

Abstract
The cyclic nucleotides cAMP and cGMP are common signaling molecules synthesized in neurons following the activation of adenylyl or guanylyl cyclase. In the striatum, the synthesis and degradation of cAMP and cGMP is highly regulated as these second messengers have potent effects on the activity of striatonigral and striatopallidal neurons. This review will summarize the literature on cyclic nucleotide signaling in the striatum with a particular focus on the impact of cAMP and cGMP on the membrane excitability of striatal medium-sized spiny output neurons (MSNs). The effects of non-selective and selective phosphodiesterase (PDE) inhibitors on membrane activity and synaptic plasticity of MSNs will also be reviewed. Lastly, this review will discuss the implications of the effects PDE modulation on electrophysiological activity of striatal MSNs as it relates to the treatment of neurological disorders such as Huntington’s and Parkinson’s disease.
Keywords: cyclic AMP, cyclic GMP, phosphodiesterase, electrophysiology
Introduction
Synthesis and degradation of cAMP and cGMP in striatal neurons
A variety of striatal neurotransmitters exert their actions via stimulation of membrane bound G-protein coupled receptors (GPCRs). Activation of GPCRs by neurotransmitters such as dopamine (DA) or adenosine stimulates the production of intracellular second messengers such as adenosine 3′-5′-cyclic monophosphate (cAMP), which is synthesized from adenosine triphosphate (ATP) following activation of adenylyl cyclase (AC) (see Figure 1). In most cases neurotransmitter receptors are functionally coupled to AC via either a stimulatory (Golf, Gs) or inhibitory G-protein (Gi) (see [1] for review). Additionally, the resulting influx of Ca2+ into the cell during activation of N-methyl-d-aspartate (NMDA) receptors or voltage-gated Ca2+ channels can mediate a calmodulin-dependent activation or inhibition of specific isoforms of AC [2]. Indeed, it is plausible that cAMP synthesis is controlled via a variety of neurotransmitter interactions targeting both GPCRs and ligand- and voltage-gated ion channels. Downstream effector pathways regulated by AC-cAMP signaling include cAMP-dependent protein kinase (PKA), GTP exchange protein activated by cAMP (EPAC), and cyclic nucleotide-gated channels (CNGC).
Figure 1. Schematic diagram of the roles of DA-AC-cAMP-PKA and NO-sGC-cGMP-PKG signaling and PDE function in the regulation of MSN membrane excitability.
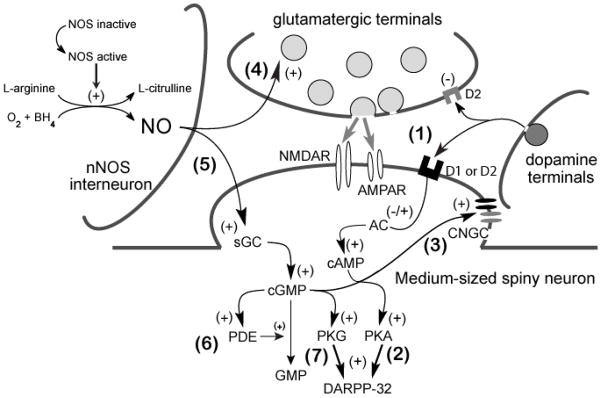
(1) DA released from DA terminals binds to D1-like or D2-like DA receptors on the postsynaptic MSN and leads to either stimulation or inhibition of AC, respectively. (2) Increases in cAMP tone will activate PKA which can phosphorylate DARPP-32 to have complex effects on downstream signaling pathways, neurotransmitter receptors, and voltage-gated ion channels (see [1] for review). (3) cAMP (along with cGMP) can also stimulate CNGCs in the plasma membrane which can lead to cation influx and membrane depolarization. (4) Tonic NO signaling increases glutamatergic transmission across corticostriatal synapses potentially via nitrosylation of presynaptic release proteins or a sGC-cGMP-dependent mechanism. (5) NO transmission can also activate sGC in the postsynaptic MSN and stimulate cGMP production. cGMP can activate CNGC (see step 3), stimulate PDEs (6), or activate PKG (7). Transient activation of NO-sGC-cGMP signaling in the intact striatum increases the responsiveness of MSNs to excitatory glutamatergic drive and facilitates short-term potentiation of corticostriatal synaptic transmission (see above and [57] for review). More robust stimulation of NO-sGC-cGMP-PKG signaling performed in striatal slice preparations can induce LTD of corticostriatal synaptic transmission (see [22] for review). Together these studies suggest that like DA D1/5 receptor modulation, the net impact of NO-sGC-cGMP-PKG signaling on membrane excitability may depend on the steady-state membrane potential of MSNs and interactions with glutamateric drive and NMDA receptor activation (Modified with permission from [57]).
Despite receiving less attention relative to cAMP, guanosine 3′-5′-cyclic monophosphate (cGMP) has been shown to play important roles in critical biological processes such as cognition and memory, hemodynamic regulation, immune response, and motor function. cGMP is synthesized in striatal medium-sized spiny output neurons (MSNs) and cholinergic interneurons following stimulation of membrane bound particulate guanylyl cyclase (pGC) or soluble guanylyl cyclase (sGC). Activation of pGC is mediated by natriuretic peptides, whereas activation of sGC is mediated by the gaseous neuromodulator nitric oxide (NO) [3–5]. The NO-sensitive sGC isoform is the primary enzyme responsible for cGMP synthesis [5]. In the striatum, NO is produced in a subclass of aspiny interneurons expressing neuronal NO synthase (nNOS) [6–10]. Transient elevations of cGMP produced by NO-dependent activation of sGC can modulate the function of several effector enzymes including CNGCs, cGMP-dependent protein kinases (PKGs), and various cyclic nucleotide phosphodiesterases (PDEs) [11].
PDEs regulate both spatial and temporal aspects of cAMP and cGMP signaling within distinct subcellular compartments of striatal neurons [12, 13]. Both cAMP and cGMP are metabolized via dual-substrate or subtype-specific PDEs to 5′-AMP or 5′-GMP, respectively. The PDE gene superfamily is comprised of eleven distinct families based on sequence homology, substrate specificity, and regulation [12, 14, 15]. Unique splice variants containing different N-terminal regulatory domains are found within each PDE family, giving rise to close to 100 [k1] individual proteins. PDE families can be grouped into three divisions according to substrate specificity; cAMP PDEs (PDE4, PDE7 and PDE8), cGMP PDEs (PDE5, PDE6 and PDE9), and dual-substrate PDEs (PDE1, PDE2, PDE3, PDE10 and PDE11). The majority of the above PDE isoforms are expressed in striatum (e.g., PDE 1, 2, 3, 4, 8, 9, and 10) [14, 16]. In many cases, different isoforms are thought to be differentially distributed and compartmentalized within individual striatal neurons. This differential sub-cellular targeting of striatal PDEs may play a critical role in determining the impact of cyclic nucleotide signaling on neuronal membrane excitability and synaptic plasticity. Therefore, drugs targeting specific PDE isoforms have the potential to elevate cyclic nucleotide concentrations within specific sub-compartments of the cell to enhance cyclic nucleotide signaling. For many years non-selective PDE inhibitors such as IBMX have been broadly used to investigate various physiological effects of cyclic nucleotides in brain regions such as striatum. The discovery and production of isoform specific PDE inhibitors will facilitate more selective manipulations of cyclic nucleotide tone within given cell populations, and potentially, within different subcellular compartments. Increasing numbers of studies have begun to evaluate the role of specific PDE isoforms on striatal function and behavior. This can be achieved pharmacologically using isoform-specific PDE inhibitors or genetically using mice lacking specific PDE isoforms. Some of these studies have examined the ability of PDE manipulations to affect measures of neuronal membrane excitability and synaptic plasticity of neuronal pathways in striatum. Outcomes from these studies (reviewed below) will be useful in assessing the potential of PDE inhibitors as therapeutic agents in a variety of neurological disorders characterized by impaired neuronal function and synaptic plasticity in striatal MSNs.
Cyclic nucleotide signaling
As mentioned above, cAMP and cGMP signaling can affect a variety of downstream targets via the activation of PKA and PKG, respectively (see Figure 1). The activation of these kinases is thought to lead to the phosphorylation of numerous other proteins including ion channels and phosphatases involved in modulating neuronal membrane excitability. For example, in the striatum, activation of DA and cAMP regulated phosphoprotein MW 32 kDa (DARPP-32) is thought to play a critical role in mediating many of the neuromodulatory effects of DA on local striatal GABAergic and glutamatergic transmission [1]. The PKA- or PKG-mediated phosphorylation of DARPP-32 on Thr-34 activates the inhibitory phosphatase action of DARPP-32 and drives the dephosphorylation and inhibition of protein phosphatase-1 (PP1). This enables PKA to induce the phosphorylation of other target substrates such as cAMP response element binding protein (CREB). Interestingly, activation of CREB initiates gene transcription thought to be involved in long-term synaptic plasticity and memory formation [17–19].
