

Abstract
Lysophosphatidic acid (LPA) induces actin rearrangement, focal adhesion assembly, and cell migration through the activation of small G protein Rho and its downstream effectors. These diverse cellular responses are mediated by its associated G protein-coupled receptors. However, the mechanisms and specificity by which these LPA receptors mediate LPA actions are still poorly understood. Here we show that LPA stimulation promotes the interaction of the LPA2 receptor with a focal adhesion molecule, TRIP6 (thyroid receptor interacting protein 6)/ZRP-1 (zyxin-related protein 1). TRIP6 directly binds to the carboxyl-terminal tail of the LPA2 receptor through its LIM domains. LPA-dependent recruitment of TRIP6 to the plasma membrane promotes its targeting to focal adhesions and co-localization with actin stress fibers. In addition, TRIP6 associates with the components of focal complexes including paxillin, focal adhesion kinase, c-Src, and p130cas in an agonist-dependent manner. Overexpression of TRIP6 augments LPA-induced cell migration; in contrast, suppression of endogenous TRIP6 expression by a TRIP6-specific small interfering RNA reduces it in SKOV3 ovarian cancer cells. Strikingly, the association with TRIP6 is specific to the LPA2 receptor but not LPA1 or LPA3 receptor, indicating a specific role for TRIP6 in regulating LPA2 receptor-mediated signaling. Taken together, our results suggest that TRIP6 functions at a point of convergence between the activated LPA2 receptor and downstream signals involved in cell adhesion and migration.
Lysophosphatidic acid (LPA)1 is a bioactive growth factor-like phospholipid, which mediates diverse biological responses such as mitogenesis, differentiation, cell survival, angiogenesis, inflammation, and cell migration (1). Although the functions of LPA were recognized in the mid-1980s, its associated receptors have just been cloned and characterized in the past few years (1). The first three LPA receptors that have been identified belong to the membrane-bound G protein-coupled receptors, including the LPA1/EDG2, LPA2/EDG4, and LPA3/EDG7 receptors of the endothelial differentiation gene family (2–4). Most recently, the G protein-coupled orphan receptor, p2y9/GPR23, has been recognized as the fourth LPA receptor, which is structurally distinct from the other LPA receptors (5). These membrane-bound LPA receptors couple to Gq, Gi/o, or G12/13 proteins and share similar functions in mediating LPA actions (1). Intriguingly, LPA has recently been identified as an agonist of the nuclear peroxisome proliferator-activated receptor γ (6). Thus, some of the LPA signaling pathways are probably differentially regulated by different LPA receptors.
LPA modulates cell adhesion and migration in many cell types by inducing actin cytoskeletal rearrangement, the assembly of focal complexes, and the formation of focal adhesions through a Rho-dependent, integrin-mediated signaling pathway (7, 8). Reciprocal activation of Rho and Rac coordinates the dynamic processes of cell migration (9). The assembly of focal complexes requires focal adhesion kinase (FAK), Src family kinases, paxillin, and p130cas (Crk-associated substrate) (10). These proteins form complexes with downstream signaling molecules, Grb2 and Crk, and trigger adhesion-induced cellular responses including mitogenic signaling, cell locomotion, and cell survival (11). Thus far, the detailed mechanisms by which LPA receptors mediate LPA-induced cell migration are not clear and remain to be explored.
Recently, members of the zyxin family have been shown to localize at focal adhesions and associate with the Cas family, p130cas and CasL/HEF1 (12). The zyxin family members, including zyxin, LPP (lipoma preferred partner), and TRIP6/ZRP-1, contain three zinc finger LIM domains at their carboxyl terminus, a proline-rich region, and nuclear export signals at their N terminus (12–15). The LIM domain (named by the initials of three homeodomain proteins, Lin-11, Isl-1, and Mec-3) has been demonstrated to be a protein-protein interaction motif that is critically involved in their functions (16). Zyxin has been shown to associate with the actin cytoskeleton and is postulated to function in integrin-mediated signaling (17). These zyxin family members localize at focal adhesions but may shuttle between plasma membrane, cytosol, and nucleus and relay unidentified signals between focal adhesions and nucleus (18–20). Since zyxin and TRIP6 associate with Cas family members, they may cooperate to regulate cell motility (12).
The LPA1, LPA2, and LPA3 receptors share high homology in amino acid sequences except for the carboxyl-terminal region, suggesting that the cytoplasmic tail of these receptors may specifically regulate their functions in LPA signaling. In an attempt to identify the molecules that specifically involve in the function and regulation of the LPA2 receptor, we used the carboxyl-terminal tail of the LPA2 receptor as the bait in a yeast two-hybrid screening. Here we demonstrate that the LPA2 receptor, but not LPA1 or LPA3 receptor, associates with TRIP6 by LPA stimulation. The LPA-dependent recruitment of TRIP6 to the plasma membrane promotes its targeting to focal adhesions and co-localization with actin. TRIP6 then serves as an adaptor for the assembly of focal complexes, thereby regulating LPA-induced cell migration.
EXPERIMENTAL PROCEDURES
Plasmid Construction
The clones containing full-length cDNA sequences of the LPA1 receptor, LPA2 receptor, and TRIP6 were obtained from the I.M.A.G.E. consortium through the American Type Culture Collection (ATCC). One guanine base near the 3′ end of the coding sequences of the LPA2 receptor, which was found deleted in the I.M.A.G.E. clone 755526 (21, 22), has been corrected by PCR. The full-length LPA3 receptor cDNA was amplified by reverse transcriptase-PCR using total RNA of SKOV3 ovarian cancer cells as the template. To construct the mammalian expression vectors, different cDNA fragments encoding the LPA1–3 receptors, a LPA2 receptor mutant lacking the carboxyl-terminal tail (aa 296–351), TRIP6 and different truncated TRIP6 mutants were amplified by PCR and inserted into pCMV-Tag2A (Stratagene), pCMV-Tag3A (Stratagene), pEGFP (Clontech), or pHcRed1 (Clontech), respectively, such that these proteins were tagged in-frame with a FLAG epitope, a Myc epitope, a green fluorescence protein (GFP), or a far red fluorescence protein (HcRed1) at their N termini. The entire sequences of each cDNA clone were verified by automatic DNA sequencing. For in vitro binding between TRIP6 and the LPA2 receptor, cDNA fragments encoding TRIP6 or the cytoplasmic tail of the LPA2 receptor (aa 296–351) (designated LPA2R-CT) were inserted inframe into pGEX-6P-3 and pGEX-6P-1 (Amersham Biosciences), respectively.
To inhibit the expression of endogenous TRIP6, the pSUPER vector was constructed as described (23) and was used to direct the expression of a small interfering RNA (siRNA) of TRIP6 (designated pSUPER-siTRIP6) in mammalian cells, which specifically targets the 19-nt sequences of TRIP6, 5′-GAAGCTGGTTCACGACATG-3′.
