

Abstract
Influenza is a global human health threat, and there is an immediate need for new antiviral therapies to circumvent the limitations of vaccination and current small molecule therapies. During viral transcription, influenza incorporates the 5′-end of the host cell’s mRNA in a process that requires the influenza endonuclease. On the basis of recently published endonuclease crystallized structures, a three-dimensional pharmacophore was developed and used to virtually screen 450,000 compounds for influenza endonuclease inhibitors. Of 264 compounds tested in a FRET-based endonuclease-inhibition assay, 16 inhibitors (IC50 < 50 μM) that span 5 molecular classes novel to this endonuclease were found (6.1% hit rate). To determine cytotoxicity and antiviral activity, subsequent cellular assays were performed. Two compounds suppress viral replication with negligible cell toxicity.
Keywords: Antiviral, influenza A, endonuclease, pharmacophore, virtual screening
In the United States, influenza causes as many as 40,000 deaths annually.1 Worldwide, cases of severe illness can total between three and five million with as many as half a million deaths.2 Vaccination can be an effective prophylactic in healthy adults, but there is generally a six-month lag time between the recognition of a new viral strain and the dissemination of an effective vaccine.3 Small molecule antivirals are an alternative. Drugs targeting neuraminidase (oseltamivir and zanamivir) and the M2-ion channel (adamantanes) are in clinical use, although emerging resistance is threatening their long-term efficacy.4,5 As a result, there is an immediate need for novel chemotherapies.
During viral mRNA transcription, influenza is unable to synthesize a 5′-mRNA cap and instead carries out a unique cap-snatching process.6 Cap snatching is performed by a heterotrimeric RNA-dependent RNA polymerase (RdRp) comprising subunits PA, PB1, and PB2. The process is initiated when the PB2 subunit binds to the 5′-end of host mRNA. Bound mRNA is cleaved 10–14 nucleotides downstream by the N-terminal domain of the PA-subunit (PAN), an endonuclease. The cleaved 5′-oligonucleotide contains the host cap and is a primer for viral mRNA synthesis, which is catalyzed by the PB1 subunit, a polymerase.
The PAN endonuclease is an appealing influenza target.7 PAN specific siRNA down-regulates viral mRNA production and blocks viral production in cell culture.8 It is highly conserved in all influenza A viruses, and PAN inhibitors should have broad efficacy with reduced occurrence of resistance. Additionally, the lack of a PAN human counterpart facilitates design of highly selective, nontoxic inhibitors. Finally, PAN inhibitors target a different enzyme than currently marketed drugs, creating the potential for combination therapies; given rapid viral mutation and adaptation, multidrug therapies may be necessary in the future.9
Several classes of PAN inhibitors demonstrating anti-influenza activity in cell and enzyme-based assays have been published. These include 2,4-dioxobutanoic acids,10,11 flutimide derivatives,12 a tetramic acid series,13 phenethylphenylphthalimides,14 green tea catehcins15 liverwort marchantins and perrottetins,16 4–5-dihydroxypyrimidines,17 and most recently, a series of 3-hydroxyquinolin-2(1H)-ones.18 Across classes, endonuclease inhibition potencies range from mid nanomolar to hundreds of micromolar. Each class includes two or three oxygen atoms, with a similar spatial arrangement, that are required for activity. As hypothesized,13 the spatial constraint originates from binding to a two metal ion active site and is consistent with crystal structures of the PAN subunit bound to several different ligands.19,20
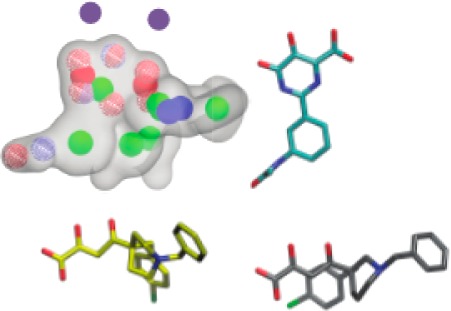
To discover new PAN inhibitors, we constructed a three-dimensional pharmacophore model, which avoids the limitations of docking scoring functions that may be exacerbated in the highly charged dimetal active site. After aligning the alpha-carbon atoms of nine publically available endonuclease-ligand complexes,19,20 the metal binding oxygen atoms of the corresponding ligands were superimposed, and five pharmacophore models were produced. A validation database was constructed by seeding known inhibitors that were not included in the pharmacophore models into the roughly 1300 compound National Cancer Institutes Diversity Set III, which we assumed was composed of nonbinders. The validation database was screened using each model, and receiver operating characteristic (ROC) curves were generated. Models were vetted by calculating the area under the ROC curve, or AUC, which is equivalent to the probability that the model will rank an inhibitor ahead of a decoy. The best performing pharmacophore model is shown in Figure 1a and produced an AUC of 0.899, with a p-value less than 0.01. Pharmacophore development and validation were performed with OpenEye software (OEChem version 3.1.2; OpenEye Scientific Software, Inc.), under a freely available academic license, as described in the Supporting Information.21,22
Figure 1.
