showed that Pin4C overexpression did not significantly change the levels of these chaperones. There was a slight reduction in Hsp104 (to 83% of the normal level). However, this reduction was not sufficient to destabilize [PSI+], since [PSI+] remains stable even in strains expressing only 50% of the normal Hsp104.34 However, fluorescence microscopy of cells expressing the endogenously-encoded Hsp104-GFP and overexpressing Pin4-dsRED revealed that, upon the formation of large Pin4C foci, most cellular Hsp104 shifted to these foci, leaving only ~33% of the normal Hsp104 level in the cytoplasm (Fig. 2D), and association of Hsp104 with Pin4C was also confirmed by co-immunoprecipitation with anti-RFP antibody. This drop in available Hsp104 could be sufficient to promote destabilization of the prion. If so, overexpression of Hsp104 should have reversed the [PSI+]-curing by excess Pin4C. To test this, we used the T160M allele of Hsp104, which can maintain [PSI+] and does not cure [PSI+] even when overexpressed.35 Indeed, [PSI+] became stable when Hsp104T160M was co-overexpressed with Pin4C, even though Pin4C continued to aggregate (Fig. 2E).
Pin4C overexpression and aggregation also lead to dramatic changes in the cellular distribution of Sis1. While normally concentrated in the nucleus, in cells with Pin4C aggregates, Sis1 shifted from the nucleus to Pin4C foci, suggesting that titration of this co-chaperone could also contribute to the [PSI+]-curing effect of Pin4C. Indeed, Sis1 co-overexpression significantly reduced [PSI+] loss in Pin4C overexpressing cells (Fig. 2E). Furthermore, Sis1 overexpression prevented both the appearance of large Pin4C-dsRED foci and the enlargement of Sup35-GFP foci, underscoring the link between these events.
Curing of [PSI+] in Cyc8C Overexpressing Cells is Mediated by an Increase of Hsp104 Levels: Evidence for the Induction Model for Negative Prion Interactions
Studies of negative prion-prion interactions conducted in several other labs indicate that enlargement of prion aggregates is a common step in the process of prion elimination by another prion or aggregation-prone protein (see next section). However, titration of chaperones is not always the cause. Indeed, we found that Cyc8, the prion-forming protein for [OCT+],29 works via another mechanism.27 Like Pin4C, Cyc8C engages in both positive and negative interactions with [PSI+], but while Pin4C also cures [URE3], Cyc8C only cures [PSI+]. The analysis of chaperone protein levels revealed another striking difference between Cyc8C and Pin4C. Overexpression of Cyc8C caused an 8-fold increase of Hsp104 levels, while the levels of Sis increased only slightly. Furthermore, excess Hsp104 did not coalesce into Cyc8C aggregates, but remained in the cytoplasm, indicative of its availability to interact with [PSI+] aggregates.
According to earlier studies, such overproduction of Hsp104 is sufficient to cure [PSI+].36 Curing of [PSI+] by excess Hsp104 has been explained by a direct interaction of the [PSI+]-forming protein, Sup35, and Hsp104. Hsp104 binds directly to Sup35, but, unlike Sup35–Hsp104 interactions mediated by Ssa and Sis1, this direct binding of Hsp104 does not promote [PSI+] fragmentation (Fig. 1F). The overexpressed Hsp104 appears to outcompete Hsp70/Hsp40 for binding to [PSI+] and is then unable to fragment it.37,38 On the contrary, Ure2, like most other prion-forming proteins, does not bind Hsp104 in the absence of Ssa1, and is thus not cured by Hsp104 overexpression,33,39 so overexpression of Cyc8C is not expected to cure [URE3]. Thus we conclude that the curing specificity of Cyc8C is consistent with the induction model.
Other Negative Prion-Prion Interactions: Applicability of Titration and Induction Models and the Possibility of Curing by Lateral Association of Prion Aggregates
Below we discuss which of the models best explains other negative prion interactions both uncovered in our study and described elsewhere.