Yeast Two-hybrid Screening
For the yeast two-hybrid screening, a cDNA fragment encoding LPA2R-CT was amplified by PCR and inserted in-frame at the 3′-end of Gal4 DNA binding domain of the pAS2–1 yeast shuttle vector (Clontech). The entire cDNA sequences of LPA2R-CT were verified by automatic DNA sequencing. This pAS-LPA2R-CT was used as the bait to screen a HeLa cell cDNA library (cDNA constructed in pGAD GH) (Clontech). After co-transformation of pAS-LPA2R-CT and the library plasmids into yeast strain Y190, cells were screened on the plates lacking tryptophan, leucine and histidine supplemented with 25 mM 3-amino-1,2,4-triazole at 30 °C for 3–6 days. Histidine-positive colonies were further screened for positive interaction by β-galactosidase assays. Plasmids harboring cDNA were isolated from positive yeast colonies, transformed into Escherichia coli HB101 by electroporation, and further selected on M9 plates. The positive cDNA clones were sequenced. To construct pAS-LPA3R-CT, a cDNA fragment encoding LPA3R-CT (aa 294–353) was amplified by PCR, inserted into pAS 2-1, and verified by DNA sequencing. To construct pAS-LPA1R-CT, an EcoRI/SalI cDNA fragment encoding amino acids 301–364 of the LPA1 receptor was removed from pCMV-FLAG-LPA2R and inserted into pAS2-1.
Cellular Co-immunoprecipitation and in Vitro GST Pull-down
To examine cellular interaction between TRIP6 and LPA receptors, the vector expressing GFP-tagged or Myc-tagged TRIP6 (wild-type or truncation mutants) was transiently transfected into HEK 293T cells without or with the FLAG-LPA receptor-expressing vector. After starvation in 0.1% fatty acid-free bovine serum albumin (BSA)-containing Dulbecco’s modified Eagle’s medium overnight, cells were incubated without or with 2 μM LPA for 10 min and harvested in co-immunoprecipitation buffer (1% Triton X-100, 10% glycerol, 150 mM NaCl, 10 mM HEPES, 1 mM EDTA, 1 mM EGTA) supplemented with a mixture of protease inhibitors and phosphatase inhibitors. The lysates were briefly sonicated to partially disrupt membrane fractions. Lysates were clarified by centrifugation at 14,000 × g for 10 min. The LPA receptors were immunoprecipitated with anti-FLAG M2 monoclonal antibody-conjugated agarose (Sigma), resolved by SDS-PAGE, and transferred to nitrocellulose membrane for immunoblotting. The co-immunoprecipitated GFP-TRIP6 and Myc-TRIP6 (wild-type or mutants) were detected with an anti-GFP or anti-Myc polyclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Similar experiments were performed to examine co-immunoprecipitation of TRIP6 with paxillin, p130cas, Myc- FAK, or HA-c-Src in HEK 293T cells. To detect the interaction between endogenous TRIP6 and transfected LPA2 receptors, NIH/3T3 cells were transiently transfected with pCMV-Tag3A or pCMV-Myc-LPA2R. After treatment or not with 2 μM LPA for 10 min, Myc-LPA2R from the whole cell lysates was immunoprecipitated with anti-Myc 9E10 monoclonal antibody-conjugated agarose beads (Babco). Co-immunoprecipitated endogenous TRIP6 was detected with a TRIP6-specific polyclonal antibody against amino acids 403–417.
To assess the direct binding of TRIP6 with LPA2R-CT in vitro, GST, GST-LPA2R-CT, and GST-TRIP6 were expressed in (BL21) (DE3) (LysS) E. coli and purified by immobilizing the proteins on glutathione- Sepharose 4B beads (Amersham Biosciences). GST-TRIP6 was further digested with PreScission Protease (Amersham Biosciences) to cleave GST. One μg of TRIP6 protein was incubated with 1 μg of GST or GST-LPA2R-CT in the binding buffer (20 mM Tris, pH 7.4, 1 mM EDTA, 150 mM NaCl, 0.5% Nonidet P-40) for 3 h at 4 °C. TRIP6 pulled down by GST proteins was resolved by SDS-PAGE and detected with a TRIP6-specific antibody (Transduction Laboratories).
LPA-induced Haptotactic Cell Migration
The haptotactic migration assay was performed in 12-well modified Boyden chambers with 8 μM pore-sized PET track-etched membranes (Fisher). SKOV3 cells (American Type Culture Collection) at passages 20–40 were transiently transfected with the expression vector of GFP, GFP-TRIP6, GFP-TRIP6(CT3) (aa 398–476), GFP-TRIP6(ΔC1) (aa 1–278), or GFP-TRIP6(ΔC2) (aa 1–350). Cells were trypsinized and washed with Dulbecco’s modified Eagle’s medium containing 1% fatty acid-free BSA to remove residual LPA. Cells at a density of 150,000/well were then placed in the upper chamber of the transwells. The membrane was coated with 10 μg/ml fibronectin overnight. Two μM LPA was added or not in the lower chamber, and cells were allowed to migrate for 6 h. The nonmigrated cells from the top surface were removed with cotton swabs. The filter was then fixed with 3% formaldehyde, cut out and mounted on the glass slide. GFP-positive cells on the whole filter were counted by fluorescence microscopy (Axioplan 2; Zeiss). Meanwhile, an aliquot of cells was plated on the coverslips. GFP-positive cells in each field were counted to ensure comparable transfection efficiency among all of the samples. To assess the inhibition of TRIP6 function by siRNA, pEGFP was co-transfected with either pSUPER or pSUPER-siTRIP6 into SKOV3 cells. LPA-induced haptotactic cell migration was assayed as described above. Meanwhile, an aliquot of cells was harvested. The expression of GFP and endogenous TRIP6 in the whole cell lysates was determined by immunoblotting probed with an anti-GFP polyclonal antibody (Santa Cruz Biotechnology) and a TRIP6-specific monoclonal antibody (Transduction Laboratories), respectively.
Immunocytochemistry
To examine LPA-induced co-localization of TRIP6 with the LPA2 receptor, SKOV3 cells were transiently co-transfected with the plasmids expressing HcRed1-TRIP6 and GFP-LPA2R. Cells were starved in Dulbecco’s modified Eagle’s medium containing 0.1% BSA overnight and then treated without or with 2 μM LPA for 10 min. The images of GFP-LPA2R and HcRed1-TRIP6 were directly captured by fluorescence microscopy (Axioplan 2; Zeiss). To examine LPA-induced co-localization of TRIP6 with actin or focal adhesion molecules, NIH/3T3 fibroblasts or SKOV3 cells expressing wild-type or mutant GFP-TRIP6 without or with Myc-FAK were plated on coverslips in 6-well plates. After serum starvation overnight, cells were treated without or with 2 μM LPA for 15–20 min and fixed in 3% formaldehyde. After permeabilization with 0.2% Triton X-100, cells were blocked in phosphate- buffered saline containing 2% BSA. To visualize actin cytoskeleton, NIH/3T3 cells were stained with TRITC-phalloidin (Sigma) for 30 min. For immunostaining of vinculin, paxillin, and Myc-FAK, SKOV3 cells were incubated with an anti-vinculin monoclonal antibody (Sigma), anti-paxillin polyclonal antibody (Santa Cruz Biotechnology), or anti-Myc 9E10 monoclonal antibody (Santa Cruz Biotechnology), respectively, for 1 h, and then with the Texas Red-X anti-mouse or anti-rabbit secondary antibody (Molecular Probes, Inc., Eugene, OR) for another 1 h.