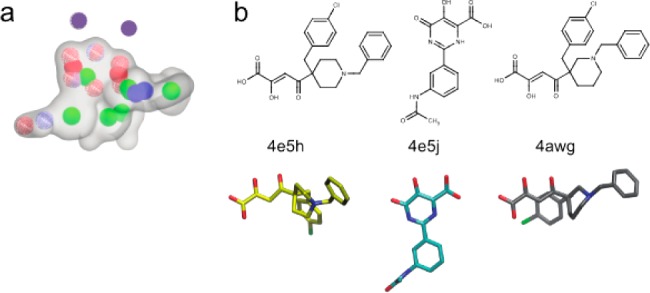
(a) Best performing pharmacophore model. Model volume is represented by a transparent gray surface. Blue, red, and red/blue wire frame spheres represent hydrogen bond donors, acceptors, and acceptors/donors, respectively. Green, red, and blue spheres represent ring centers, anions, and cations, respectively. Magenta spheres (not a model element) represent the positions of the active-site metal cations. (b) Inhibitors included in the model are shown in 2- and 3-dimensional representations and labeled with their PDB IDs. The 3-dimensional representations are aligned to the pharmacophore.
The best performing pharmacophore was used to screen the ChemBridge Express Pick screening library. The library was filtered to remove undesirable compounds, and database size was reduced from roughly 450,000 compounds to approximately 100,000 compounds. A conformational ensemble of each compound was generated. Each conformation was aligned to the pharmacophore and assigned a score based on the extent of overlap. The highest scoring conformation was used to rank each compound. Virtual library filtering and screening are detailed more extensively in the Supporting Information.
Then, 237 compounds with likely metal binding activity were selected from the virtual screens for experimental characterization. Endonuclease inhibition was measured in a FRET-based assay using a recombinant pH1N1 2009 PAN construct14 (assay detailed in the Supporting Information). Twenty compounds produced at least 50% inhibition at 200 μM, of which, 6 had dose response IC50 values below 50 μM, ranging from 0.74 to 46 μM. To develop structure–activity relationships around these 6 hits, an additional 27 analogues were purchased from ChemBridge and assayed for inhibition activity. Endonuclease inhibition activity of the analogues spanned a range of IC50 values, from 0.6 μM to greater than 100 μM. Compounds with an IC50 value less than or equal to 50 μM were further characterized by measuring their cytotoxicities. Cytotoxicity was determined in Madin-Darby canine kidney (MDCK) cells by measuring the CC50, or the compound concentration that kills half the population of MDCK cells (assay detailed in the Supporting Information). Antiviral activity was determined for nontoxic compounds whose CC50 values were greater than 50 μM. Antiviral activity was taken as the IC50, or the compound concentration at which half the population of PR8 H1N1 infected MDCK cells survive (assay detailed in the Supporting Information).15
In total, 16 of the 264 compounds tested inhibited endonuclease activity with IC50 values less than or equal to 50 μM. On the basis of their metal binding scaffolds, the inhibitors can be grouped into 5 classes: (1) 1,4-disubstituted-2,4-dioxobutanamides, (2) 1,6-disubstituted-1,3,4,6-hexanetetrones, (3) catechols, (4) spirodihydronapthalenones, and (5) trihydroxyphenyl compounds. In the Supporting Information, the most potent inhibitor from each class is shown aligned to the pharmacophore and docked to the active site.
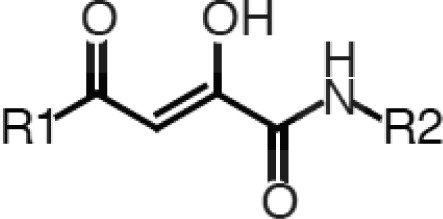
The 2,4-dioxobutanamides (Table 1) are similar to the series of 2,4-dioxobutanoic acids reported by Merck.10,11 Compounds from this series likely bind in a manner analogous to the 4-phenyl-2,4-dioxobutanoates19,20 but have greater opportunities for derivatization at the amide R2 position. The series indicates a preference for smaller groups at R2, which likely reflects active-site steric requirements, consistent with our docking results (Figure S5, Supporting Information). While compounds in this series have good endonuclease inhibition activity, they exhibit neither cell toxicity nor antiviral activity, suggesting poor cell permeability. Additionally, internal ligand strain may be an issue (Figures S3 and S4, Supporting Information).