Curing of [URE3] by Pin3 overexpression is another example of a negative prion interaction with distinct specificity, revealed in our study.27 While efficiently eliminating [URE3], excess Pin3 does not destabilize [PSI+]. As in the case of Pin4C overexpression discussed above, in Pin3 overexpressing cells, both Hsp104 and Sis1 coalesced into fluorescent foci, indicative of co-aggregation with Pin3 aggregates. But because Pin3 overexpression simultaneously slightly increased cellular concentrations of both Hsp104 and Sis1, this chaperone titration lead to only a slight reduction of cytoplasmic levels of Hsp104 (to 74%), and to an insignificant reduction of Sis1 levels. Consistent with our data, such a small drop in Hsp104 levels is obviously insufficient to cure [PSI+],34 but it may be sufficient for [URE3] destabilization. Indeed, experimental evidence suggests that the number of [URE3] propagons transmissible to daughter cells is less than the number of [PSI+] propagons, which could explain why [URE3] is more sensitive to the reduction of aggregate fragmentation and, specifically, to Hsp104 inactivation.25,40,41 Thus, we hypothesize that curing of [URE3] by Pin3 is caused by Hsp104 titration (Fig. 1D).
Several negative prion interactions were recently reported by the Nakamura and Yoshida labs.25 They report curing of one or several yeast prions ([URE3] and, sometimes, [PSI+] or [PIN+]) by overexpression of Gpg1, a non-Q/N-rich G-protein γ-subunit mimic42; by overexpression of mutant alleles of Rnq1 in [PIN+] strains; and by a Rnq1-Δ100 deletion or an array of rnq1 point mutations in the presence of [PIN+].20-22,43 Also, overexpression of two more proteins identified in our screen for [PIN+],12 Lsm4 and New1, was recently shown to cause cells to lose prions.44,45 Similar to our observations for Pin4C,27 all the above negative interactions are associated with enlargement of aggregates proposed to result from inefficient fragmentation and poor transmission to daughter cells.25,42-45 However, while this is consistent with Hsp104 deficiency, a model postulating aggregate enlargement by direct lateral association of heterologous aggregates was favored (Fig. 1C).43,44
The key arguments that favor lateral association over titration in the studies from the Nakamura and Yoshida labs are: (1) co-localization of prion aggregates with the aggregates of the curing protein; (2) no reduction in cellular levels of Hsp104 as determined by Western analyses of total cell lysates, and no reduction in Hsp104 function as determined by thermotolerance. However, detection of co-localization, while consistent with the lateral association model, does not prove it. Indeed, we observed occasional co-localization of [PSI+] and [PIN+] foci in cells that stably propagated both prions.17 Also, normal or nearly normal cellular levels of Hsp104 may be detected even when cytoplasmic levels are sharply reduced27 (see above), and retention of thermotolerance activity, which depends upon the increased levels of Hsp104 induced during exposure to heat, is possible despite of reduced levels of cytoplasmic Hsp104 prior to temperature increase. Finally changes in the appearance of [PSI+] aggregates in the course of curing by Gpg1 and New142,45 strikingly resemble changes of [PSI+] in the presence of the Hsp104-inactivating chemical GuHCl observed in our earlier study.46 Indeed when we re-examined curing of [PSI+] by Gpg1, we found strong coalescence of Hsp104 into cytoplasmic foci, indicative of association with Gpg1 aggregates.27
In conclusion, the key arguments in our work favoring Hsp104 titration (Fig. 1D) over lateral association (Fig. 1C) to explain curing of [PSI+] by overexpressed Pin4C were: (1) the requirement for continuous Sup35 synthesis for [PSI+] aggregate enlargement; (2) the drop in Hsp104 cytoplasmic concentration to levels likely to destabilize [PSI+]; (3) the rescue of [PSI+] destabilization by excess Hsp104 or Sis1; and (4) the lack of co-localization of [PSI+] and Pin4C aggregates. Of these arguments, only rescue of [PSI+] destabilization by excess Hsp104 or Sis1 could be consistent with the lateral association model, e.g., if this association made aggregates less accessible for fragmentation. The other arguments strongly favor the titration model. For similar reasons, we also favor the titration model for two other [PSI+] curing proteins tested in our work, Pin3 and Gpg1. For curing of [PSI+] by overexpressed Cyc8C, there is a key argument in favor of the induction model: an 8-fold increase in the Hsp104 level was observed and this increase is sufficient for curing. Thus there is strong support for two modes of negative prion-prion interactions, neither of which is based on the direct interaction of prion-forming proteins. We expect that as more prion proteins are identified, additional examples of indirect and new examples of direct interactions will be uncovered.
