

Abstract
Genotoxic stress triggers apoptosis through multiple signaling pathways. Recent studies have demonstrated a specific induction of E2F1 accumulation and a role for E2F1 in apoptosis upon DNA damage. Induction of E2F1 is mediated by phosphorylation events that are dependent on DNA damage-responsive protein kinases, such as ATM. How ATM phosphorylation leads to E2F1 stabilization is unknown. We now show that 14-3-3τ, a phosphoserine-binding protein, mediates E2F1 stabilization. 14-3-3τ interacts with ATM-phosphorylated E2F1 during DNA damage and inhibits E2F1 ubiquitination. Depletion of 14-3-3τ or E2F1, but not E2F2 or E2F3, blocks adriamycin-induced apoptosis. 14-3-3τ is also required for expression and induction of E2F1 apoptotic targets, such as p73, Apaf-1, and caspases, during DNA damage. Together, these data demonstrate a novel function for 14-3-3τ in the regulation of E2F1 protein stability and apoptosis during DNA damage.
Proper responses to genotoxic stress are vital to maintain genomic stability and prevent the development of cancer. The involvement of E2F1 in the DNA damage response has been recognized (1–5). E2F1 is a member of the E2F transcriptional factor family, which regulates a very diverse array of genes and plays an important role in the regulation of cell cycle progression and other biological processes (1, 6–8). Among E2F family members, E2F1 is unique in its ability to trigger apoptosis (9–12) and its induction in response to DNA damage (2, 4). E2F1 transactivates p73 expression during adriamycin treatment (4) and is required for etoposide-induced apoptosis in murine thymocytes (2). E2F1 also induces the expression of several other genes involved in apoptosis, such as p14ARF (10, 13), Apaf-1 (12), and caspase-3, -7, -8, and -9 (14). The proapoptotic activity of E2F1 is probably mediated through the induction of these genes.
The signaling events that lead to E2F1 induction upon DNA damage have also been delineated. ATM (ataxia telangiectasia mutated) and ATR (ATM and Rad3-related) phosphorylate E2F1 at Ser31 but do not phosphorylate E2F2 or E2F3, and this specificity accounts for the selective induction of E2F1 among the E2F family during DNA damage (2). E2F1 is also phosphorylated by Chk2 (3). Together, these phosphorylation events lead to stabilization and activation of E2F1. In addition to phosphorylation, acetylation has also been recognized to play a role in the activation and stabilization of E2F1 protein during DNA damage (4, 15). Thus, it appears that several DNA damage signaling pathways are involved in the induction of E2F1. However, the mechanism by which these modifications lead to E2F1 stabilization remains unclear.
We now provide evidence that a member of the 14-3-3 family proteins, 14-3-3τ, plays a critical role in the regulation of E2F1 stability. 14-3-3 proteins are a family of dimeric phosphoserine/phosphothreonine-binding proteins, which are involved in a very diverse spectrum of signaling pathways (16). 14-3-3τ binds to Ser31-phosphorylated E2F1 and inhibits the ubiquitination of E2F1 during DNA damage. It is required for the expression and induction of several E2F1 apoptotic target genes as well as apoptosis during DNA damage. Our data suggest a model in which binding of 14-3-3τ to E2F1 interferes with the function of an E31 ligase and therefore inhibits the ubiquitination and degradation of E2F1.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
HEK293 and U2OS cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. The transfection was performed by the calcium phosphate method or the Gene Pulser Xcell electroporation system (Bio-Rad) according to the manufacturer’s instructions.
Yeast Two-hybrid Screen
The N terminus of E2F1 (amino acids 1–109) in pAS2-1 vector was used as a bait to screen a HeLa cDNA library in pGADGH as described (17).
Recombinant Plasmids
pCMV-SPORT6-14-3-3τ was obtained from ResGen. FLAG-tagged 14-3-3τ expression vector was constructed by excising the 14-3-3τ cDNA from pCMV-SPORT6-14-3-3τ by XhoI digestion and inserted into pCMV-Tag 2C. The KpnI/XhoI fragment of pCMV-SPORT6–14-3-3τ was moved to pCMV-Tag2 vector to construct FLAG-tagged ΔN14-3-3τ expression vector. The construction of pSUPER-siE2F1, siE2F2, and siE2F3 has been described (18). The 19-nucleotide target sequence for si14-3-3τ is 5′-GGACTATCGGGAG-AAAGTG-3′, and the sequence for si14-3-3τ-2 is 5′-CGAGGAG-CGCAACCTGCTC-3′.
Immunoprecipitation and Western Blot Analysis
Cells were harvested with TNN buffer for immunoprecipitation as previously described (17). Anti-FLAG (M2) beads were purchased from Sigma. Anti-HA beads (HA.11 affinity matrix) were purchased from Covance. Specific signals were detected with appropriate antibodies. The antibodies specific to E2F1 (C-20 and KH95), E2F2 (C-20), E2F3 (C-18 and N-20), 14-3-3τ (C-17 and H-8), GST (B-14), and proliferating cell nuclear antigen (PC-10) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). β-Actin antibody was purchased from Sigma. Anti-green fluorescence protein (GFP) antibody was purchased from Clontech. The monoclonal antibody for 14-3-3τ was purchased from EMD Biosciences. The monoclonal antibody for poly(ADP-ribose) polymerase (PARP) was purchased from Pharmingen.