Table 1. 1,4-Disubstituted-2,4-dioxobutanamides.
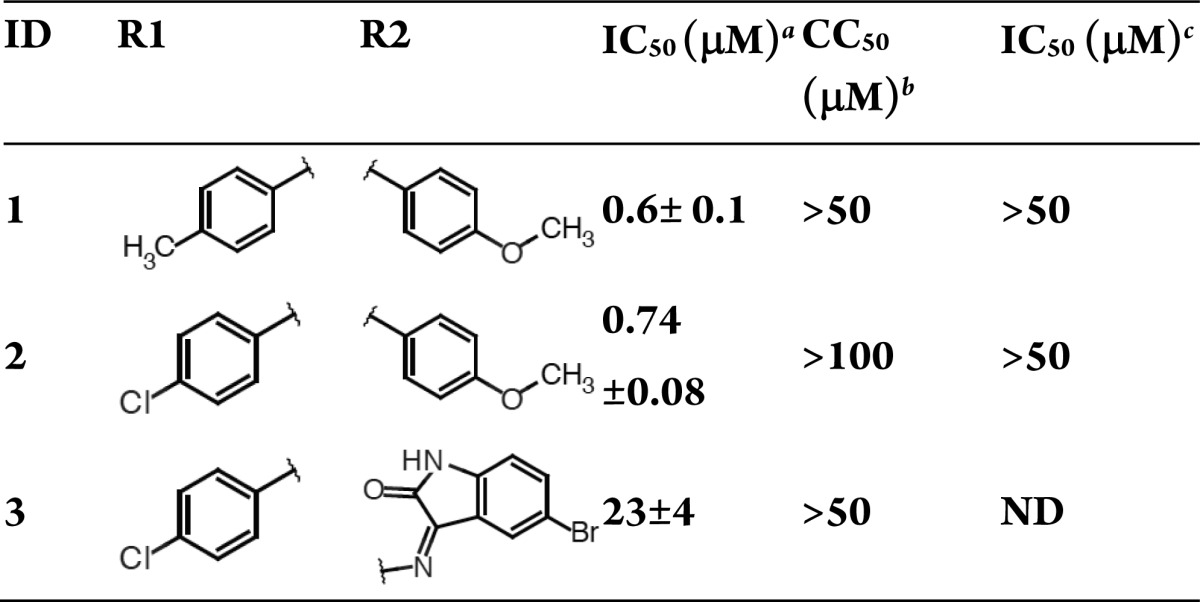
H1N1 endonuclease inhibition.
MDCK cytotoxicity.
Inhibition of the cytopathic effect in MDCK antiviral inhibition. Averages and standard errors over 3 independent measurements are reported.
The 1,6-disubstituted-1,3,4,6-hexanetetrones (Table 2) and butanamides are closely related, and reflection through the plane bisecting the butanamide carbon atoms 1 and 2 yields the hexanetetrone scaffold. The structural similarities imply that, like the butanamides, internal ligand strain may penalize binding. A roughly 2-fold preference for the tolyl substituent suggests that the R1 and R2 groups may occupy predominantly apolar-binding pockets, which is consistent with our docking results (Figure S5, Supporting Information). The most promising of the two, compound 4, pairs low cell toxicity with reasonable endonuclease inhibition and antiviral activity.
Table 2. 1,6-Disubstituted-1,3,4,6-hexanetetrones.