Acknowledgments
This work was supported by National Institutes of Health grants 7R01 GM070934-06 (to ILD) and 5R01GM056350-16 (to SWL).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/prion/article/26021
References
- 1.Wickner RB, Edskes HK, Bateman DA, Kelly AC, Gorkovskiy A, Dayani Y, Zhou A. Amyloids and yeast prion biology. Biochemistry. 2013;52:1514–27. doi: 10.1021/bi301686a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol. 2011;3:a006833. doi: 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newby GA, Lindquist S. Blessings in disguise: biological benefits of prion-like mechanisms. Trends Cell Biol. 2013;23:251–9. doi: 10.1016/j.tcb.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Toyama BH, Weissman JS. Amyloid structure: conformational diversity and consequences. Annu Rev Biochem. 2011;80:557–85. doi: 10.1146/annurev-biochem-090908-120656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du Z. The complexity and implications of yeast prion domains. Prion. 2011;5:311–6. doi: 10.4161/pri.5.4.18304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross ED, Toombs JA. The effects of amino acid composition on yeast prion formation and prion domain interactions. Prion. 2010;4:60–5. doi: 10.4161/pri.4.2.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michelitsch MD, Weissman JS. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc Natl Acad Sci U S A. 2000;97:11910–5. doi: 10.1073/pnas.97.22.11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137:146–58. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halfmann R, Alberti S, Krishnan R, Lyle N, O’Donnell CW, King OD, Berger B, Pappu RV, Lindquist S. Opposing effects of glutamine and asparagine govern prion formation by intrinsically disordered proteins. Mol Cell. 2011;43:72–84. doi: 10.1016/j.molcel.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–67. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics. 1997;147:507–19. doi: 10.1093/genetics/147.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN(+)] Cell. 2001;106:171–82. doi: 10.1016/S0092-8674(01)00427-5. [PIN+] [DOI] [PubMed] [Google Scholar]
- 13.Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell. 2000;5:163–72. doi: 10.1016/S1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 14.Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI(+)] prion. Cell. 2001;106:183–94. doi: 10.1016/S0092-8674(01)00440-8. [DOI] [PubMed] [Google Scholar]
- 15.Taneja V, Maddelein ML, Talarek N, Saupe SJ, Liebman SW. A non-Q/N-rich prion domain of a foreign prion, [Het-s], can propagate as a prion in yeast. Mol Cell. 2007;27:67–77. doi: 10.1016/j.molcel.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Derkatch IL, Liebman SW. Prion-prion interactions. Prion. 2007;1:161–9. doi: 10.4161/pri.1.3.4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc Natl Acad Sci U S A. 2004;101:12934–9. doi: 10.1073/pnas.0404968101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwimmer C, Masison DC. Antagonistic interactions between yeast [PSI(+)] and [URE3] prions and curing of [URE3] by Hsp70 protein chaperone Ssa1p but not by Ssa2p. Mol Cell Biol. 2002;22:3590–8. doi: 10.1128/MCB.22.11.3590-3598.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bradley ME, Liebman SW. Destabilizing interactions among [PSI(+)] and [PIN(+)] yeast prion variants. Genetics. 2003;165:1675–85. doi: 10.1093/genetics/165.4.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurahashi H, Shibata S, Ishiwata M, Nakamura Y. Selfish prion of Rnq1 mutant in yeast. Genes Cells. 2009;14:659–68. doi: 10.1111/j.1365-2443.2009.01297.x. [DOI] [PubMed] [Google Scholar]
- 21.Kurahashi H, Ishiwata M, Shibata S, Nakamura Y. A regulatory role of the Rnq1 nonprion domain for prion propagation and polyglutamine aggregates. Mol Cell Biol. 2008;28:3313–23. doi: 10.1128/MCB.01900-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shibata S, Kurahashi H, Nakamura Y. Localization of prion-destabilizing mutations in the N-terminal non-prion domain of Rnq1 in Saccharomyces cerevisiae. Prion. 2009;3:250–8. doi: 10.4161/pri.3.4.10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edskes HK, Wickner RB. Conservation of a portion of the S. cerevisiae Ure2p prion domain that interacts with the full-length protein. Proc Natl Acad Sci U S A. 2002;99(Suppl 4):16384–91. doi: 10.1073/pnas.162349599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edskes HK, Gray VT, Wickner RB. The [URE3] prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc Natl Acad Sci U S A. 1999;96:1498–503. doi: 10.1073/pnas.96.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurahashi H, Oishi K, Nakamura Y. A bipolar personality of yeast prion proteins. Prion. 2011;5:305–10. doi: 10.4161/pri.5.4.18307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vitrenko YA, Gracheva EO, Richmond JE, Liebman SW. Visualization of aggregation of the Rnq1 prion domain and cross-seeding interactions with Sup35NM. J Biol Chem. 2007;282:1779–87. doi: 10.1074/jbc.M609269200. [DOI] [PubMed] [Google Scholar]