RESULTS
The Carboxyl-terminal Tail of the LPA2 Receptor, but Not LPA1 or LPA3 Receptor, Interacts with the LIM Domains of TRIP6
Among the amino acid sequences of all three LPA receptors, the carboxyl-terminal region of these receptors is much less homologous. The overall identity of the cytoplasmic tails is 27% between the LPA1 and LPA2 receptors and 17% between the LPA2 and LPA3 receptors. This diversity could account for the receptor specificity in mediating LPA signaling. In an attempt to identify the molecules that are specifically involved in LPA2 receptor-mediated signaling, a fusion protein containing the carboxyl-terminal tail (aa 296–351) of the LPA2 receptor (designated LPA2R-CT) and the Gal4 DNA binding domain was used as the bait to screen a HeLa cell cDNA library. A total of 4 million clones were screened, and 28 positive clones encoding the carboxyl sequences of TRIP6 (aa 220–476 and 308–476) were isolated (Fig. 1A). These two clones, designated TRIP6(ΔN1) and TRIP6(ΔN2), encode the carboxyl LIM domains 1–3 and LIM domains 2 and 3 of TRIP6, respectively. The interaction was further verified by selective growth of yeast cells co-expressing both LPA2R-CT and TRIP6(ΔN1) (Fig. 1B). We further examined the ability of the carboxyl-terminal tails of LPA1 and LPA3 receptors to bind to TRIP6. Strikingly, TRIP6 only interacted with the LPA2 receptor but not LPA1 or LPA3 receptor in yeast (Fig. 1B).
Fig. 1. TRIP6 interacts with the LPA2 receptor, but not LPA1 or LPA3 receptor in yeast.

A, the schematic structures of TRIP6 and two TRIP6(ΔN) clones identified as LPA2 receptor-interacting proteins in a yeast two-hybrid screening. TRIP6 contains a nuclear export signal (aa 100–107), a N-terminal proline-rich region, and three C-terminal LIM domains. TRIP6(ΔN1) (aa 220–476) and TRIP6(ΔN2) (aa 308–476) contain LIM domains 1–3 and LIM domains 2 and 3, respectively. B, interaction of TRIP6(ΔN1) with LPA2R-CT in yeast. The cDNA fragments encoding the carboxyl-terminal tail of LPA1R (aa 301–364), LPA2R (aa 296–351), and LPA3R (aa 294–353) were inserted into pAS2–1, respectively. pAS or each pAS-LPAR-expressing vector was transformed into yeast Y190 cells with either pGAD or pGAD-TRIP6 encoding TRIP6(ΔN1). The interaction of TRIP6(ΔN1) with LPA2R-CT was verified by selective growth of transformants on a plate lacking tryptophan, leucine, and histidine supplemented with 3-amino-1,2,4-triazole.
Next, we examined whether TRIP6 directly interacts with the LPA2 receptor. In this experiment, a GST fusion protein of LPA2R-CT was used to pull down the purified full-length TRIP6 in vitro. As shown in Fig. 2, TRIP6 was pulled down by GST-LPA2R-CT but not GST, indicating a direct interaction between TRIP6 and the carboxyl-terminal tail of the LPA2 receptor.
Fig. 2. TRIP6 interacts with the carboxyl-terminal tail of the LPA2 receptor in vitro.
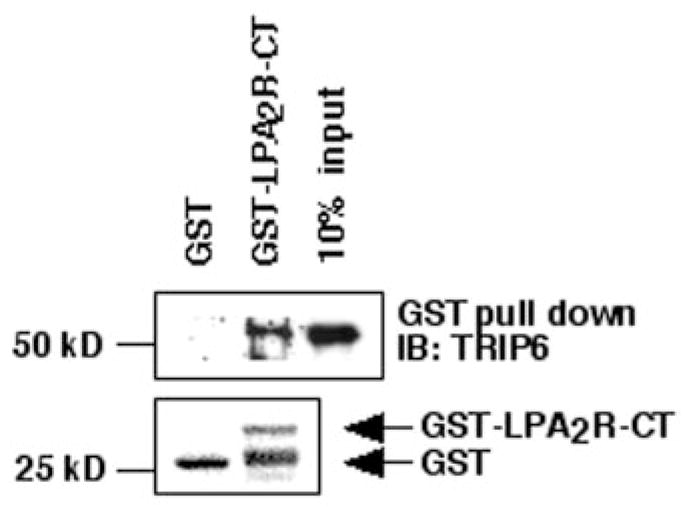
One μg of purified TRIP6 was incubated with 1 μg of GST or GST-LPA2R-CT (aa 296–351) as described under “Experimental Procedures.” TRIP6 pulled down by GST-LPA2R-CT was detected by immunoblotting (IB) with an anti-TRIP6 antibody. GST-LPA 2R and GST were visualized by Ponceau S staining. The third lane shown is a 10% input of TRIP6 used in this experiment.
To examine the association of TRIP6 with different LPA receptors in cells, co-immunoprecipitation of GFP-TRIP6 with the FLAG-tagged LPA1, LPA2, or LPA3 receptor or a LPA2 receptor mutant lacking the cytoplasmic tail (designated LPA2R(ΔC)) was performed in HEK 293T cells, which express a barely detectable level of endogenous TRIP6. As shown in Fig. 3A, LPA stimulation for 10 min promoted co-immunoprecipitation of TRIP6 with the LPA2 receptor but not LPA1 or LPA3 receptor. In contrast, deletion of the carboxyl-terminal tail of LPA2R abolished its interaction with TRIP6 (Fig. 3A). We then performed co-immunoprecipitation of a Myc-tagged LPA2 receptor with endogenous TRIP6 in NIH/3T3 fibroblasts. The result showed that endogenous TRIP6 associated with the transfected Myc-tagged LPA2 receptor in a LPA-dependent manner (Fig. 3B). Taken together from cellular co-immunoprecipitation, yeast two-hybrid assays and in vitro GST pull-down experiments, it is clear that TRIP6 specifically binds to the LPA2 receptor but not other LPA receptors.
Fig. 3. The LIM domains of TRIP6 bind to the LPA2 receptor, but not LPA1R, LPA3R, or a LPA2R mutant lacking its carboxyl-terminal tail.