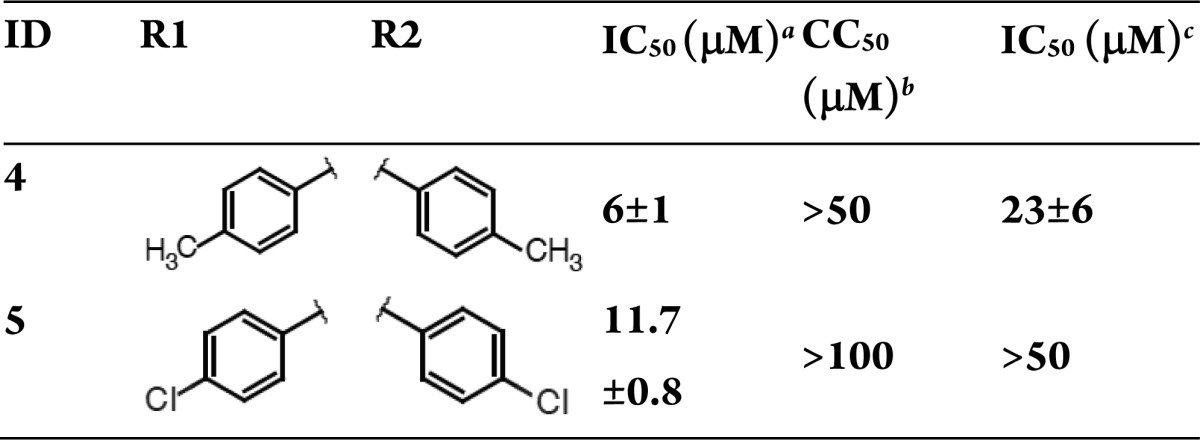
H1N1 endonuclease inhibition.
MDCK cytotoxicity.
Inhibition of the cytopathic effect in MDCK antiviral inhibition. Averages and standard errors over 3 independent measurements are reported.
The catechol compounds (Table 3) are homologous to the phenethylphenylphthalimides14 and green tea catechins.15 Compounds 6, 7, 9, and 10 share a similar 3,4-dihydroxy-N-(phenylmethylidene) benzohydrazide scaffold, while a piperazine bridges two substituted phenyl groups in 8. The metal binding oxygen atoms in this class are preorganized around the phenyl ring reducing internal ligand strain. Comparing the benzohydrazide derivatives indicates a preference for smaller phenylmethylidene substituents, with a roughly 3-fold preference for the dichlorinated substituent (6 vs 7). However, both compounds 6 and 7 have liabilities, with 6 being cytotoxic and 7 lacking cytotoxicity and antiviral activity. Compound 8 is cytotoxic, and compound 9 lacks antiviral activity and cytotoxicity. Compound 10 was poorly soluble, and