As mentioned above, PKG is also a potent activator of striatal DARPP-32 [1]. Studies performed in striatal brain slices have shown that the NO-sGC-cGMP mediated stimulation of PKG results in increased DARPP-32 (Thr-34) phosphorylation in MSNs [20, 21]. The PKG-mediated phosphorylation of DARPP-32 is rapid, transient and requires a coincidental increase in intracellular calcium levels induced following glutamatergic stimulation of NMDA, AMPA and metabotropic glutamate 5 receptors [20, 21]. Acting in concert, the PKA/PKG effector pathways play a key role in regulating long-term changes in striatal synaptic efficacy [22], and as such, are likely to be critically involved in the planning and execution of complex motor behaviors [23].
The remainder of this review will describe how regulation of cAMP/cGMP signaling modifies striatal MSN membrane excitability and corticostriatal synaptic plasticity. The role of specific PDE subtypes in regulating these key processes and the potential utility of sub-type specific PDE inhibitors/activators for reversing pathophysiological changes in membrane excitability and synaptic plasticity will be discussed.
Modulation of striatal neuronal excitability by cyclic nucleotides and PDEs
Striatal projection cells (i.e., MSNs) express very high levels of ACs, sGCs, and various PDEs. Indeed, numerous PDEs appear to play a complex role in the regulation of both cGMP and cAMP signaling cascades in both striatonigral and striatopallidal MSNs [13]. Several PDE isoforms/families are highly expressed in the striatum including the dual substrate enzymes PDE1B, PDE2A, and PDE10A. The cAMP specific enzymes PDE3A, PDE3B, PDE4D, PDE7B, and PDE8B are also prominently expressed in striatum. The cGMP specific enzyme PDE9A is moderately expressed in the striatum and appears to regulate tonic levels of cGMP [14].
Activation of AC and sGC in the striatum is controlled by a variety of different neurotransmitters (e.g., DA, 5HT, neuropeptide Y, adenosine, glutamate) which act through GPCRs and stimulation of NO synthesis, respectively [1]. Numerous studies from our lab and others have shown that under physiological conditions, drugs that augment cAMP or cGMP levels in MSNs (e.g., PDE inhibitors or cyclase activators) facilitate spontaneous and evoked corticostriatal transmission [24–26]. Conversely, drugs that decrease cyclic nucleotide production (e.g., cyclase inhibitors) or block protein kinase activity have opposite effects [24, 26, 27]. For example, pioneering studies by Michael Levine’s group revealed that bath application of the AC activator forskolin enhanced the amplitude and duration of cortically-evoked glutamatergic synaptic responses in a dose-dependent manner [24]. The facilitatory effects of forskolin on glutamatergic transmission were blocked by bath application of PKA inhibitors and mimicked by similar application of a PKA activator. Forskolin was also shown to enhance the excitatory effects of local perfusion of glutamate receptor agonists on membrane activity [24]. Furthermore, application of a PKA inhibitor alone decreased the amplitude of cortically-evoked excitatory post synaptic potentials (EPSPs) [24]. Studies in organotypic co-cultures of corticostriatal networks have also shown that PKA inhibition can eliminate NMDA receptor-mediated plateau depolarizations [28]. Taken together, these outcomes indicate that activation of AC-cAMP-PKA signaling in striatal MSNs facilitates corticostriatal transmission and potentiates the excitatory effects of AMPA and NMDA receptor activation. It is also now clear that excitatory responses induced by NMDA are potentiated by D1 and attenuated by D2 receptor stimulation [29–34]. Given that the D1 family of DA receptors is positively coupled to AC and D2-like receptors are negatively coupled to AC [1], it is very likely that stimulation of postsynaptic AC-cAMP-PKA signaling by D1-like receptors is a primary signaling pathway by which DA facilitates corticostriatal transmission. Conversely, D2 receptor activation produces the opposite effect in part by suppressing AC-cAMP-PKA signaling cascades in both pre- and postsynaptic elements (Figure 1).
Our laboratory has focused on how striatal NO-sGC-cGMP signaling modulates the membrane properties of MSNs and their responsiveness to excitatory corticostriatal synaptic transmission. Striatal NO is synthesized by a subclass of relatively aspiny GABAergic interneurons which express nNOS [35]. These NO interneurons make sparse synaptic contacts with the more distal dendrites of sGC-expressing MSNs [36]; although see also [37, 38]. Given the gaseous and diffusible nature of NO, synaptic connections may not be required for NO-mediated activation of the postsynaptic sGC receptor. In any event, several studies performed in intact rats found that striatal NO-sGC-cGMP signaling acts to enhance the membrane excitability of striatal MSNs and facilitate corticostriatal transmission [25–27, 35, 39, 40]. Early work suggested that reverse dialysis of NO generating compounds increased the basal firing rate and burst duration of striatal neurons recorded in anesthetized rats [35]. Conversely, drugs designed to deplete NO such as nNOS inhibitors or NO scavengers have been shown to suppress spontaneous firing [39], possibly via decreasing the amplitude of spontaneously occurring, glutamate-driven depolarized plateau potentials (i.e., up events) and by hyperpolarizing the membrane potential of MSNs [26]. NO scavengers also decrease the responsiveness of striatal MSNs to both depolarizing current injections [35] and cortical stimulation [26]. Similar inhibitory effects are observed with systemic or intracellular administration of sGC inhibitors [26, 27, 40]. The inhibitory effects of nNOS and sGC inhibitors on the membrane excitability of striatal MSNs recorded in vivo were observed to be at least partially reversed by cGMP or cGMP analogues, indicating that NO and cGMP act to enhance MSN activity and increase their responsiveness to corticostriatal input [26].
Consistent with observations with NO generators and cGMP, we have found that systemic, intrastriatal, and intracellular administration of various selective and non-selective PDE inhibitors increases membrane excitability and responsiveness of MSNs to corticostriatal drive [25–27; See Table 1]. For example, intracellular application of the PDE inhibitor zaprinast depolarized the membrane potential of MSNs and increased the duration of depolarized up states which are known to be driven by cortical input [26]. Likewise, intracellular injection of the PDE10A inhibitor papaverine depolarized the average membrane potential and increased the duration of the depolarized up states (unpublished observations). We also found that reverse dialysis of the PDE10A inhibitors papaverine or TP-10 increased cortically-evoked spike activity of MSNs and total number of spikes elicited per stimulation [25]. Intrastriatal infusion of TP-10 also decreased the onset latency of cortically-evoked spikes and reduced the variance in latency to spike, suggesting that when activated, PDE10A may filter asynchronous or weak cortical input to MSNs by suppressing cyclic nucleotide production [25]. Together, these findings indicate that inhibition of striatal PDE10A activity, and the resulting increase in cAMP/cGMP tone, increases the responsiveness of MSNs to glutamatergic corticostriatal transmission (Table 1).
Table 1.
Effects of specific PDE inhibitors and genetic deletion on striatal function and related behavioural measures
| PDE | Substrate | Selective Inhibitors | Effects of specific PDE manipulations | Refs |
|---|---|---|---|---|
|
PDE 1 PDE1B |
cGMP>cAMP | Vinpocetine/IC224 |
|
[99–102] |
|
PDE 2 PDE2A |
cGMP/cAMP | EHNA/BAY 60-7550 |
|
[103–105] |
|
PDE 3 PDE3A,B |
cAMP | Milrinone/Cilostamide |
|
[12–16, 106] |
|
PDE 4 PDE4D |
cAMP | Rolipram |
|
[12–16, 45, 107, 116, 119] |
|
PDE 7 PDE7B |
cAMP | – |
|
[108, 109] |
|
PDE 8 PDE8B |
cAMP | – |
|
[12–16, 110] |
|
PDE 9 PDE9A |
cGMP | BAY 73-6691/PF-4447943 |
|
[14, 111, 112] |
|
PDE 10 PDE10A |
cGMP/cAMP | Papaverine/TP-10 (PF-545920)/PQ-10/MP-10/THPP-1 |
|
[13, 25, 41–46, 74, 76, 93, 113–115, 117–119] |
It is well known that striatal output is relayed by MSNs making up one of two pathways: 1) the “direct” pathway (striatonigral MSNs) or, 2) the “indirect” pathway (striatopallidal MSNs). Given that there is no evidence for significant differential expression of PDE10A mRNA or protein in striatopallidal or striatonigral MSNs [41–44], we predicted that PDE10A inhibitors would have identical effects on identified striatopallidal and striatonigral MSNs. We found that a robust increase in cortically-evoked activity was apparent in putative striatopallidal MSNs following systemic administration of TP-10 [25]. Surprisingly, TP-10 administration did not affect cortically-evoked activity in antidromically-identified striatonigral MSNs. Based on these findings, we predict that PDE10A inhibition has a greater facilitatory effect on corticostriatal synaptic activity in striatopallidal as compared to striatonigral MSNs [25]. This hypothesis is strongly supported by studies by Snyder and colleagues demonstrating that papaverine increases DARPP-32 and GluR1 (AMPA) receptor phosphorylation much more robustly in identified striatopallidal versus striatonigral MSNs [45]. However, it should also be noted that other studies have shown that PDE10A inhibition has similar effects on gene expression in striatopallidal as compared to striatonigral MSNs [46; see also Table 1]. Thus, TP-10 was found to elevate p-CREB in all MSNs and similar increases in substance P and enkephalin mRNA were also observed with this compound [46]. Together, the above studies suggest that while PDE10A inhibition may exert more widespread and potent changes in responsiveness to excitatory synaptic drive in striatopallidal MSNs, it can also induce potent effects on intracellular signaling and gene expression in striatonigral MSNs.