A, the GFP-TRIP6 expression vector was transiently transfected into HEK 293T cells alone or with one of the vectors expressing FLAG-tagged LPA receptors (LPA1R, LPA2R, LPA3R, or LPA2R(ΔC) lacking its C-terminal tail). Cells were starved in Dulbecco’s modified Eagle’s medium containing 0.1% fatty acid-free BSA overnight and then treated without or with 2 μM LPA for 10 min. The LPA receptors from the whole cell lysates were immunoprecipitated (IP) with anti-FLAG M2 mouse antibody-conjugated agarose beads, and the immunoblot (IB) was probed with an anti-GFP polyclonal antibody to detect co-immunoprecipitated GFP-TRIP6. The same blot was then stripped and reprobed with an anti-FLAG polyclonal antibody to detect immunoprecipitated receptors. Results shown are LPA receptor monomers (~40 kDa) and multiple modified forms of LPA receptors (~50–150 kDa). The bottom panel shows the expression of GFP-TRIP6 in the whole cell lysates. B, LPA-dependent co-immunoprecipitation of endogenous TRIP6 with transfected Myc-LPA2R in NIH/3T3 cells. NIH/3T3 cells were transiently transfected with either pCMV-Tag3A (mock) or pCMV-Myc-LPA2R. The LPA2 receptor was immunoprecipitated with anti-Myc 9E10 monoclonal antibody-conjugated agarose beads from the unstimulated cells and cells treated with 2 μM LPA for 10 min. The co-immunoprecipitated endogenous TRIP6 was detected with a TRIP6- specific polyclonal antibody. C, co-immunoprecipitation of FLAG-LPA 2R with GFP-TRIP6(CT2) (aa 339–476) or GFP-TRIP6(CT3) (aa 398–476), which contain LIM domains 2 and 3 and LIM3 domain, respectively. This experiment was carried out without or with LPA in HEK 293T cells as described above. D, co-immunoprecipitation of FLAG-LPA2R with Myc-TRIP6 or Myc-TRIP6(ΔC1) (aa 1–278) lacking the entire LIM domains. A similar experiment was performed in HEK 293T cells as described above. Myc-tagged TRIP6 and TRIP6(ΔC1) were detected with an anti-Myc polyclonal antibody.
The yeast two-hybrid screening identified two LPA2R-interacting TRIP6 clones, which encode LIM domains 1–3 and domains 2 and 3, respectively, suggesting that TRIP6 may associate with the LPA2 receptor through its LIM domains. To identify the receptor-binding domain of TRIP6 in cells, a number of vectors expressing different TRIP6 truncation mutants have been generated. Their molecular structures are depicted in Fig. 4E. Indeed, we found that TRIP6(CT2) mutant (aa 339–476), which contains LIM domains 2 and 3, was able to co-immunoprecipitate with the LPA2 receptor (Fig. 3C). Even the TRIP6(CT3) mutant (aa 398–476), containing the LIM3 domain alone, was capable of binding to the LPA2 receptor (Fig. 3C). However, we found that LPA still induced a weak but detectable binding between the LPA2 receptor and a TRIP6(ΔC2) mutant (aa 1–350), which lacks LIM domains 2 and 3 (data not shown). Thus, it appears that all of the LIM domains of TRIP6 contribute to its binding to the LPA2 receptor. Different from the wild-type TRIP6, these truncated LIM domains interact with the LPA2 receptor in an agonist-independent manner, suggesting that the N-terminal region of TRIP6 regulates this agonist-dependent receptor binding. We further examined the receptor binding ability of a TRIP6(ΔC1) mutant (aa 1–278) lacking the entire LIM domains. As shown in Fig. 3D, LPA stimulation for 10 min promoted co-immunoprecipitation of the LPA2 receptor with wild-type TRIP6 but not the TRIP6(ΔC1) mutant. The LPA2R binding ability of different TRIP6 mutants is summarized in Fig. 4E.
Fig. 4. LPA promotes co-localization of TRIP6 with the LPA2 receptor and TRIP6 targeting to focal adhesions.
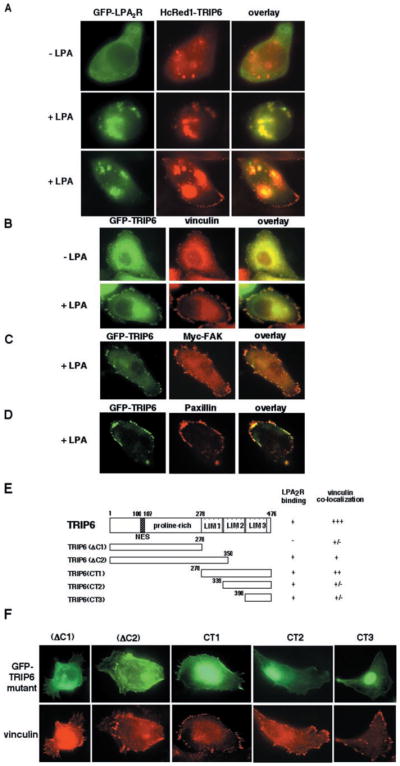
A, SKOV3 cells were co-transfected with the expression plasmids of GFP-LPA2R and HcRed1-TRIP6. After serum starvation overnight, cells were treated without or with 2 μM LPA for 10 min. The GFP fluorescence of LPA2R and the far red fluorescence of TRIP6 were visualized directly by fluorescence microscopy. B–D, SKOV3 cells transiently expressing GFP-TRIP6 alone or with Myc-FAK were treated without or with 2 μM LPA for 15 min. Cells were fixed, and GFP-TRIP6 was visualized by fluorescence microscopy. The immunostaining of endogenous vinculin (B), Myc-FAK (C), and endogenous paxillin (D) was performed as described under “Experimental Procedures.” E, the molecular structures of different TRIP6 truncation mutants and a summary of their capacity to bind to the LPA2 receptor and co-localize with vinculin. F, SKOV3 cells expressing GFP-TRIP6 mutants were starved overnight and treated with 2 μM LPA for 15 min. The endogenous vinculin was detected with an anti-vinculin antibody, and GFP-TRIP6 mutants were visualized by fluorescence microscopy.