neither cell toxicity nor antiviral activity could be determined. The order of magnitude drop in IC50 of compound 10 indicates both that the active site can accommodate larger molecules (provided the appropriate metal binding groups are present and that there is a preference for the smaller compounds in the series, consistent with structural data.19,20
Table 3. Catechols.
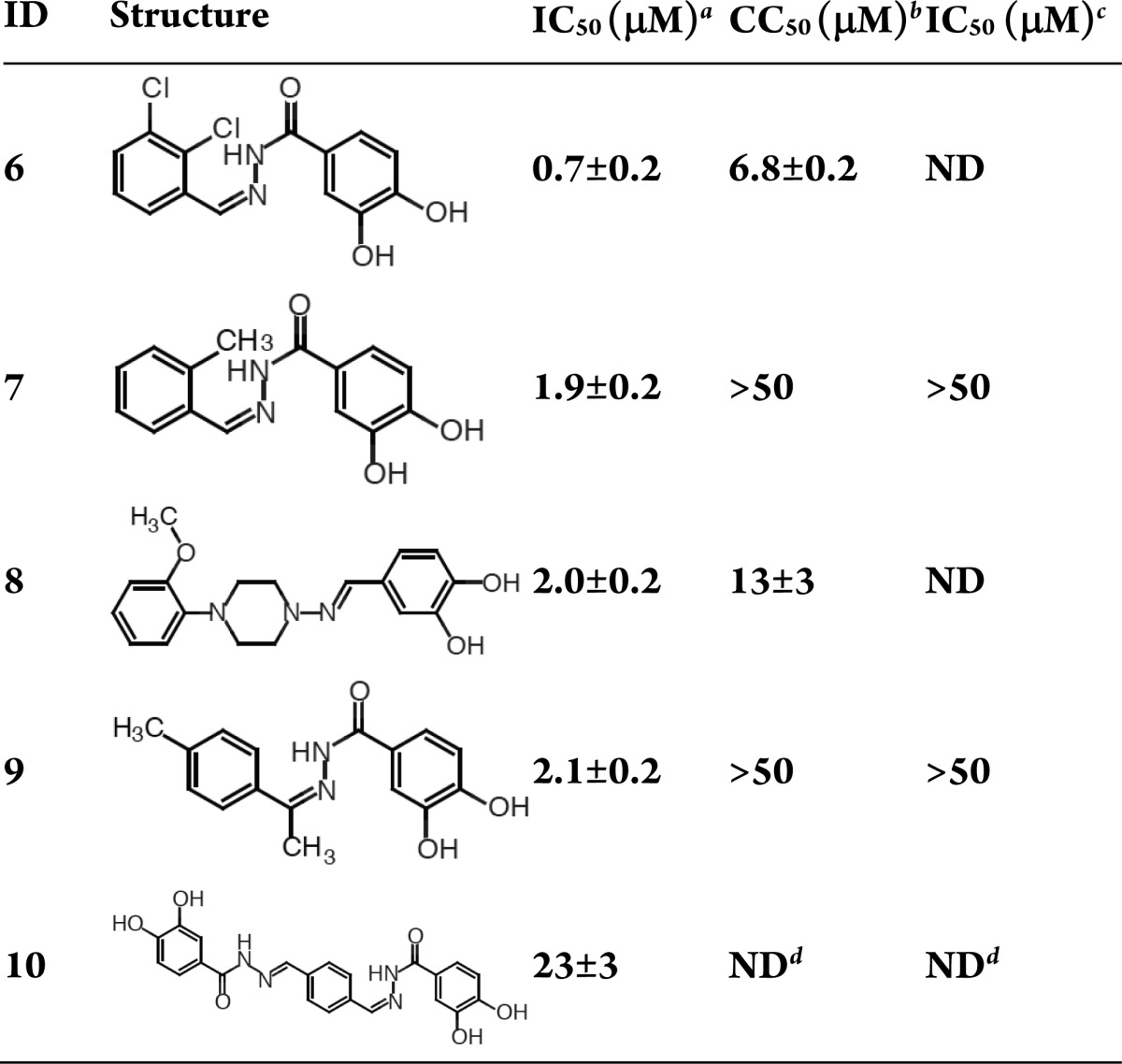
H1N1 endonuclease inhibition.
MDCK cytotoxicity.
Inhibition of the cytopathic effect in MDCK antiviral inhibition.
Solubility issues hampered cytotoxicity and antiviral assays. Averages and standard errors over 3 independent measurements are reported.
Like the catechols, the spirodihydronaphthalenones (Table 4) contain a dihydroxy phenyl metal binding group. Comparing compounds 11 and 12 shows a roughly 6-fold preference for the unsubstituted cyclohexyl ring. Both compounds lack cytotoxicity and antiviral activity.
Table 4. Spirodihydronaphthalenones.
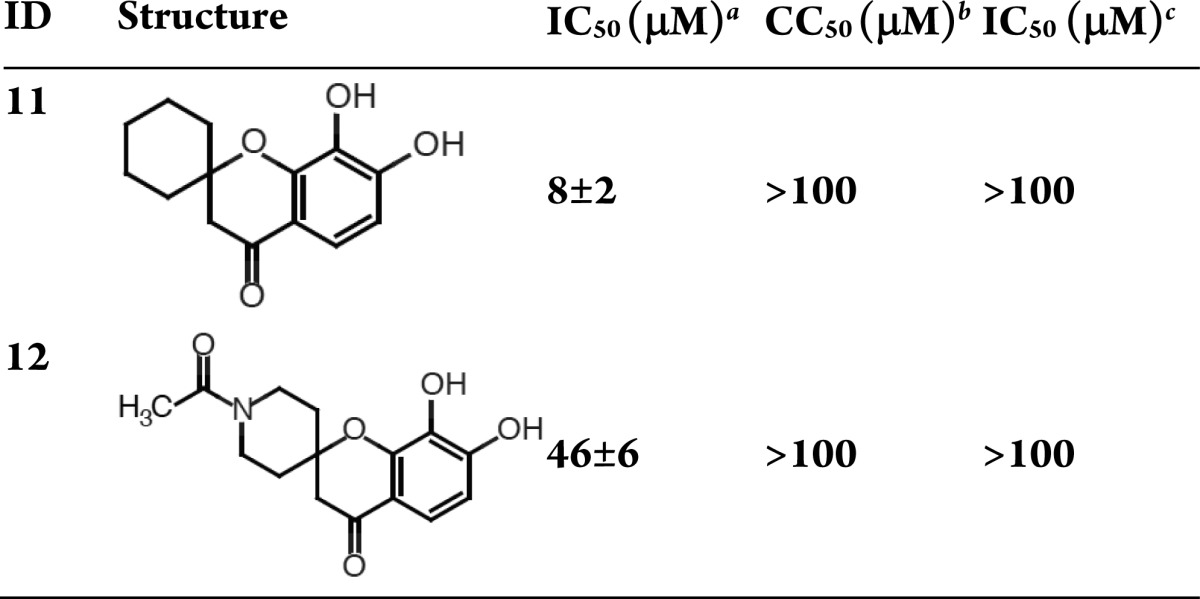
H1N1 endonuclease inhibition.