In support of the above conclusion, we found that TP-10 administration increased the incidence of antidromic responses observed in identified striatonigral MSNs [25]. Therefore, it is likely that PDE10A inhibition increases the axonal, and potentially, terminal excitability of striatonigral MSNs. This finding is not surprising given that PDE10A is transported to the axons and terminals of MSNs and, based on Western blot analysis, the terminal concentration of the protein appears to be at least as high as in striatum [43]. Taken together, these findings strongly indicate that PDE10A inhibition increases striatal output from both striatopallidal and striatonigral MSNs (Table 1). However, it is likely that the facilitatory effect on striatonigral MSNs is mediated via direct excitatory effects on axons, and potentially, nerve terminals.
Modulation of striatal synaptic plasticity by cyclic nucleotides and PDEs
Corticostriatal synaptic plasticity is believed to occur in a bidirectional manner and both long-term depression (LTD) and long-term potentiation (LTP) can be observed in direct and indirect pathway MSNs under appropriate conditions [47]. A variety of studies have also shown that interactions between striatal DA and NO signaling play a critical role in the regulation of short and long-term synaptic plasticity [27, 36, 39, 48, 49]. In many cases this is thought to occur via cyclic nucleotide-dependent actions on protein kinase and phosphatase activities [20, 50]. In the intact animal, NO release can be evoked by high frequency stimulation (30 Hz) of corticostriatal pathway in a manner that depends on NMDA and DA D1/5 receptor activation [40]. This facilitation of NO signaling may also act in a retrograde manner to potentiate presynaptic glutamate release [39, 51]. Consistent with this, we found that systemic administration of a nNOS inhibitor abolished excitatory responses observed in striatal MSNs during high frequency stimulation of the frontal cortex [27]. Furthermore, inhibition of nNOS activity was shown to increase the magnitude of D2 receptor-mediated short-term depression of cortically-evoked spike activity induced during phasic stimulation of frontal cortical afferents [39]. Therefore, corticostriatal transmission may be preferentially detected and amplified under some conditions by nNOS interneurons in a feed-forward excitatory manner which may initiate the synchronization of local network activity with synaptically-driven events. In addition, disruption of NO signaling would be expected to impede the integration of corticostriatal transmission and short-term plasticity within striatal networks [26, 39, 40]. The above interpretation is supported by studies showing that pharmacological or genetic downregulation of striatal nNOS activity has profound effects on striatal output as measured in electrophysiological [35, 52] and behavioral studies [23].
In addition to the above in vivo studies showing that NO-sGC-cGMP signaling can facilitate corticostriatal transmission and short-term synaptic plasticity, studies performed in brain slice preparations from both rats and mice have frequently reported inhibitory effects (i.e., LTD) of NO-sGC-cGMP signaling on excitatory corticostriatal transmission [22]. A potential parsimonious explanation for this apparent discrepancy between in vivo and in vitro studies is that high frequency stimulation of the corticostriatal pathway can be integrated differently in intact versus reduced striatal preparations in a manner mediated by NO-sGC-cGMP signaling. This differential processing by NO is not likely due to differences in down-stream effector molecules such as cGMP versus protein nitrosylation/nitration, as similar mechanisms are implicated in studies using in vivo and in vitro preparations (e.g., sGC and PDEs play a key role in NO-cGMP mediated effects in all of these studies). Studies showing that stimulation protocols known to produce LTD of corticostriatal neurotransmission in vitro, actually lead to LTP in vivo [53], instead suggest that these differences may be related to changes in neuronal properties and network states that have been observed in comparisons across intact and reduced preparations (e.g., see [54]). Interestingly, a similar reversal of LTD to LTP in vitro occurs following removal of magnesium from the bath perfusate [55]. It is therefore likely that typical electrical stimulation protocols (e.g., 100 Hz tetanic high frequency stimulation), when applied to the intact preparation versus a brain slice, will induce a greater activation of glutamatergic drive onto postsynaptic AMPA receptors and more robust depolarization-induced removal of the voltage-dependent magnesium block of NMDA receptors, commonly leading to an LTP-like state. Conversely, similar stimulation of corticostriatal signaling in brain slices commonly leads to LTD in the absence of the removal of magnesium block of NMDA receptors or other depolarizing stimuli. This is supported further by studies showing that LTD-induction in the mature striatum is not NMDA receptor dependent, whereas LTP requires a state where activation of these channels can occur such as during DA D1/5 receptor co-activation (Reviewed in [56]). Similar to LTP, stimulation of striatal NOS activity also requires NMDA and D1/5 receptor co-activation (Reviewed in [57]), thus it is more readily apparent how corticostriatal pathway activation could lead to a NO-sGC-cGMP-dependent LTP-like state, versus an LTD-like phenomenon. Nonetheless, it is possible that NO signaling may act to facilitate and stabilize the dominant state of short- or long-term synaptic plasticity occurring across corticostriatal synapses (i.e., primarily potentiation when postsynaptic NMDA receptors are activated, or depression in the absence of this activation). Future investigations using novel genetic and optical approaches should be useful for determining the role of NO in bidirectional synaptic plasticity at both excitatory and inhibitory synapses onto identified striatonigral and striatopallidal MSNs.
Impact of cyclic nucleotide signaling and phosphodiesterase function on the activation of transmembrane ion channels and receptors
Considerable evidence indicates that cyclic nucleotides control membrane excitability via direct effects on cyclic nucleotide gated ion channels or indirectly via activation of PKA and PKG, and subsequent phosphorylation of transmembrane ion channels, neurotransmitter receptors, and other signaling molecules [see [1, 5] for review]. One example of a transmembrane ion channel that is directly modulated by cyclic nucleotides is the hyperpolarization-activated cyclic nucleotide-gated (HCN) channel [see [58], for review]. HCN channels are activated at resting membrane potentials (i.e., hyperpolarized states) and this activation is often facilitated by direct interaction with cyclic nucleotides. HCN channels are permeable to Na+ and K+. Channel opening induces membrane depolarization and reduces membrane resistance, resulting in complex changes in neuronal excitability and synaptic plasticity. While HCN channels are expressed only in low levels in the striatum [59] they have been shown to potently regulate the membrane activity of at least some striatal neuron populations [60].
As discussed above, both PKA and PKG can affect the activity and trafficking of transmembrane ion channels and neurotransmitter receptors through activation of the DARPP-32 signaling cascade [1, 61] or via direct mechanisms [62]. While direct effects of PKG have not been well studied, PKA has been shown to phosphorylate the NMDA NR1 and AMPA GluA1 receptor subunits, leading to their insertion in the cell membrane and an increase in inward depolarizing currents [62]. PKA can also phosphorylate GABA β1 and β3 subunits leading to decreased channel activity and Cl-influx. These actions of PKA are largely permitted by DARPP-32 inhibition of PP-1 [61]. PKA has also been shown to phosphorylate Nav1 and Cav1 channels leading to decreased channel activity, whereas opposite effects are observed with Cav1.2 and Cav1.3 subunit phosphorylation [62]. Interestingly, PKA and PKG appear to have opposite effects on neuronal Na+/K+ ATPase activity with the former having inhibitory and the later having excitatory effects [61, 63].