- 27.Yang Z, Hong JY, Derkatch IL, Liebman SW. Heterologous gln/asn-rich proteins impede the propagation of yeast prions by altering chaperone availability. PLoS Genet. 2013;9:e1003236. doi: 10.1371/journal.pgen.1003236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nehlin JO, Carlberg M, Ronne H. Yeast galactose permease is related to yeast and mammalian glucose transporters. Gene. 1989;85:313–9. doi: 10.1016/0378-1119(89)90423-X. [DOI] [PubMed] [Google Scholar]
- 29.Patel BK, Gavin-Smyth J, Liebman SW. The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion. Nat Cell Biol. 2009;11:344–9. doi: 10.1038/ncb1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Satpute-Krishnan P, Serio TR. Prion protein remodelling confers an immediate phenotypic switch. Nature. 2005;437:262–5. doi: 10.1038/nature03981. [DOI] [PubMed] [Google Scholar]
- 31.Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem. 2003;278:49636–43. doi: 10.1074/jbc.M307996200. [DOI] [PubMed] [Google Scholar]
- 32.Winkler J, Tyedmers J, Bukau B, Mogk A. Chaperone networks in protein disaggregation and prion propagation. J Struct Biol. 2012;179:152–60. doi: 10.1016/j.jsb.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 33.Romanova NV, Chernoff YO. Hsp104 and prion propagation. Protein Pept Lett. 2009;16:598–605. doi: 10.2174/092986609788490078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wegrzyn RD, Bapat K, Newnam GP, Zink AD, Chernoff YO. Mechanism of prion loss after Hsp104 inactivation in yeast. Mol Cell Biol. 2001;21:4656–69. doi: 10.1128/MCB.21.14.4656-4669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hung GC, Masison DC. N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics. 2006;173:611–20. doi: 10.1534/genetics.106.056820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;268:880–4. doi: 10.1126/science.7754373. [psi+] [DOI] [PubMed] [Google Scholar]
- 37.Helsen CW, Glover JR. Insight into molecular basis of curing of [PSI+] prion by overexpression of 104-kDa heat shock protein (Hsp104) J Biol Chem. 2012;287:542–56. doi: 10.1074/jbc.M111.302869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winkler J, Tyedmers J, Bukau B, Mogk A. Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J Cell Biol. 2012;198:387–404. doi: 10.1083/jcb.201201074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moriyama H, Edskes HK, Wickner RB. [URE3] prion propagation in Saccharomyces cerevisiae: requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol Cell Biol. 2000;20:8916–22. doi: 10.1128/MCB.20.23.8916-8922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eaglestone SS, Ruddock LW, Cox BS, Tuite MF. Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI(+)] of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2000;97:240–4. doi: 10.1073/pnas.97.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ripaud L, Maillet L, Cullin C. The mechanisms of [URE3] prion elimination demonstrate that large aggregates of Ure2p are dead-end products. EMBO J. 2003;22:5251–9. doi: 10.1093/emboj/cdg488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishiwata M, Kurahashi H, Nakamura Y. A G-protein gamma subunit mimic is a general antagonist of prion propagation in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2009;106:791–6. doi: 10.1073/pnas.0808383106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurahashi H, Pack CG, Shibata S, Oishi K, Sako Y, Nakamura Y. [PSI(+)] aggregate enlargement in rnq1 nonprion domain mutants, leading to a loss of prion in yeast. Genes Cells. 2011;16:576–89. doi: 10.1111/j.1365-2443.2011.01511.x. [DOI] [PubMed] [Google Scholar]
- 44.Oishi K, Kurahashi H, Pack CG, Sako Y, Nakamura Y. A bipolar functionality of Q/N-rich proteins: Lsm4 amyloid causes clearance of yeast prions. Microbiologyopen. 2013;2:415–30. doi: 10.1002/mbo3.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inoue Y, Kawai-Noma S, Koike-Takeshita A, Taguchi H, Yoshida M. Yeast prion protein New1 can break Sup35 amyloid fibrils into fragments in an ATP-dependent manner. Genes Cells. 2011;16:545–56. doi: 10.1111/j.1365-2443.2011.01510.x. [DOI] [PubMed] [Google Scholar]
- 46.Zhou P, Derkatch IL, Liebman SW. The relationship between visible intracellular aggregates that appear after overexpression of Sup35 and the yeast prion-like elements [PSI(+)] and [PIN(+)] Mol Microbiol. 2001;39:37–46. doi: 10.1046/j.1365-2958.2001.02224.x. [PIN+] [DOI] [PubMed] [Google Scholar]