MDCK cytotoxicity.
Inhibition of the cytopathic effect in MDCK antiviral inhibition. Averages and standard errors over 3 independent measurements are reported.
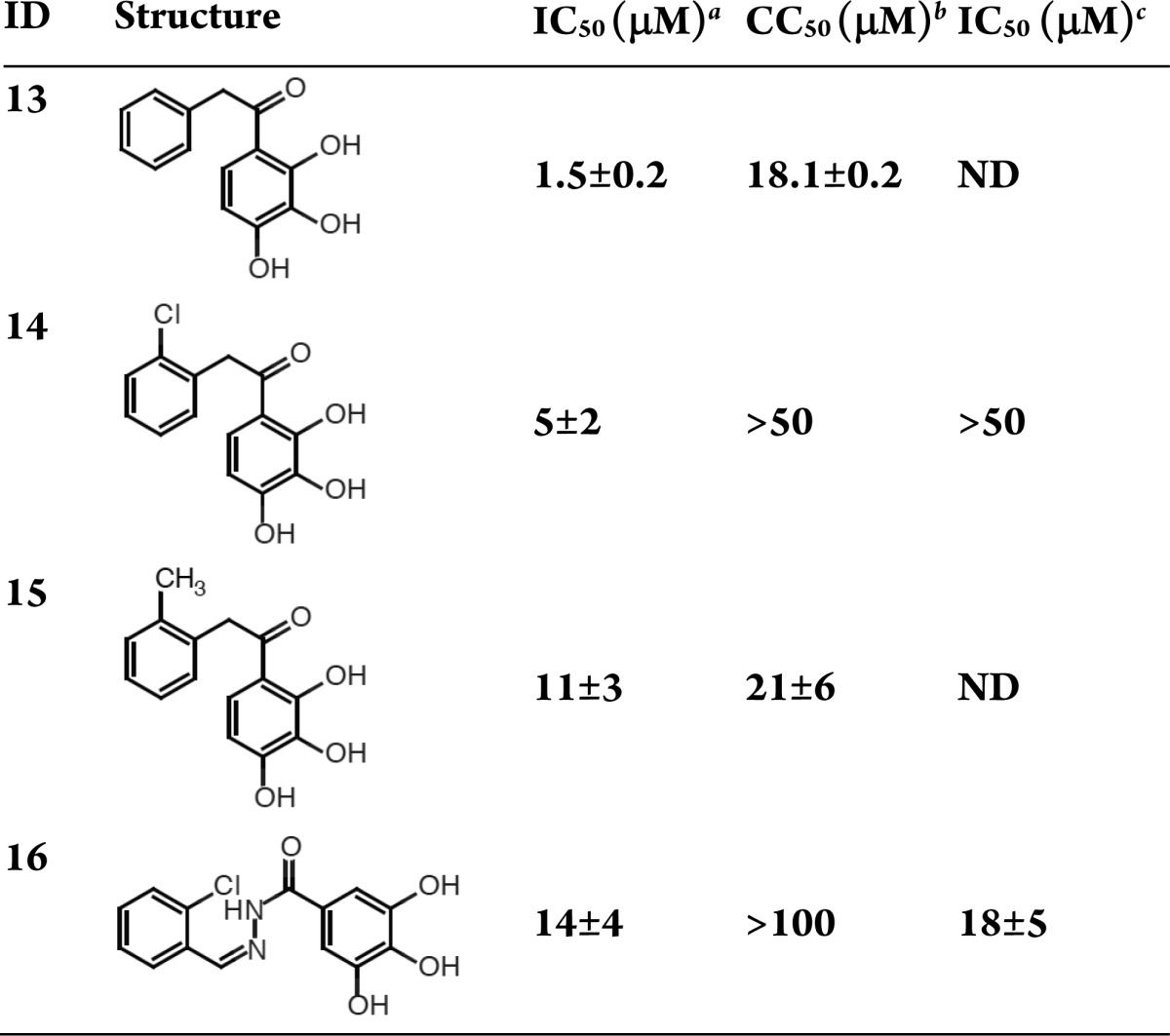
With the exception of compound 14, which is neither cytotoxic nor active against virally infected cells, the 2,3,4-trihydroxyl compounds (Table 5) are cytotoxic, despite their reasonable endonuclease inhibition activity. In contrast, compound 16, an oxidized analogue of the N-(phenylmethylidene)benzohydrazide scaffolds (compounds 6, 7, 9, and 10 in Table 3), has comparable enzyme inhibition and antiviral activities with negligible cytotoxicity.