Manipulating cyclic nucleotide signaling and phosphodiesterase function for the treatment of Huntington’s and Parkinson’s disease
It is likely that numerous different PDE isoforms act collectively in striatal neurons to regulate intracellular availability of cyclic nucleotides within specific subcellular compartments. Because of this tight control of cAMP and cGMP signaling, PDEs are in position to regulate the function of a host of signaling effector molecules involved in fine-tuning the electrical activity patterns of striatal neurons. Given that these activity patterns are thought to be abnormal in diseases of the basal ganglia such as Huntington’s disease (HD) and Parkinson’s disease (PD), this key function of PDEs has tremendous ramifications for the treatment of neurological disorders. In addition to abnormal firing patterns, aberrant forms of synaptic plasticity are thought to exist within the striatum in a number of prevalent movement disorders. Therefore, effective treatments for diseases of the basal ganglia, in which normal planning and initiation or suppression of motor behaviors are impaired, will likely be capable of reversing abnormal striatal neuronal excitability and synaptic plasticity. Recent advances towards this goal are detailed below. Studies examining the biochemical, electrophysiological, and behavioral effects of genetic PDE deletions and isoform-selective PDE inhibitors are summarized in Table 1.
Huntington’s disease
HD is an inherited, neurodegenerative disorder characterized by the presence of excessive trinucleotide repeats in the Huntingtin gene resulting in programmed cell death in cortical and striatal regions [64]. Hallmark pathology of HD includes loss of corticostriatal pyramidal cells and striatal MSNs, particularly striatopallidal cells of the indirect output pathway. Striatopallidal MSNs are thought to degenerate as a result of overexpression of huntingtin protein produced by the HD gene, however, the exact pathophysiology leading to cell loss remains unknown [64]. Preclinical studies of HD models have shown age-dependent abnormalities in the modulation of corticostriatal transmission by DA [65, 66]. This pathophysiology is characterized by early symptomatic increases in glutamate release and progressive decreases in glutamate release in late stages of the disease [67]. Drugs that downregulate DA transmission (e.g., tetrabenazine, DA receptor antagonists) have been shown to reverse these pathophysiological changes in HD models [67] and reduce chorea and motor symptoms in patients with HD [68].
In rodent models of HD, striatal cAMP levels decrease as the disease progresses from presymptomatic to symptomatic stages [69]. Other studies revealed that the mutant huntingtin protein impairs cAMP signaling and CREB function [69] and that cAMP levels are reduced in the CSF of HD patients compared to controls [70, 71]. Additionally, nNOS mRNA is much lower in the striatum of HD patients compared to control subjects [72] indicating that NO synthesis is likely to be depressed. Given this, the above changes are likely to lead to reduced cGMP tone and PKG hypoactivity in sGC-containing MSNs. Further recent studies have reported that PDE10A and PDE1B mRNA and protein are reduced in the striatum in mouse models of HD [73, 74], but see also [75]. Complementary outcomes have been reported in postmortem studies comparing time-matched brain samples from controls and HD patients [76]. It is currently unclear to what extent the above mentioned decreases in markers of cAMP/cGMP and PDE signaling result from disease-induced MSN death versus alterations in striatal function induced by mutant huntingtin. In the case of nNOS, however, cell death is likely not a major factor contributing to decreased striatal nNOS mRNA levels as a selective sparing of somatostatin/NPY/nNOS expressing interneurons is consistently observed in postmortem tissue from HD patients and in animal models [72, 77–80].
As mentioned above, agents such as PDE inhibitors, which augment cAMP/cGMP tone in striatal MSNs, increase measures of spontaneous and evoked corticostriatal transmission in normal animals. These agents can also induce LTD of corticostriatal transmission (reviewed in [22]). Despite these studies, it is not known whether decreases in cyclic nucleotide levels in the HD striatum can lead to abnormal glutamatergic transmission observed in early and late stages of this disease. However, the PDE10A inhibitor TP-10 has been reported to have neuroprotective effects in HD models [81]. In these preclinical studies quinolinic acid lesions of the striatum and pathology in the cortex and striatum observed in R6/2 mice were all significantly attenuated by administration of TP-10 [81, 82]. Together these findings indicate that elevations in cyclic nucleotide tone can act to offset some of the pathophysiological disturbances occurring in corticostriatal pathways in HD. It is also possible that enduring abnormalities in the synthesis and catabolism of cyclic nucleotides may contribute to the abnormalities in corticostriatal transmission observed in preclinical models and patients with HD. Indeed, treatment with PDE inhibitors may boost cyclic nucleotide tone and alleviate homeostatic imbalances between different PDE isoforms which may be up- or downregulated as a result of pathophysiological changes (e.g., hyperdopaminergia; glutamatergic dysregulation) in the HD striatum. An understanding of how these complex phenomena may contribute to the HD phenotype will require further studies aimed at investigating the role of different PDE isoforms in regulating MSN neuron activity and corticostriatal transmission in identified projection pathways. Finally, the exact role of cAMP and cGMP in this PDE regulation will also need to be characterized in striatonigral and striatopallidal MSNs in presymptomatic and symptomatic HD models.
Parkinson’s disease and related disorders
PD is a neurodegenerative disorder first described by James Parkinson almost two centuries ago [83] and defined by loss of DAergic neurons in the substantia nigra pars compacta [84]. The DAergic denervation in PD results in dysregulation of neuronal activity within the striatum and downstream basal ganglia nuclei, which is associated with motor dysfunction. One cardinal pathophysiological feature of PD is an increase in overall firing activity in striatopallidal pathway MSNs and a decrease in activity in the striatonigral pathway [85]. In terms of synaptic plasticity, corticostriatal transmission is abnormally enhanced following chronic DA depletion and striatal LTD is absent in rodent models of PD potentially due to loss of D2 receptor activation [47, 86]. The gold standard pharmacotherapy for PD rests on treatment with the DAergic precursor levodopa (L-DOPA); however prolonged treatment with L-DOPA can result in the development of severe motor side effects such as dyskinesias and other motor fluctuations [87].
Given the above, there is clearly a necessity to identify novel non-dopaminergic mechanisms as new therapeutic targets for PD. Among these, sGC-cGMP-PKG signaling is emerging as a promising candidate for second messenger-based therapies for the amelioration of PD symptoms [88]. Numerous findings indicate that cGMP is an important cellular intermediary which controls functional interactions between DA and glutamate in the normal and parkinsonian striatum [20, 89]. Indeed, converging evidence from animal models indicates that alterations in cGMP homeostasis may contribute to pathophysiological changes in basal ganglia circuits observed in PD. Specifically, the regulation of corticostriatal synaptic transmission by sGC-cGMP signaling is likely to be disrupted in the parkinsonian striatum as an upregulation of sGC expression and activity (i.e., cGMP synthesis) have been reported in MPTP-treated mice [90, 91]. Interestingly, the net effect of these complex neuroadaptations induced by striatal DA depletion can be mimicked by acute D2 (but not D1) receptor antagonism [92, 93]. Given that the vast majority of D2 receptors are localized in MSNs of the so-called “striatopallidal or indirect pathway”, the above studies suggest that alterations in cGMP signaling observed after striatal DA-depletion are cell-type and synapse specific (i.e., occurring in cortico-striatopallidal pathways). Consistent with the work of Chalimoniuk and colleagues, together with our collaborators we recently showed that striatal tissue levels of cGMP are abnormally elevated in 6-OHDA lesioned rats [94]. We also found that systemic administration of the sGC inhibitor ODQ, using a dose which normalized the DA depletion-induced elevations in striatal cGMP levels, reversed the abnormal elevations in striatal MSN firing. In addition, systemic and intrastriatal infusion of a sGC inhibitor significantly reduced the increase in cytochrome oxidase (i.e., marker of neuronal activation) levels observed in the subthalamic nucleus of 6-OHDA lesioned animals [94], implicating increased striatal cGMP tone in the pathophysiological overactivation of striatopallidal output observed in the parkinsonian striatum. Moreover, in the same study, sGC inhibition significantly improved deficits in forelimb stepping performance in 6-OHDA lesioned rats and completely reversed similar measures of forelimb akinesia in MPTP-treated mice [94]. Taken together, the above studies indicate that sGC-cGMP-PKG signaling can be modulated as an approach for treating PD, and potentially, other neurological disorders associated with overactive corticostriatal transmission.
As with HD (see above), it is plausible that PDE inhibitors may be useful for treating dyskinetic side effects observed in patients with PD who have been treated with L-DOPA for an extended period (>5–10 years). This is supported by recent studies performed in animal models of L-DOPA-induced dyskinesia. These reports have shown that the non-selective PDE inhibitor zaprinast reverses decreases in striatal cyclic nucleotide levels and abnormal involuntary movements induced by L-DOPA administration [89, 95]. Studies in striatal brain slice preparations have revealed that dyskinetic animals lack the ability to reverse previously induced LTP (i.e. there is a lack of bidirectional plasticity) within the corticostriatal pathway [96]. Because this persistent LTP of corticostriatal transmission is reversed by PDE inhibitors [95], it is likely that deficits in cyclic nucleotide signaling contribute in an important way to abnormalities in synaptic plasticity observed in models of L-DOPA-induced dyskinesia.