LPA Induces Co-localization of TRIP6 with the LPA2 Receptor and Promotes TRIP6 Targeting to Focal Adhesions
To investigate the potential functional role of TRIP6 in LPA2 receptor-mediated signaling, localization of TRIP6 with the LPA2 receptor was first examined in SKOV3 ovarian cancer cells expressing a GFP fusion protein of the LPA2 receptor and a HcRed1 fusion protein of TRIP6. The protein distribution was visualized directly by fluorescence microscopy. As shown in Fig. 4A, GFP-LPA2R was diffusely present or formed small clusters on the plasma membrane and in the cytoplasm by serum starvation overnight. Similarly, HcRed1-TRIP6 was predominantly cytosolic with some aggregates in the unstimulated cells. However, the distribution of these TRIP6 aggregates did not co-localize with the LPA2R clusters. After exposure of cells to LPA for 10 min, a large portion of HcRed1-TRIP6 appeared in plasma membrane clusters that coincided precisely with the distribution of GFP-LPA2R, indicative of LPA-dependent translocation of TRIP6 to the receptor (Fig. 4A, middle panel). Strikingly, LPA-induced cell rounding was greatly enhanced by overexpression of the LPA2 receptor, consistent with previous reports for the role of the LPA2 receptor in LPA-induced morphological changes (24). In addition to the membrane-bound clusters, the complexes of GFP-LPA2R and HcRed1-TRIP6 were also present in the cytosol, suggesting that TRIP6 may be internalized with the activated receptor (Fig. 4A, bottom panel). In the meantime, LPA also promoted TRIP6 targeting to focal adhesions at the leading edge, where, however, the LPA2 receptor was not present (Fig. 4A, bottom panel). Thus, TRIP6 must have dissociated from the LPA2 receptor before targeting to focal adhesions.
The LIM domain-containing proteins, including the family members of paxillin and zyxin, have been shown to localize at focal adhesions mainly through the LIM domains; however, the non-LIM region also has some weak targeting capacity (19, 25, 26). To further investigate LPA-dependent targeting of TRIP6 to focal adhesions, co-localization of TRIP6 with the focal adhesion molecules, including vinculin, FAK, and paxillin, were examined in SKOV3 cells expressing GFP-TRIP6 in the absence or presence of LPA. These SKOV3 cells have been shown to express endogenous LPA1, LPA2, and LPA3 receptors at high levels (27, 28). As shown in Fig. 4B, GFP fluorescence of TRIP6 was diffusely present in the cytoplasm after serum starvation overnight, although a small amount of TRIP6 was found at focal adhesions. After LPA stimulation for 15 min, a large portion of TRIP6 was depleted from the cytosol and recruited to focal adhesions at the periphery of the plasma membrane, where it was co-localized with vinculin (Fig. 4B), FAK (Fig. 4C), and paxillin (Fig. 4D).
To identify the domains of TRIP6 required for LPA-induced targeting of TRIP6 to focal adhesions, we examined co-localization of vinculin with a number of TRIP6 truncation mutants. The molecular structures of these mutants and their abilities to co-immunoprecipitate with the LPA2 receptor are shown in Fig. 4E. We found that the focal adhesion targeting capacity of TRIP6 was retained when LIM2 and -3 domains were deleted (Fig. 4F, TRIP6(ΔC2)), but was almost completely lost when the entire LIM domains were removed (Fig. 4F, TRIP6(ΔC1)). The TRIP6(CT1) mutant, containing all three LIM domains, was able to co-localize with vinculin, whereas the TRIP6(CT2) mutant, containing LIM domains 2 and 3, and the TRIP6(CT3) mutant, containing LIM3 domain alone, only showed a rudimentary capacity to localize to focal adhesions by LPA stimulation (Fig. 4F). All of these results indicate that the LIM1 domain of TRIP6 is the minimal requirement for LPA-induced TRIP6 targeting to focal adhesions; however, other domains appear to cooperate to allow maximal accumulation of TRIP6 at these sites. Since focal adhesion targeting domains of TRIP6 partially overlap with its LPA2R-binding domains, the membrane-bound TRIP6 might dissociate from the LPA2 receptor through a preferential binding to focal adhesion molecules. It should be noted that not only LPA, but also other ligands such as thrombin, platelet-derived growth factor, and sphingosine 1-phosphate, are able to promote translocation of TRIP6 to focal adhesions (data not shown). Thus, it appears to be a general phenomenon by stimulation with different growth factors. Nonetheless, the interaction of TRIP6 with the LPA2 receptor seems to provide an additional mechanism for recruiting TRIP6 to the plasma membrane and focal adhesions.
TRIP6 Co-localizes with Actin by LPA Stimulation
Next, we examined LPA-induced actin cytoskeletal rearrangement by staining actin with TRITC-phalloidin in NIH/3T3 fibroblasts expressing wild-type or mutant TRIP6. These NIH/3T3 cells express endogenous LPA1–3 receptors by reverse transcriptase- PCR analysis (data not shown). As shown in Fig. 5A, LPA stimulation for 20 min triggered the assembly of actin and co-localization of TRIP6 with actin stress fibers. We further performed deletion analysis to identify the regions of TRIP6 required for actin co-localization. The molecular structures of these mutants and their capacity to co-localize with actin were shown in Fig. 5B. We found that co-localization with actin was retained but was significantly reduced when LIM domains 2 and 3 or the entire LIM domains were deleted (Fig. 5C, TRIP6(ΔC2) and TRIP6(ΔC1)). Among the three LIM domain-containing proteins, TRIP6(CT1), containing LIM domains 1–3, showed the best capacity to co-localize with actin, whereas TRIP6(CT2), containing LIM domains 2 and 3, only remained a weak capacity. The TRIP6(CT3) mutant, containing LIM3 domain alone, almost completely lost its capacity to co-localize with actin (Fig. 5C). Thus, LIM domains 1 and 2 cooperate to allow the most efficient targeting of TRIP6 to actin stress fibers; however, the proline-rich region of TRIP6 also contributes to this capacity.
Fig. 5. TRIP6 co-localizes with actin by LPA treatment.

A, NIH/3T3 fibroblasts transiently expressing GFP-TRIP6 were treated without or with 2 μM LPA for 20 min. Actin was stained with TRITC-phalloidin. B, the molecular structures of different TRIP6 truncation mutants and a summary of their capacity to co-localize with actin. C, NIH/3T3 fibroblasts expressing different GFP-TRIP6 truncation mutants were treated with 2 μM LPA for 20 min. The top panel shows GFP fluorescence of different TRIP6 mutants, and the bottom panel shows actin stained with TRITC-phalloidin.
TRIP6 Associates with Paxillin, p130cas, FAK, and c-Src in an Agonist-dependent Manner
TRIP6 directly binds to the carboxyl-terminal tail of the LPA2 receptor, raising the possibility that TRIP6 may play an important role in linking LPA2 receptor-mediated signaling to the assembly of focal complexes and cell migration. The components of focal complexes known to be involved in cell migration include p130cas, FAK, paxillin, and c-Src. To assess whether TRIP6 associates with these molecules by LPA stimulation, we examined LPA-dependent co-immunoprecipitation between TRIP6 and paxillin, p130cas, FAK, or c-Src in HEK 293T cells. Our results showed that LPA induced co-immunoprecipitation of TRIP6 with paxillin (Fig. 6A), p130cas (Fig. 6B), Myc-FAK (Fig. 6C), and HA-c-Src (Fig. 6D). Thus, LPA-promoted recruitment of TRIP6 to the plasma membrane transforms it into an adaptor for the formation of multiple protein complexes. These findings further suggest a potential regulatory role for TRIP6 in LPA-induced cell migration and downstream signaling events.