Table 5. Trihydroxyphenyl Compounds.
H1N1 endonuclease inhibition.
MDCK cytotoxicity.
Inhibition of the cytopathic effect in MDCK antiviral inhibition. Averages and standard errors over 3 independent measurements are reported.
In summary, this letter reported the development and application of a 3-dimensional structure-guided pharmacophore that led to the discovery of 16 new endonuclease inhibitors, spanning 5 molecular classes with IC50 values less than 50 μM. Ten of these compounds have IC50 values below 10 μM, and three have IC50 values below 1 μM. Each compound was further characterized by determining toxicity and antiviral activity in MDCK cells. Two compounds have promising profiles. Compound 4 is a 1,3,4,6-hexanetetrone that inhibits the endonuclease with an IC50 of 6 μM, allows infected cell survival with an IC50 of 23 μM, and shows negligible toxicity. Similarly, compound 16, a N-(phenylmethylidene)benzohydrazide, has negligible cell toxicity, allows infected cell survival with an IC50 of 18 μM and inhibits endonuclease catalysis with an IC50 of 14 μM. The compound classes reported here should facilitate future discovery and design efforts.
Supporting Information Available
Details describing pharmacophore model development, library filtering and screening, endonuclease inhibition assays, cytotoxicity assays, antiviral activity assays, pharmacophore alignment and docking, and evaluation of dioxobutanamide internal ligand strain. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ These authors contributed equally to this work.
The work was carried out with financial support from the NIH DP2 OD007237 and XSEDE grant CHE060073N, to R.E.A. Additional support from the National Biomedical Computation Resource, Center for Theoretical Biological Physics, and KeckII Center is gratefully acknowledged.
The authors declare no competing financial interest.
This paper was posted ASAP on November 6, 2013. References 21 and 22 were added and the revised version was reposted on November 13, 2013.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Seasonal Influenza (Flu). http://www.cdc.gov/flu (accessed 3/5/2012).
- Influenza (Seasonal). http://www.who.int/mediacentre/factsheets/fs211/en/index.html (accessed 3/5/2013).
- Nichol K. L.; Wuorenma J.; von Sternberg T. Benefits of influenza vaccination for low-, intermediate-, and high-risk senior citizens. Arch. Intern. Med. 1998, 158, 1769–1776. [DOI] [PubMed] [Google Scholar]
- de Jong M. D.; Tran T. T.; Truong H. K.; Vo M. H.; Smith G. J.; Nguyen V. C.; Bach V. C.; Phan T. Q.; Do Q. H.; Guan Y.; Peiris J. S.; Tran T. H.; Farrar J. Oseltamivir resistance during treatment of influenza A (H5N1) infection. N. Engl. J. Med. 2005, 353, 2667–2672. [DOI] [PubMed] [Google Scholar]
- Leang S. K.; Deng Y. M.; Shaw R.; Caldwell N.; Iannello P.; Komadina N.; Buchy P.; Chittaganpitch M.; Dwyer D. E.; Fagan P.; Gourinat A. C.; Hammill F.; Horwood P. F.; Huang Q. S.; Ip P. K.; Jennings L.; Kesson A.; Kok T.; Kool J. L.; Levy A.; Lin C.; Lindsay K.; Osman O.; Papadakis G.; Rahnamal F.; Rawlinson W.; Redden C.; Ridgway J.; Sam I. C.; Svobodova S.; Tandoc A.; Wickramasinghe G.; Williamson J.; Wilson N.; Yusof M. A.; Kelso A.; Barr I. G.; Hurt A. C. Influenza antiviral resistance in the Asia-Pacific region during 2011. Antiviral Res. 2013, 97, 206–210. [DOI] [PubMed] [Google Scholar]
- Plotch S. J.; Bouloy M.; Ulmanen I.; Krug R. M. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 1981, 23, 847–858. [DOI] [PubMed] [Google Scholar]
- Das K.; Aramini J. M.; Ma L. C.; Krug R. M.; Arnold E. Structures of influenza A proteins and insights into antiviral drug targets. Nat. Struct. Mol. Biol. 2010, 17, 530–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa M.; Kadowaki S. E.; Watanabe I.; Kadowaki Y.; Takei M.; Fukuda H. PA subunit of RNA polymerase as a promising target for anti-influenza virus agents. Antiviral Res. 2008, 78, 194–201. [DOI] [PubMed] [Google Scholar]