In line with the above studies, the PDE10A inhibitor papaverine has been shown to reduce stereotypies observed following apomorphine administration [97] and hyperactivity induced by the psychostimulants phencyclidine and amphetamine [93]. We have also observed that co-administration of the PDE10A inhibitor TP-10 with L-DOPA reduces the severity of hyperkinesias and dystonia observed in dyskinetic 6-OHDA-lesioned rats (unpublished observation). Given these observations, it is likely that selective PDE inhibitors will be useful pharmacological tools for reversing pathophysiological and behavioral correlates of hyperdopaminergia induced by L-DOPA therapy, potentially, because these agents are able to restore bidirectional corticostriatal plasticity and activate striatopallidal pathways.
A related PD-like neurological disorder, autosomal-dominant striatal degeneration (ADSD), is a movement disorder characterized by bradykinesia, dysarthria, and muscle rigidity, but lacking the presence of tremors associated with PD. ADSD is a genetic disorder caused by a complex frameshift mutation in the PDE8B gene and results in loss of enzyme activity and cAMP metabolism [98]. Together with the above studies of PDE function in HD and PD, the involvement of PDE8B in the etiology of ADSD highlights the importance of cyclic nucleotides and PDEs in regulation of motor function.
Conclusions
Despite increasing attention directed towards the role of cAMP and cGMP signaling in neurological disorders and side effects of current drug treatments, little is known about the function of many of the PDE families, isoforms, and splice variants as it pertains to the regulation of striatal neuronal excitability and synaptic transmission. Given the strong expression of AC, sGC, and numerous PDEs in striatal neurons, it is likely that future studies examining the impact of selective pathway inhibitors and activators on the regulation of neuronal excitability and synaptic plasticity in naïve animals and models of basal ganglia dysfunction will be invaluable for the development of novel pharmacotherapies for the treatment of these devastating diseases affecting basal ganglia function.
Acknowledgments
We thank Dr. Kuei Y. Tseng for helpful comments regarding the content and organization of this manuscript. This work was supported by the Parkinson’s Disease Foundation, Brain Research Foundation, Cure Huntington’s Disease Initiative, and United States Public Health grants NS047452 and NS047452-S1 (ARW).
Abbreviations
- cAMP
adenosine 3′-5′-cyclic monophosphate
- ATP
adenosine triphosphate
- AC
adenylyl cyclase
- ADSD
autosomal-dominant striatal degeneration
- CREB
cAMP response element binding protein
- PKA
cAMP-dependent protein kinase
- PKG
cGMP-dependent protein kinase
- CNGC
cyclic nucleotide-gated channel
- PDE
cyclic nucleotide phosphodiesterase
- DA
dopamine
- DARPP-32
dopamine and cAMP regulated phosphoprotein MW 32 kDa
- cGMP
guanosine 3′-5′-cyclic monophosphate
- GPCRs
G-protein coupled receptors
- EPAC
GTP exchange protein activated by cAMP
- HD
Huntington’s disease
- L-DOPA
levodopa
- LTD
long-term depression
- LTP
long-term potentiation
- MSN
medium-sized spiny output neuron
- nNOS
neuronal nitric oxide synthase
- NO
nitric oxide
- NMDA
N-methyl-d-aspartate
- PD
Parkinson’s disease
- pGC
particulate guanylyl cyclase
- PP1
protein phosphatase-1
- sGC
soluble guanylyl cyclase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Greengard P. The neurobiology of slow synaptic transmission. Science. 2001;294(5544):1024–30. doi: 10.1126/science.294.5544.1024. [DOI] [PubMed] [Google Scholar]
- 2.Mons N, Decorte L, Jaffard R, Cooper DM. Ca2+-sensitive adenylyl cyclases, key integrators of cellular signalling. Life Sci. 1998;62(17–18):1647–52. doi: 10.1016/s0024-3205(98)00122-2. [DOI] [PubMed] [Google Scholar]
- 3.Boehning D, Snyder SH. Novel neural modulators. Annu Rev Neurosci. 2003;26:105–31. doi: 10.1146/annurev.neuro.26.041002.131047. [DOI] [PubMed] [Google Scholar]
- 4.Bredt DS. Nitric oxide signaling specificity--the heart of the problem. J Cell Sci. 2003;116(Pt 1):9–15. doi: 10.1242/jcs.00183. [DOI] [PubMed] [Google Scholar]
- 5.Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27(11):2783–802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bredt DS, Hwang PM, Snyder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347(6295):768–70. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- 7.Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci U S A. 1990;87(2):682–5. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Vente J, Markerink-van Ittersum M, van Abeelen J, Emson PC, Axer H, Steinbusch HW. NO-mediated cGMP synthesis in cholinergic neurons in the rat forebrain: effects of lesioning dopaminergic or serotonergic pathways on nNOS and cGMP synthesis. Eur J Neurosci. 2000;12(2):507–19. doi: 10.1046/j.1460-9568.2000.00927.x. [DOI] [PubMed] [Google Scholar]
- 9.Hope BT, Michael GJ, Knigge KM, Vincent SR. Neuronal NADPH diaphorase is a nitric oxide synthase. Proc Natl Acad Sci U S A. 1991;88(7):2811–4. doi: 10.1073/pnas.88.7.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vincent SR, Das S, Maines MD. Brain heme oxygenase isoenzymes and nitric oxide synthase are co-localized in select neurons. Neuroscience. 1994;63(1):223–31. doi: 10.1016/0306-4522(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 11.Murad F. Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med. 2006;355(19):2003–11. doi: 10.1056/NEJMsa063904. [DOI] [PubMed] [Google Scholar]
- 12.Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol Ther. 2006;109(3):366–98. doi: 10.1016/j.pharmthera.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 13.Menniti FS, Faraci WS, Schmidt CJ. Phosphodiesterases in the CNS: targets for drug development. Nat Rev Drug Discov. 2006;5(8):660–70. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- 14.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58(3):488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 15.Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007;100(3):309–27. doi: 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- 16.Kleppisch T. Phosphodiesterases in the central nervous system. Handb Exp Pharmacol. 2009;(191):71–92. doi: 10.1007/978-3-540-68964-5_5. [DOI] [PubMed] [Google Scholar]
- 17.Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16(1):89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 18.Deisseroth K, Tsien RW. Dynamic multiphosphorylation passwords for activity-dependent gene expression. Neuron. 2002;34(2):179–82. doi: 10.1016/s0896-6273(02)00664-5. [DOI] [PubMed] [Google Scholar]
- 19.Frank DA, Greenberg ME. CREB: a mediator of long-term memory from mollusks to mammals. Cell. 1994;79(1):5–8. doi: 10.1016/0092-8674(94)90394-8. [DOI] [PubMed] [Google Scholar]
- 20.Nishi A, Watanabe Y, Higashi H, Tanaka M, Nairn AC, Greengard P. Glutamate regulation of DARPP-32 phosphorylation in neostriatal neurons involves activation of multiple signaling cascades. Proc Natl Acad Sci U S A. 2005;102(4):1199–204. doi: 10.1073/pnas.0409138102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsou K, Snyder GL, Greengard P. Nitric oxide/cGMP pathway stimulates phosphorylation of DARPP-32, a dopamine- and cAMP-regulated phosphoprotein, in the substantia nigra. Proc Natl Acad Sci U S A. 1993;90(8):3462–5. doi: 10.1073/pnas.90.8.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30(5):211–9. doi: 10.1016/j.tins.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 23.Del Bel EA, Guimaraes FS, Bermudez-Echeverry M, Gomes MZ, Schiaveto-de-souza A, Padovan-Neto FE, et al. Role of nitric oxide on motor behavior. Cell Mol Neurobiol. 2005;25(2):371–92. doi: 10.1007/s10571-005-3065-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colwell CS, Levine MS. Excitatory synaptic transmission in neostriatal neurons: regulation by cyclic AMP-dependent mechanisms. J Neurosci. 1995;15(3 Pt 1):1704–13. doi: 10.1523/JNEUROSCI.15-03-01704.