Fig. 6. LPA induces the association of TRIP6 with paxillin, p130cas, FAK, and c-Src in HEK 293T cells.

A and B, HEK 293T cells were transiently transfected with an empty vector or a Myc-TRIP6-expressing vector. Cells were starved overnight and then treated without or with 2 μM LPA for 10 min. TRIP6 from the whole cell lysates was immunoprecipitated with anti-Myc 9E10 antibody-conjugated agarose beads and resolved by SDS-PAGE. The endogenous paxillin (A) and p130cas (B) co-immunoprecipitated with Myc-TRIP6 were detected with their specific antibodies. C, HEK 293T cells transiently expressing GFP-TRIP6 without or with Myc-FAK were incubated in the absence or presence of 2 μM LPA for 10 min. Myc-FAK was immunoprecipitated (IP) with anti-Myc 9E10 antibody-conjugated agarose beads. The immunoblot (IB) was probed with an anti-GFP antibody to detect GFP-TRIP6. D, LPA-dependent co-immunoprecipitation of HA-c-Src and FLAG-TRIP6 was performed in HEK 293T cells as described above. TRIP6 was immunoprecipitated with anti-FLAG M2 antibody-conjugated agarose beads, and the co-immunoprecipitated c-Src was detected with an anti-HA antibody. The bottom panel of each figure is an immunoblot showing the expression of endogenous paxillin (A), endogenous p130cas (B), transfected GFP-TRIP6 (C), and HA-c-Src (D) in the whole cell lysates.
TRIP6 Regulates LPA-induced Cell Migration in SKOV3 Cells
To assess the effect of TRIP6 on LPA-induced haptotactic cell migration, SKOV3 cells were transiently transfected with the expression vector of GFP, GFP-TRIP6, GFP-TRIP6( CT3), GFP-TRIP6(ΔC1), or GFP-TRIP6(ΔC2). The TRIP6(CT3) mutant, containing LIM3 domain alone, was able to bind to the LPA2 receptor (Fig. 3C) but did not localize at focal adhesions (Fig. 4F) or co-localize with actin (Fig. 5C); thus, it might specifically block the function of the LPA2 receptor in cell migration. We found that in the absence of any ligand, the basal number of migrated cells was similar among all of the transfectants. LPA treatment for 6 h induced 3.45-fold increase of cell migration, which was further enhanced to 6.84- fold by TRIP6 but was decreased to 2.11-fold by TRIP6(CT3) (Fig. 7A). The TRIP6(ΔC1) mutant lacks the entire LIM domains for receptor binding (Fig. 3D) and focal adhesion targeting (Fig. 4F). However, it reduced LPA-induced cell migration to 2.41-fold (Fig. 7A), suggesting that the N-terminal proline-rich region of TRIP6 is also important for its function in cell migration. The TRIP6(ΔC2) mutant, which shows a low receptor- binding ability (data not shown) and a weak focal adhesion targeting capacity (Fig. 4F), did not significantly alter LPA-induced cell migration (Fig. 7A). Interestingly, GFP-TRIP6 and GFP-TRIP6(ΔC2), but not GFP, GFP-TRIP6(CT3), or GFP-TRIP6( ΔC1), showed striking punctate patterns in about 5–10% of the migrated cells, suggesting that TRIP6 formed protein complexes with some unidentified signaling molecules (Fig. 7A). It is likely that this complex formation is dependent on integrin, since it could be reproduced by plating TRIP6- expressing cells on fibronectin-coated coverslips (data not shown). The TRIP6(CT3) mutant specifically binds to the LPA2 receptor and partially reduces LPA-induced cell migration, suggesting a role for the LPA2 receptor in mediating LPA-induced cell migration. However, other LPA receptors appear to contribute to this event as well.
Fig. 7. TRIP6 enhances LPA-induced but not thrombin-induced cell migration, whereas suppression of endogenous TRIP6 expression by a TRIP6-specific siRNA inhibits it in SKOV3 cells.

A, SKOV3 cells transiently expressing GFP, GFP-TRIP6, GFP-TRIP6( CT3), GFP-TRIP6(ΔC1), or GFP-TRIP6(ΔC2) were subjected to haptotactic cell migration assays as described under “Experimental Procedures.” Two μM LPA was added or not in the lower chamber of the transwells, and cells were allowed to migrate for 6 h. Nonmigrated cells from the top surface were removed with a Q-tip. The filter was fixed, and GFP-positive cells migrated to the bottom surface were counted by fluorescence microscopy. LPA-induced cell migration was determined as the -fold increase of migrated cells with LPA exposure versus without LPA exposure. The bottom panel shows images of some migrated cells. B, thrombin-induced cell migration was performed in SKOV3 cells transiently expressing GFP or GFP-TRIP6 as described above. 1 units/ml of thrombin was added or not in the bottom chamber of the transwells. C, pEGFP was transiently co-transfected with pSUPER or pSUPER-siTRIP6 into SKOV3 cells. An LPA-induced haptotactic cell migration assay was carried out as described above. In the meantime, equal amounts of cells were harvested and dissolved in SDS lysis buffer. The bottom panel is an immunoblot showing the expression of endogenous TRIP6 in cells transfected with pSUPER (lane 1) or pSUPER-siTRIP6 (lane 2). This blot was reprobed with an anti-GFP antibody to ensure equal expression of GFP in each sample. Data shown in each figure are the mean ± S.E. of 3–7 independent experiments.
Previously, it has been shown that displacing zyxin from its normal subcellular location perturbs cell migration (29). In addition, overexpression of LPP increases EGF-induced cell migration in vascular smooth muscle cells (30). These observations suggest an intrinsic role for these zyxin family members in cell migration. To investigate whether TRIP6 has similar effects on cell migration by stimulation with other ligands, thrombin-induced cell migration was carried out in SKOV3 cells overexpressing either GFP or GFP-TRIP6. We found that thrombin-induced haptotactic cell migration was not significantly altered by overexpression of TRIP6 in SKOV3 cells (Fig. 7B). Other ligands, such as platelet-derived growth factor and sphingosine 1-phosphate, mildly induced haptotactic cell migration in SKOV3 cells, which was not affected by TRIP6 either (data not shown). These results suggest that although TRIP6 can be targeted to focal adhesions by different growth factors and is probably one of the components directly involved in cell migration, TRIP6 specifically augments LPA-induced cell migration presumably through LPA-promoted interaction with the LPA2 receptor.