- Moscona A. Global transmission of oseltamivir-resistant influenza. N. Engl. J. Med. 2009, 360, 953–956. [DOI] [PubMed] [Google Scholar]
- Tomassini J.; Selnick H.; Davies M. E.; Armstrong M. E.; Baldwin J.; Bourgeois M.; Hastings J.; Hazuda D.; Lewis J.; McClements W.; et al. Inhibition of cap (m7GpppXm)-dependent endonuclease of influenza virus by 4-substituted 2,4-dioxobutanoic acid compounds. Antimicrob. Agents Chemother. 1994, 38, 2827–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings J. C.; Selnick H.; Wolanski B.; Tomassini J. E. Anti-influenza virus activities of 4-substituted 2,4-dioxobutanoic acid inhibitors. Antimicrob. Agents Chemother. 1996, 40, 1304–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomassini J. E.; Davies M. E.; Hastings J. C.; Lingham R.; Mojena M.; Raghoobar S. L.; Singh S. B.; Tkacz J. S.; Goetz M. A. A novel antiviral agent which inhibits the endonuclease of influenza viruses. Antimicrob. Agents Chemother. 1996, 40, 1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes K. E.; Ermert P.; Fassler J.; Ives J.; Martin J. A.; Merrett J. H.; Obrecht D.; Williams G.; Klumpp K. Use of a pharmacophore model to discover a new class of influenza endonuclease inhibitors. J. Med. Chem. 2003, 46, 1153–1164. [DOI] [PubMed] [Google Scholar]
- Iwai Y.; Takahashi H.; Hatakeyama D.; Motoshima K.; Ishikawa M.; Sugita K.; Hashimoto Y.; Harada Y.; Itamura S.; Odagiri T.; Tashiro M.; Sei Y.; Yamaguchi K.; Kuzuhara T. Anti-influenza activity of phenethylphenylphthalimide analogs derived from thalidomide. Bioorg. Med. Chem. 2010, 18, 5379–5390. [DOI] [PubMed] [Google Scholar]
- Kuzuhara T.; Iwai Y.; Takahashi H.; Hatakeyama D.; Echigo N. Green tea catechins inhibit the endonuclease activity of influenza A virus RNA polymerase. PLoS Curr. 2009, RRN1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai Y.; Murakami K.; Gomi Y.; Hashimoto T.; Asakawa Y.; Okuno Y.; Ishikawa T.; Hatakeyama D.; Echigo N.; Kuzuhara T. Anti-influenza activity of marchantins, macrocyclic bisbibenzyls contained in liverworts. PLoS One 2011, 6, e19825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman B. M.; Jake Slavish P.; DuBois R. M.; Boyd V. A.; White S. W.; Webb T. R. Identification of influenza endonuclease inhibitors using a novel fluorescence polarization assay. ACS Chem. Biol. 2012, 7, 526–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagong H. Y.; Parhi A.; Bauman J. D.; Patel D.; Vijayan R. S. K.; Das K.; Arnold E.; LaVoie E. J. 3-Hydroxyquinolin-2(1H)-ones as inhibitors of influenza A endonuclease. ACS Med. Chem. Lett. 2013, 4, 547–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBois R. M.; Slavish P. J.; Baughman B. M.; Yun M. K.; Bao J.; Webby R. J.; Webb T. R.; White S. W. Structural and biochemical basis for development of influenza virus inhibitors targeting the PA endonuclease. PLoS Pathog. 2012, 8, e1002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalinski E.; Zubieta C.; Wolkerstorfer A.; Szolar O. H.; Ruigrok R. W.; Cusack S. Structural analysis of specific metal chelating inhibitor binding to the endonuclease domain of influenza pH1N1 (2009) polymerase. PLoS Pathog. 2012, 8, e1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins P. C.; Skillman A. G.; Warren G. L.; Ellingson B. A.; Stahl M. T. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model 2010, 50, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins P. C.; Nicholls A. Conformer generation with OMEGA: learning from the data set and the analysis of failures. J. Chem. Inf. Model 2012, 52, 2919–2936. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.