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Threlfell S, Sammut S, Menniti FS, Schmidt CJ, West AR. Inhibition of Phosphodiesterase 10A Increases the Responsiveness of Striatal Projection Neurons to Cortical Stimulation. J Pharmacol Exp Ther. 2009;328(3):785–95. doi: 10.1124/jpet.108.146332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.West AR, Grace AA. The Nitric Oxide-Guanylyl Cyclase Signaling Pathway Modulates Membrane Activity States and Electrophysiological Properties of Striatal Medium Spiny Neurons Recorded In Vivo. J Neurosci. 2004;24(8):1924–35. doi: 10.1523/JNEUROSCI.4470-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sammut S, Threlfell S, West AR. Nitric oxide-soluble guanylyl cyclase signaling regulates corticostriatal transmission and short-term synaptic plasticity of striatal projection neurons recorded in vivo. Neuropharmacology. 2010;58(3):624–31. doi: 10.1016/j.neuropharm.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tseng KY, Snyder-Keller A, O’Donnell P. Dopaminergic modulation of striatal plateau depolarizations in corticostriatal organotypic cocultures. Psychopharmacology (Berl) 2007;191(3):627–40. doi: 10.1007/s00213-006-0439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brady AM, O’Donnell P. Dopaminergic modulation of prefrontal cortical input to nucleus accumbens neurons in vivo. J Neurosci. 2004;24(5):1040–9. doi: 10.1523/JNEUROSCI.4178-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cepeda C, Buchwald NA, Levine MS. Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci U S A. 1993;90(20):9576–80. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chergui K, Lacey MG. Modulation by dopamine D1-like receptors of synaptic transmission and NMDA receptors in rat nucleus accumbens is attenuated by the protein kinase C inhibitor Ro 32–0432. Neuropharmacology. 1999;38(2):223–31. doi: 10.1016/s0028-3908(98)00187-7. [DOI] [PubMed] [Google Scholar]
- 32.Levine MS, Li Z, Cepeda C, Cromwell HC, Altemus KL. Neuromodulatory actions of dopamine on synaptically-evoked neostriatal responses in slices. Synapse. 1996;24(1):65–78. doi: 10.1002/syn.890240102. [DOI] [PubMed] [Google Scholar]
- 33.Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. 1998;18(24):10297–303. doi: 10.1523/JNEUROSCI.18-24-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jocoy EL, V, Andre M, Cummings DM, Rao SP, Wu N, Ramsey AJ, et al. Dissecting the contribution of individual receptor subunits to the enhancement of N-methyl-d-aspartate currents by dopamine D1 receptor activation in striatum. Front Syst Neurosci. 2011;5:28. doi: 10.3389/fnsys.2011.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.West AR, Galloway MP, Grace AA. Regulation of striatal dopamine neurotransmission by nitric oxide: effector pathways and signaling mechanisms. Synapse. 2002;44(4):227–45. doi: 10.1002/syn.10076. [DOI] [PubMed] [Google Scholar]
- 36.Calabresi P, Centonze D, Gubellini P, Marfia GA, Bernardi G. Glutamate-triggered events inducing corticostriatal long-term depression. J Neurosci. 1999;19(14):6102–10. doi: 10.1523/JNEUROSCI.19-14-06102.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gittis AH, Nelson AB, Thwin MT, Palop JJ, Kreitzer AC. Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. J Neurosci. 2010;30(6):2223–34. doi: 10.1523/JNEUROSCI.4870-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ibanez-Sandoval O, Tecuapetla F, Unal B, Shah F, Koos T, Tepper JM. A novel functionally distinct subtype of striatal neuropeptide Y interneuron. J Neurosci. 2011;31(46):16757–69. doi: 10.1523/JNEUROSCI.2628-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ondracek JM, Dec A, Hoque KE, Lim SA, Rasouli G, Indorkar RP, et al. Feed-forward excitation of striatal neuron activity by frontal cortical activation of nitric oxide signaling in vivo. Eur J Neurosci. 2008;27(7):1739–54. doi: 10.1111/j.1460-9568.2008.06157.x. [DOI] [PubMed] [Google Scholar]
- 40.Sammut S, Park DJ, West AR. Frontal cortical afferents facilitate striatal nitric oxide transmission in vivo via a NMDA receptor and neuronal NOS-dependent mechanism. J Neurochem. 2007;103(3):1145–56. doi: 10.1111/j.1471-4159.2007.04811.x. [DOI] [PubMed] [Google Scholar]
- 41.Coskran TM, Morton D, Menniti FS, Adamowicz WO, Kleiman RJ, Ryan AM, et al. Immunohistochemical localization of phosphodiesterase 10A in multiple mammalian species. J Histochem Cytochem. 2006;54(11):1205–13. doi: 10.1369/jhc.6A6930.2006. [DOI] [PubMed] [Google Scholar]
- 42.Sano H, Nagai Y, Miyakawa T, Shigemoto R, Yokoi M. Increased social interaction in mice deficient of the striatal medium spiny neuron-specific phosphodiesterase 10A2. J Neurochem. 2008;105(2):546–56. doi: 10.1111/j.1471-4159.2007.05152.x. [DOI] [PubMed] [Google Scholar]
- 43.Seeger TF, Bartlett B, Coskran TM, Culp JS, James LC, Krull DL, et al. Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 2003;985(2):113–26. doi: 10.1016/s0006-8993(03)02754-9. [DOI] [PubMed] [Google Scholar]
- 44.Xie Z, Adamowicz WO, Eldred WD, Jakowski AB, Kleiman RJ, Morton DG, et al. Cellular and subcellular localization of PDE10A, a striatum-enriched phosphodiesterase. Neuroscience. 2006;139(2):597–607. doi: 10.1016/j.neuroscience.2005.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishi A, Kuroiwa M, Miller DB, O’Callaghan JP, Bateup HS, Shuto T, et al. Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J Neurosci. 2008;28(42):10460–71. doi: 10.1523/JNEUROSCI.2518-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strick CA, James LC, Fox CB, Seeger TF, Menniti FS, Schmidt CJ. Alterations in gene regulation following inhibition of the striatum-enriched phosphodiesterase, PDE10A. Neuropharmacology. 2010;58(2):444–51. doi: 10.1016/j.neuropharm.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 47.Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321(5890):848–51. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calabresi P, Gubellini P, Centonze D, Sancesario G, Morello M, Giorgi M, et al. A Critical Role of the Nitric Oxide/cGMP Pathway in Corticostriatal Long-Term Depression. 1999:2489–99. doi: 10.1523/JNEUROSCI.19-07-02489.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doreulee N, Sergeeva OA, Yanovsky Y, Chepkova AN, Selbach O, Godecke A, et al. Cortico-striatal synaptic plasticity in endothelial nitric oxide synthase deficient mice. Brain Res. 2003;964(1):159–63. doi: 10.1016/s0006-8993(02)04121-5. [DOI] [PubMed] [Google Scholar]
- 50.Calabresi P, Gubellini P, Centonze D, Picconi B, Bernardi G, Chergui K, et al. Dopamine and cAMP-regulated phosphoprotein 32 kDa controls both striatal long-term depression and long-term potentiation, opposing forms of synaptic plasticity. J Neurosci. 2000;20(22):8443–51. doi: 10.1523/JNEUROSCI.20-22-08443.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.West AR, Galloway MP. Endogenous nitric oxide facilitates striatal dopamine and glutamate efflux in vivo: role of ionotropic glutamate receptor-dependent mechanisms. Neuropharmacology. 1997;36(11–12):1571–81. doi: 10.1016/s0028-3908(97)00148-2. [DOI] [PubMed] [Google Scholar]
- 52.West AR, Grace AA. Striatal nitric oxide signaling regulates the neuronal activity of midbrain dopamine neurons in vivo. J Neurophysiol. 2000;83(4):1796–808. doi: 10.1152/jn.2000.83.4.1796. [DOI] [PubMed] [Google Scholar]
- 53.Charpier S, Deniau JM. In vivo activity-dependent plasticity at cortico-striatal connections: evidence for physiological long-term potentiation. Proc Natl Acad Sci U S A. 1997;94(13):7036–40. doi: 10.1073/pnas.94.13.7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pare D, Shink E, Gaudreau H, Destexhe A, Lang EJ. Impact of spontaneous synaptic activity on the resting properties of cat neocortical pyramidal neurons In vivo. J Neurophysiol. 1998;79(3):1450–60. doi: 10.1152/jn.1998.79.3.1450. [DOI] [PubMed] [Google Scholar]