To verify the physiological function of TRIP6 in LPA-induced cell migration, we further knocked down the expression of endogenous TRIP6 by a TRIP6-specific siRNA. In this experiment, the pEGFP expression vector was transiently transfected into SKOV3 cells with either the pSUPER empty vector (23) or pSUPER-siTRIP6 expressing TRIP6 siRNA. A haptotactic cell migration assay was performed as described above, and GFP-positive cells migrated to the bottom side of the filter were counted by fluorescence microscopy. Our result showed that suppression of endogenous TRIP6 expression by TRIP6- specific siRNA significantly attenuated LPA-induced cell migration from 4.49- to 1.67-fold (Fig. 7C). The reduction of endogenous TRIP6 expression in cells transfected with pSUPER-siTRIP6 (lane 2) versus pSUPER (lane 1) was 50% (Fig. 7C). It should be noted that the extent of TRIP6 siRNA-mediated knockdown of endogenous TRIP6 would be much greater, since the best transfection efficiency of SKOV3 cells we could reach was ~50%. Taken together, these results suggest a pivotal role for TRIP6 in linking LPA2 receptors and the downstream signals required for cell adhesion and migration.
DISCUSSION
To gain insights into the understanding of the mechanisms by which the LPA2 receptor mediates LPA signaling, we have identified TRIP6 as a novel LPA2 receptor-interacting protein and showed that TRIP6 regulates LPA-induced cell migration in SKOV3 ovarian cancer cells. TRIP6 is a newly identified zyxin family member, which has been postulated to be involved in cell migration (12). However, so far its function has not yet been fully understood. In this paper, we demonstrated that TRIP6 associated with the membrane-bound and internalized LPA2 receptor by LPA stimulation but was dissociated from the LPA2 receptor when it was translocated to focal adhesions. Moreover, LPA induced co-localization of TRIP6 with actin and promoted its association with the major signaling components known to regulate cell adhesion and migration, including c-Src, FAK, p130cas, and paxillin. Using deletion analysis, we showed that LIM1 domain was the minimal requirement for its focal adhesion targeting, whereas both LIM domains 1 and 2 and the N-terminal proline-rich region contributed to its co-localization with actin.
In this report, we provided evidence for the direct involvement of TRIP6 in cell migration by showing that suppression of endogenous TRIP6 expression by a TRIP6-specific siRNA attenuated LPA-induced cell migration. Cell migration is a dynamic process that requires a tight coordination between cell attachment and detachment from extracellular matrix. Thus, a moderate strength of cell adhesion would be the most favorable for cell migration (31). This process requires an appropriate regulation of the assembly and disassembly of focal complexes. Therefore, overexpression of one focal adhesion component may alter cell migration depending on the formation and turnover of focal complexes. It may also be dependent on different cellular contexts. This might explain why previously it has been shown that zyxin and TRIP6 interacted with p130cas and CasL/HEF1; however, overexpression of TRIP6 in 10T1/2 cells slowed cell migration, perhaps by preventing the interaction of p130cas and the downstream signaling molecule such as Crk (12). In contrast, recently LPP has been shown to promote EGF-induced cell migration in vascular smooth muscles (30). Different from these reports, here we demonstrated that overexpression of TRIP6 enhanced LPA-induced, but not thrombin-induced, cell migration in SKOV3 ovarian cancer cells. It should be noted that our results do not exclude the involvement of TRIP6 in other growth factor-mediated cell migration. Instead, our results suggest that TRIP6 plays a role in actin dynamics and is a component of focal complexes involved in cell adhesion and migration. Nonetheless, it seems plausible that such receptor binding of TRIP6 promotes its recruitment to the plasma membrane and positions it in proximity to other signaling molecules, thereby enhancing LPA-promoted cell migration. On the other hand, TRIP6 may serve as an adaptor by recruiting unidentified proteins to the receptor-occupied protein complexes and activate LPA-induced downstream signaling. Since TRIP6 only interacts with the LPA2 receptor and not other LPA receptors, it is of further interest that different LPA receptors may mediate LPA signaling through different mechanisms. For example, it has recently been shown that the LPA1 receptor couples to a Gi-phosphoinositide 3-kinase-Tiam1 pathway to activate Rac, and thereby stimulates cell motility (32). Another report demonstrates that the LPA3 receptor is required for LPA-induced, laminin-mediated cell migration in some ovarian cancer cells (33).
The LIM domains of TRIP6 display a high degree of sequence homology with LPP and zyxin, which also contain three LIM domains at their carboxyl terminus (13). The overall identity of all three LIM domains is 71.9% between TRIP6 and LPP and 61.5% between TRIP6 and zyxin. Although all three proteins possess the proline-rich sequences at their N terminus, the identity of this region is much less homologous. Given the high degree of sequence homology in their LIM domains, LPP and zyxin may also bind to the LPA2 receptor. Indeed, we found that both LPP and zyxin were able to co-immunoprecipitate with the LPA2 receptor, but not the LPA1 or LPA3 receptor, in an agonist-dependent manner.2 Whether LPP and zyxin also play a role in LPA2 receptor-mediated signaling remains to be elucidated.
The function of LPA in cell migration plays a central role not only in wound healing and embryonic development but also in the progression of tumors from a noninvasive to an invasive and metastatic phenotype (1). In particular, the levels of LPA and the expression of LPA2 and LPA3 receptors are elevated in ovarian cancer cells but not in normal ovarian epithelial cells (28, 34). These observations implicate a potential role for LPA signaling in ovarian cancer progression. The present studies provide evidence for a physiological role of TRIP6 in regulating LPA-induced cell migration in SKOV3 ovarian cancer cells. Furthermore, TRIP6 may internalize with the activated LPA2 receptor and shuttle between focal adhesions, cytoplasm, and nucleus, such that it may relay LPA2 receptor-mediated signals from the plasma membrane to nucleus and regulate LPA-dependent gene expression. The interaction between TRIP6 and the LPA2 receptor but not other LPA receptors might help in understanding the specific role of the LPA2 receptor in ovarian tumor progression in the future.
Acknowledgments
We thank Dr. Wen-Cheng Xiong for providing the Myc-FAK expression vector and helpful discussion, Dr. Graeme B. Bolger for the HeLa cell cDNA library, Dr. William A. May for pSUPER plasmid, and Dr. Candece L. Gladson for technical help in the cell migration assay.
Footnotes
This work was supported by National Institutes of Health Grant CA100848 (to F.-T. L.) and a pilot project development award from the University of Alabama at Birmingham Ovarian Cancer SPORE (to F.-T. L.).
The abbreviations used are: LPA, lysophosphatidic acid; FAK, focal adhesion kinase; GFP, green fluorescent protein; siRNA, small interfering RNA; BSA, bovine serum albumin; TRITC, tetramethylrhodamine isothiocyanate; EGF, epidermal growth factor; aa, amino acids; LPA1R, LPA2R, and LPA3R, LPA1, LPA2, and LPA3 receptor, respectively.
F.-T. Lin, unpublished results.