- 55.Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992;12(11):4224–33. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Surmeier DJ, Plotkin J, Shen W. Dopamine and synaptic plasticity in dorsal striatal circuits controlling action selection. Curr Opin Neurobiol. 2009;19(6):621–8. doi: 10.1016/j.conb.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.West AR, Tseng KY. Nitric Oxide-Soluble Guanylyl Cyclase-Cyclic GMP Signaling in the Striatum: New Targets for the Treatment of Parkinson’s Disease? Front Syst Neurosci. 2011;5:55. doi: 10.3389/fnsys.2011.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benarroch EE. HCN channels: function and clinical implications. Neurology. 2013;80(3):304–10. doi: 10.1212/WNL.0b013e31827dec42. [DOI] [PubMed] [Google Scholar]
- 59.Monteggia LM, Eisch AJ, Tang MD, Kaczmarek LK, Nestler EJ. Cloning and localization of the hyperpolarization-activated cyclic nucleotide-gated channel family in rat brain. Brain Res Mol Brain Res. 2000;81(1–2):129–39. doi: 10.1016/s0169-328x(00)00155-8. [DOI] [PubMed] [Google Scholar]
- 60.Wilson CJ. The mechanism of intrinsic amplification of hyperpolarizations and spontaneous bursting in striatal cholinergic interneurons. Neuron. 2005;45(4):575–85. doi: 10.1016/j.neuron.2004.12.053. [DOI] [PubMed] [Google Scholar]
- 61.Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–96. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- 62.Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30(5):228–35. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 63.Munhoz CD, Kawamoto EM, de Sa Lima L, Lepsch LB, Glezer I, Marcourakis T, et al. Glutamate modulates sodium-potassium-ATPase through cyclic GMP and cyclic GMP-dependent protein kinase in rat striatum. Cell Biochem Funct. 2005;23(2):115–23. doi: 10.1002/cbf.1217. [DOI] [PubMed] [Google Scholar]
- 64.MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–83. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 65.Andre VM, Cepeda C, Levine MS. Dopamine and glutamate in Huntington’s disease: A balancing act. CNS Neurosci Ther. 2010;16(3):163–78. doi: 10.1111/j.1755-5949.2010.00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cepeda C, Bamford NS, Andre VM, Levine MS. Alterations in corticostriatal synaptic function in Huntington’s and Parkinson’s diseases. In: Steiner H, Tseng KY, editors. Handbook of Basal Ganglia Structure and Function, a Decade of Progress. Elsevier Inc. Academic Press; London: 2010. pp. 607–23. [Google Scholar]
- 67.Andre VM, Cepeda C, Fisher YE, Huynh M, Bardakjian N, Singh S, et al. Differential electrophysiological changes in striatal output neurons in Huntington’s disease. J Neurosci. 2011;31(4):1170–82. doi: 10.1523/JNEUROSCI.3539-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology. 2006;66(3):366–72. doi: 10.1212/01.wnl.0000198586.85250.13. [DOI] [PubMed] [Google Scholar]
- 69.Gines S, I, Seong S, Fossale E, Ivanova E, Trettel F, Gusella JF, et al. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum Mol Genet. 2003;12(5):497–508. doi: 10.1093/hmg/ddg046. [DOI] [PubMed] [Google Scholar]
- 70.Cramer H, Kohler J, Oepen G, Schomburg G, Schroter E. Huntington’s chorea-- measurements of somatostatin, substance P and cyclic nucleotides in the cerebrospinal fluid. J Neurol. 1981;225(3):183–7. doi: 10.1007/BF00313747. [DOI] [PubMed] [Google Scholar]
- 71.Cramer H, Warter JM, Renaud B. Analysis of neurotransmitter metabolites and adenosine 3′,5′-monophosphate in the CSF of patients with extrapyramidal motor disorders. Adv Neurol. 1984;40:431–5. [PubMed] [Google Scholar]
- 72.Norris PJ, Waldvogel HJ, Faull RL, Love DR, Emson PC. Decreased neuronal nitric oxide synthase messenger RNA and somatostatin messenger RNA in the striatum of Huntington’s disease. Neuroscience. 1996;72(4):1037–47. doi: 10.1016/0306-4522(95)00596-x. [DOI] [PubMed] [Google Scholar]
- 73.Haibei H, Elizabeth AM, Andrea LOH, Geraldine TG, Eileen MD-W. Mutant huntingtin affects the rate of transcription of striatum-specific isoforms of phosphodiesterase. 2004;10A:3351–63. doi: 10.1111/j.1460-9568.2004.03796.x. [DOI] [PubMed] [Google Scholar]
- 74.Hebb ALO, Robertson HA, Denovan-Wright EM. Striatal phosphodiesterase mRNA and protein levels are reduced in Huntington’s disease transgenic mice prior to the onset of motor symptoms. Neuroscience. 2004;123(4):967–81. doi: 10.1016/j.neuroscience.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 75.Leuti A, Laurenti D, Giampa C, Montagna E, Dato C, Anzilotti S, et al. Phosphodiesterase 10A (PDE10A) localization in the R6/2 mouse model of Huntington’s disease. Neurobiol Dis. 2013;52:104–16. doi: 10.1016/j.nbd.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 76.Hu H, McCaw EA, Hebb AL, Gomez GT, Denovan-Wright EM. Mutant huntingtin affects the rate of transcription of striatum-specific isoforms of phosphodiesterase 10A. Eur J Neurosci. 2004;20(12):3351–63. doi: 10.1111/j.1460-9568.2004.03796.x. [DOI] [PubMed] [Google Scholar]
- 77.Beal MF, Swartz KJ, Finn SF, Mazurek MF, Kowall NW. Neurochemical characterization of excitotoxin lesions in the cerebral cortex. J Neurosci. 1991;11(1):147–58. doi: 10.1523/JNEUROSCI.11-01-00147.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cicchetti F, Parent A. Striatal interneurons in Huntington’s disease: selective increase in the density of calretinin-immunoreactive medium-sized neurons. Mov Disord. 1996;11(6):619–26. doi: 10.1002/mds.870110605. [DOI] [PubMed] [Google Scholar]
- 79.Ferrante RJ, Kowall NW, Beal MF, Richardson EP, Jr, Bird ED, Martin JB. Selective sparing of a class of striatal neurons in Huntington’s disease. Science. 1985;230(4725):561–3. doi: 10.1126/science.2931802. [DOI] [PubMed] [Google Scholar]
- 80.Rajput PS, Kharmate G, Norman M, Liu SH, Sastry BR, Brunicardi CF, et al. Somatostatin receptor 1 and 5 double knockout mice mimic neurochemical changes of Huntington’s disease transgenic mice. PLoS One. 2011;6(9):e24467. doi: 10.1371/journal.pone.0024467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Giampa C, Patassini S, Borreca A, Laurenti D, Marullo F, Bernardi G, et al. Phosphodiesterase 10 inhibition reduces striatal excitotoxicity in the quinolinic acid model of Huntington’s disease. Neurobiol Dis. 2009;34(3):450–6. doi: 10.1016/j.nbd.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 82.Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington’s disease. PLoS One. 2010;5(10):e13417. doi: 10.1371/journal.pone.0013417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Parkinson J. An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci. 2002;14(2):223–36. doi: 10.1176/jnp.14.2.223. discussion 2. [DOI] [PubMed] [Google Scholar]
- 84.Rodriguez-Oroz MC, Jahanshahi M, Krack P, Litvan I, Macias R, Bezard E, et al. Initial clinical manifestations of Parkinson’s disease: features and pathophysiological mechanisms. Lancet Neurol. 2009;8(12):1128–39. doi: 10.1016/S1474-4422(09)70293-5. [DOI] [PubMed] [Google Scholar]
- 85.Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12(10):366–75. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- 86.Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445(7128):643–7. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- 87.Obeso JA, Grandas F, Vaamonde J, Luquin MR, Artieda J, Lera G, et al. Motor complications associated with chronic levodopa therapy in Parkinson’s disease. Neurology. 1989;39(11 Suppl 2):11–9. [PubMed] [Google Scholar]
- 88.Calabresi P, Di Filippo M, Gallina A, Wang Y, Stankowski JN, Picconi B, et al. New synaptic and molecular targets for neuroprotection in Parkinson’s disease. Mov Disord. 2013;28(1):51–60. doi: 10.1002/mds.25096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Giorgi M, D’Angelo V, Esposito Z, Nuccetelli V, Sorge R, Martorana A, et al. Lowered cAMP and cGMP signalling in the brain during levodopa-induced dyskinesias in hemiparkinsonian rats: new aspects in the pathogenetic mechanisms. Eur J Neurosci. 2008;28(5):941–50. doi: 10.1111/j.1460-9568.2008.06387.x. [DOI] [PubMed] [Google Scholar]
- 90.Chalimoniuk M, Langfort J. The effect of subchronic, intermittent L-DOPA treatment on neuronal nitric oxide synthase and soluble guanylyl cyclase expression and activity in the striatum and midbrain of normal and MPTP-treated mice. Neurochem Int. 2007;50(6):821–33. doi: 10.1016/j.neuint.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 91.Chalimoniuk M, Langfort J, Lukacova N, Marsala J. Upregulation of guanylyl cyclase expression and activity in striatum of MPTP-induced parkinsonism in mice. Biochem Biophys Res Commun. 2004;324(1):118–26. doi: 10.1016/j.bbrc.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 92.Altar CA, Boyar WC, Kim HS. Discriminatory roles for D1 and D2 dopamine receptor subtypes in the in vivo control of neostriatal cyclic GMP. Eur J Pharmacol. 1990;181(1–2):17–21. doi: 10.1016/0014-2999(90)90240-7. [DOI] [PubMed] [Google Scholar]
- 93.Siuciak JA, Chapin DS, Harms JF, Lebel LA, McCarthy SA, Chambers L, et al. Inhibition of the striatum-enriched phosphodiesterase PDE10A: A novel approach to the treatment of psychosis. Neuropharmacology. 2006;51(2):386–96. doi: 10.1016/j.neuropharm.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 94.Tseng KY, Caballero A, Dec A, Cass DK, Simak N, Sunu E, et al. Inhibition of striatal soluble guanylyl cyclase-cGMP signaling reverses basal ganglia dysfunction and akinesia in experimental parkinsonism. PLoS One. 2011;6(11):e27187. doi: 10.1371/journal.pone.0027187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Picconi B, Bagetta V, Ghiglieri V, Paille V, Di Filippo M, Pendolino V, et al. Inhibition of phosphodiesterases rescues striatal long-term depression and reduces levodopa-induced dyskinesia. Brain. 2011;134(Pt 2):375–87. doi: 10.1093/brain/awq342. [DOI] [PubMed] [Google Scholar]
- 96.Picconi B, Centonze D, Hakansson K, Bernardi G, Greengard P, Fisone G, et al. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci. 2003;6(5):501–6. doi: 10.1038/nn1040. [DOI] [PubMed] [Google Scholar]
- 97.Kostowski W, Gajewska S, Bidzinski A, Hauptman M. Papaverine, drug-induced stereotypy and catalepsy and biogenic amines in the brain of the rat. Pharmacol Biochem Behav. 1976;5(1):15–7. doi: 10.1016/0091-3057(76)90281-1. [DOI] [PubMed] [Google Scholar]
- 98.Appenzeller S, Schirmacher A, Halfter H, Baumer S, Pendziwiat M, Timmerman V, et al. Autosomal-dominant striatal degeneration is caused by a mutation in the phosphodiesterase 8B gene. Am J Hum Genet. 2010;86(1):83–7. doi: 10.1016/j.ajhg.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Polli JW, Kincaid RL. Expression of a calmodulin-dependent phosphodiesterase isoform (PDE1B1) correlates with brain regions having extensive dopaminergic innervation. J Neurosci. 1994;14(3 Pt 1):1251–61. doi: 10.1523/JNEUROSCI.14-03-01251.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yan C, Bentley JK, Sonnenburg WK, Beavo JA. Differential expression of the 61 kDa and 63 kDa calmodulin-dependent phosphodiesterases in the mouse brain. J Neurosci. 1994;14(3 Pt 1):973–84. doi: 10.1523/JNEUROSCI.14-03-00973.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hebb ALO, Robertson HA, Denovan-Wright EM. Striatal phosphodiesterase mRNA and protein levels are reduced in Huntington’s disease transgenic mice prior to the onset of motor symptoms. Neuroscience. 2004;123(4):967–81. doi: 10.1016/j.neuroscience.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 102.Reed TM, Repaske DR, Snyder GL, Greengard P, Vorhees CV. Phosphodiesterase 1B knock-out mice exhibit exaggerated locomotor hyperactivity and DARPP-32 phosphorylation in response to dopamine agonists and display impaired spatial learning. J Neurosci. 2002;22(12):5188–97. doi: 10.1523/JNEUROSCI.22-12-05188.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lin DT, Fretier P, Jiang C, Vincent SR. Nitric oxide signaling via cGMP-stimulated phosphodiesterase in striatal neurons. Synapse. 2010;64(6):460–6. doi: 10.1002/syn.20750. [DOI] [PubMed] [Google Scholar]
- 104.Stephenson DT, Coskran TM, Kelly MP, Kleiman RJ, Morton D, O’Neill SM, et al. The distribution of phosphodiesterase 2A in the rat brain. Neuroscience. 2012;226:145–55. doi: 10.1016/j.neuroscience.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wykes V, Bellamy TC, Garthwaite J. Kinetics of nitric oxide-cyclic GMP signalling in CNS cells and its possible regulation by cyclic GMP. J Neurochem. 2002;83:37–47. doi: 10.1046/j.1471-4159.2002.01106.x. [DOI] [PubMed] [Google Scholar]
- 106.Cho CH, Cho DH, Seo MR, Juhnn YS. Differential changes in the expression of cyclic nucleotide phosphodiesterase isoforms in rat brains by chronic treatment with electroconvulsive shock. Exp Mol Med. 2000;32(3):110–4. doi: 10.1038/emm.2000.19. [DOI] [PubMed] [Google Scholar]
- 107.Siuciak JA, McCarthy SA, Chapin DS, Martin AN. Behavioral and neurochemical characterization of mice deficient in the phosphodiesterase-4B (PDE4B) enzyme. Psychopharmacology (Berl) 2008;197(1):115–26. doi: 10.1007/s00213-007-1014-6. [DOI] [PubMed] [Google Scholar]
- 108.Kurz A, Double KL, Lastres-Becker I, Tozzi A, Tantucci M, Bockhart V, et al. A53T-alpha-synuclein overexpression impairs dopamine signaling and striatal synaptic plasticity in old mice. PLoS One. 2010;5(7):e11464. doi: 10.1371/journal.pone.0011464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reyes-Irisarri E, Perez-Torres S, Mengod G. Neuronal expression of cAMP-specific phosphodiesterase 7B mRNA in the rat brain. Neuroscience. 2005;132(4):1173–85. doi: 10.1016/j.neuroscience.2005.01.050. [DOI] [PubMed] [Google Scholar]
- 110.Appenzeller S, Schirmacher A, Halfter H, Baumer S, Pendziwiat M, Timmerman V, et al. Autosomal-dominant striatal degeneration is caused by a mutation in the phosphodiesterase 8B gene. Am J Hum Genet. 2010;86(1):83–7. doi: 10.1016/j.ajhg.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wunder F, Tersteegen A, Rebmann A, Erb C, Fahrig T, Hendrix M. Characterization of the first potent and selective PDE9 inhibitor using a cGMP reporter cell line. Mol Pharmacol. 2005;68(6):1775–81. doi: 10.1124/mol.105.017608. [DOI] [PubMed] [Google Scholar]
- 112.Verhoest PR, Proulx-Lafrance C, Corman M, Chenard L, Helal CJ, Hou X, et al. Identification of a brain penetrant pde9a inhibitor utilizing prospective design and chemical enablement as a rapid lead optimization strategy. J Med Chem. 2009;52:7946–49. doi: 10.1021/jm9015334. [DOI] [PubMed] [Google Scholar]
- 113.Siuciak JA, McCarthy SA, Chapin DS, Fujiwara RA, James LC, Williams RD, et al. Genetic deletion of the striatum-enriched phosphodiesterase PDE10A: evidence for altered striatal function. Neuropharmacol. 2006;51:374–85. doi: 10.1016/j.neuropharm.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 114.Grauer SM, V, Pulito L, Navarra RL, Kelly MP, Kelley C, Graf R, et al. PDE10A inhibitor activity in preclinical models of the positive, cognitive and negative symptoms of schizophrenia. J Pharmacol Exp Ther. 2009;331:574–90. doi: 10.1124/jpet.109.155994. [DOI] [PubMed] [Google Scholar]
- 115.Schmidt CJ, Chapin DS, Cianfrogna J, Corman ML, Hajos M, Harms JF, et al. Preclinical characterization of selective PDE10A inhibitors: A new therapeutic approach to the treatment of schizophrenia. J Pharmacol Exp Ther. 2008;325:681–90. doi: 10.1124/jpet.107.132910. [DOI] [PubMed] [Google Scholar]
- 116.West AR, Galloway MP. Regulation of serotonin-facilitated dopamine release in vivo: the role of protein kinase A activating transduction mechanisms. Synapse. 1996;23:20–27. doi: 10.1002/(SICI)1098-2396(199605)23:1<20::AID-SYN3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 117.Kleiman RJ, Kimmel LH, Bove SE, Lanz TA, Harms JF, Romegialli A, et al. Chronic suppression of PDE10A alters striatal expression of genes responsible for neurotransmitter synthesis, neurotransmission and signaling pathways implicated in Huntington’s Disease. J Pharmacol Exp Ther. 2011;336:64–76. doi: 10.1124/jpet.110.173294. [DOI] [PubMed] [Google Scholar]
- 118.Smith SM, Uslaner JM, Cox CD, Huszar SL, Cannon CE, Vardigan JD, et al. The novel phosphodiesterase 10A inhibitor THPP-1 has antipsychotic-like effects in rats and improves cognition in rat and rhesus monkey. Neuropharmacol. 2013;64:215–23. doi: 10.1016/j.neuropharm.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 119.Nishi A, Snyder GL. Biochemical and behavioral profiles of phosphodiesterase inhibition in dopaminergic transmission. J Pharmacol Sci. 2010;116:6–16. doi: 10.1254/jphs.10r01fm. [DOI] [PubMed] [Google Scholar]