References
- 1.Contos JJ, Ishii I, Chun J. Mol Pharmacol. 2000;58:1188–1196. doi: 10.1124/mol.58.6.1188. [DOI] [PubMed] [Google Scholar]
- 2.Hecht JH, Weiner JA, Post SR, Chun J. J Cell Biol. 1996;135:1071–1083. doi: 10.1083/jcb.135.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.An S, Bleu T, Huang W, Hallmark OG, Coughlin SR, Goetzl EJ. FEBS Lett. 1997;417:279–282. doi: 10.1016/s0014-5793(97)01301-x. [DOI] [PubMed] [Google Scholar]
- 4.Bandoh K, Aoki J, Hosono H, Kobayashi S, Kobayashi T, Murakami-Murofushi K, Tsujimoto M, Arai H, Inoue K. J Biol Chem. 1999;274:27776–27785. doi: 10.1074/jbc.274.39.27776. [DOI] [PubMed] [Google Scholar]
- 5.Noguchi K, Ishii S, Shimizu T. J Biol Chem. 2003;278:25600–25606. doi: 10.1074/jbc.M302648200. [DOI] [PubMed] [Google Scholar]
- 6.McIntyre TM, Pontsler AV, Silva AR, St Hilaire A, Xu Y, Hinshaw JC, Zimmerman GA, Hama K, Aoki J, Arai H, Prestwich GD. Proc Natl Acad Sci U S A. 2003;100:131–136. doi: 10.1073/pnas.0135855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sah VP, Seasholtz TM, Sagi SA, Brown JH. Annu Rev Pharmacol Toxicol. 2000;40:459–489. doi: 10.1146/annurev.pharmtox.40.1.459. [DOI] [PubMed] [Google Scholar]
- 8.Panetti TS, Magnusson MK, Peyruchaud O, Zhang Q, Cooke ME, Sakai T, Mosher DF. Prostaglandins. 2001;64:93–106. doi: 10.1016/s0090-6980(01)00102-2. [DOI] [PubMed] [Google Scholar]
- 9.Rottner K, Hall A, Small JV. Curr Biol. 1999;9:640–648. doi: 10.1016/s0960-9822(99)80286-3. [DOI] [PubMed] [Google Scholar]
- 10.Seufferlein T, Rozengurt E. J Biol Chem. 1994;269:9345–9351. [PubMed] [Google Scholar]
- 11.Hanks SK, Polte TR. BioEssays. 1997;19:137–145. doi: 10.1002/bies.950190208. [DOI] [PubMed] [Google Scholar]
- 12.Yi J, Kloeker S, Jensen CC, Bockholt S, Honda H, Hirai H, Beckerle MC. J Biol Chem. 2002;277:9580–9589. doi: 10.1074/jbc.M106922200. [DOI] [PubMed] [Google Scholar]
- 13.Murthy KK, Clark K, Fortin Y, Shen SH, Banville D. J Biol Chem. 1999;274:20679–20687. doi: 10.1074/jbc.274.29.20679. [DOI] [PubMed] [Google Scholar]
- 14.Petit MM, Mols R, Schoenmakers EF, Mandahl N, Van de Ven WJ. Genomics. 1996;36:118–129. doi: 10.1006/geno.1996.0432. [DOI] [PubMed] [Google Scholar]
- 15.Sadler I, Crawford AW, Michelsen JW, Beckerle MC. J Cell Biol. 1992;119:1573–1587. doi: 10.1083/jcb.119.6.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bach I. Mech Dev. 2000;91:5–17. doi: 10.1016/s0925-4773(99)00314-7. [DOI] [PubMed] [Google Scholar]
- 17.Beckerle MC. BioEssays. 1997;19:949–957. doi: 10.1002/bies.950191104. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Gilmore TD. Biochim Biophys Acta. 2003;1593:115–120. doi: 10.1016/s0167-4889(02)00349-x. [DOI] [PubMed] [Google Scholar]
- 19.Petit MM, Meulemans SM, Van de Ven WJ. J Biol Chem. 2003;278:2157–2168. doi: 10.1074/jbc.M206106200. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Gilmore TD. Biochim Biophys Acta. 2001;1538:260–272. doi: 10.1016/s0167-4889(01)00077-5. [DOI] [PubMed] [Google Scholar]
- 21.An S, Bleu T, Hallmark OG, Goetzl EJ. J Biol Chem. 1998;273:7906–7910. doi: 10.1074/jbc.273.14.7906. [DOI] [PubMed] [Google Scholar]
- 22.Contos JJ, Chun J. Genomics. 2000;64:155–169. doi: 10.1006/geno.2000.6122. [DOI] [PubMed] [Google Scholar]
- 23.Brummelkamp TR, Bernards R, Agami R. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 24.Ishii I, Contos JJ, Fukushima N, Chun J. Mol Pharmacol. 2000;58:895–902. doi: 10.1124/mol.58.5.895. [DOI] [PubMed] [Google Scholar]
- 25.Nix DA, Fradelizi J, Bockholt S, Menichi B, Louvard D, Friederich E, Beckerle MC. J Biol Chem. 2001;276:34759–34767. doi: 10.1074/jbc.M102820200. [DOI] [PubMed] [Google Scholar]
- 26.Brown MC, Perrotta JA, Turner CE. J Cell Biol. 1996;135:1109–1123. doi: 10.1083/jcb.135.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu YL, Tee MK, Goetzl EJ, Auersperg N, Mills GB, Ferrara N, Jaffe RB. J Natl Cancer Inst. 2001;93:762–768. doi: 10.1093/jnci/93.10.762. [DOI] [PubMed] [Google Scholar]
- 28.Mills GB, Eder A, Fang X, Hasegawa Y, Mao M, Lu Y, Tanyi J, Tabassam FH, Wiener J, Lapushin R, Yu S, Parrott JA, Compton T, Tribley W, Fishman D, Stack MS, Gaudette D, Jaffe R, Furui T, Aoki J, Erickson JR. Cancer Treat Res. 2002;107:259–283. doi: 10.1007/978-1-4757-3587-1_12. [DOI] [PubMed] [Google Scholar]
- 29.Drees BE, Andrews KM, Beckerle MC. J Cell Biol. 1999;147:1549–1560. doi: 10.1083/jcb.147.7.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorenne I, Nakamoto RK, Phelps CP, Beckerle MC, Somlyo AV, Somlyo AP. Am J Physiol. 2003;285:C674–C685. doi: 10.1152/ajpcell.00608.2002. [DOI] [PubMed] [Google Scholar]
- 31.Cox EA, Huttenlocher A. Microsc Res Tech. 1998;43:412–419. doi: 10.1002/(SICI)1097-0029(19981201)43:5<412::AID-JEMT7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 32.Van Leeuwen FN, Olivo C, Grivell S, Giepmans BN, Collard JG, Moolenaar WH. J Biol Chem. 2003;278:400–406. doi: 10.1074/jbc.M210151200. [DOI] [PubMed] [Google Scholar]
- 33.Sengupta S, Xiao YJ, Xu Y. FASEB J. 2003;17:1570–1572. doi: 10.1096/fj.02-1145fje. [DOI] [PubMed] [Google Scholar]
- 34.Xu Y, Fang XJ, Casey G, Mills GB. Biochem J. 1995;309:933–940. doi: 10.1042/bj3090933. [DOI] [PMC free article] [PubMed] [Google Scholar]