GST Pull-down Assay
The full-length cDNA of 14-3-3τ was inserted into an expression vector pGEX6P1, encoding glutathione S-transferase (GST). GST, GST-E2F1, and GST-14-3-3τ proteins were induced and purified from E. coli as previously described (2). The GST portion of GST-E2F1 was excised by PreScission protease (Amersham Biosciences). 2 μg of purified GST-14-3-3τ or GST was incubated at 4 °C overnight with purified E2F1 or the cellular lysates prepared from HEK293 cells, which had been transfected with pcDNA3-HA-E2F1 (wild-type) or pcDNA3-HA-E2F1(S31A) and lysed in TNN buffer. GST-14-3-3η was pulled down with glutathione-Sepharose, and the bound E2F1 was analyzed by Western blotting as described (17).
In Vitro Peptide Binding Assay
A biotin-labeled nonphosphorylated peptide (biotin-RLLDSSQIVI) or phosphorylated peptide (biotin-RLLD-SpSQIVI; where pS represents phosphoserine) was synthesized by Sigma. Biotin-peptides (2 μg) were incubated with GST-14-3-3τ or GST protein (2 μg) in 1 ml of NETN-A buffer (20 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM EDTA, pH 8.0, and 0.5% Nonidet P-40) at 4 °C overnight. The mixtures were then incubated with either streptavidin-Sepharose (Amersham Biosciences), or glutathione-Sepharose at 4°C for 3 h and washed with NETN-B buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, pH 8.0 and 0.5% Nonidet P-40) six times. The bound GST-14-3-3τ was subjected to 10% SDS-PAGE and analyzed by Western blotting with anti-GST antibody.
ELISA
A 96-well Immulon 4 flat bottom plate (VWR) was coated with biotin-labeled nonphosphorylated or phosphorylated peptides (5 μg/ml and 50 μl/well) at 4 °C overnight. The wells were blocked with 5% bovine serum albumin/phosphate-buffered saline at room temperature for 1.5 h and then incubated with GST-14-3-3τ or GST (5 μg/ml and 50 μl/well) at 4 °C overnight. The wells were then washed with 0.5% Tween 20/phosphate-buffered saline. The bound GST or GST-14-3-3τ was detected by GST antibody followed by horseradish peroxidase-conjugated secondary antibody. The plate was developed with 3,3′,5,5′-tetramethylbenzidine (Sigma) and read at OD450/650 in a spectrophotometer (Bio-Tek Instruments, Inc.).
RT-PCR
HEK293 and U2OS cells were transfected with pSUPER or pSUPER-si14-3-3τ. The U2OS cells were treated with 300 ng/ml neocarzinostatin (NCS) for 0 or 5 h. The HEK293 cells were treated with 5 μM adriamycin for 0, 6, or 12 h. RNA was extracted using TRIzol reagent. RT-PCR was performed using the following primer pairs. We ensured linear amplification in all cases. RT-PCR primer sequences and PCR conditions are shown in Table I.
Table I.
RT-PCR primer sequences and PCR conditions
| Gene | Sequences | Annealing temperature | Cycle number | Size |
|---|---|---|---|---|
| °C | bp | |||
| p73 | 5′-CATGGTCTCGGGGTCCCACT-3′ 5′-CGTGAACTCCTCCTTGATGG-3′ |
62 | 50 | 471 |
| Apaf-1 | 5′-AATGGACACCTTCTTGGACG-3′ 5′-GCACTTCATCCTCATGAGCC-3′ |
56 | 45 | 331 |
| Caspase 3 | 5′-TCGGTCTGGTACAGATGTCG-3′ 5′-CATACAAGAAGTCGGCCTCC-3′ |
56 | 40 | 398 |
| Caspase 7 | 5′-CAAAGCCACTGACTGAGATG-3′ 5′-CAACCCAATGAATAAATGAT-3′ |
56 | 40 | 259 |
| Caspase 8 | 5′-ACAGTGAAGATCTGGCCTCC-3′ 5′-GCAGGTTCATGTCATCATCC-3′ |
56 | 50 | 375 |
| Caspase 9 | 5′-GGCAGCTGATCATAGATCTGG-3′ 5′-CTGCTTGCCTGTTAGTTCGC-3′ |
55 | 45 | 120 |
| p107 | 5′-TGGTGTCGCAAATGATGCTGG-3′ 5′-AGGAGCTGATCCAAATGCCTG-3′ |
55 | 40 | 362 |
| Cyclin E | 5′-CTCCAGGAAGAGGAAGGCAA-3′ 5′-GTAAAAGGTCTCCCTGTGAAG-3′ |
55 | 25 | 421 |
| TK-1 | 5′-ATGAGCTGCATTAACCTGCCCACT-3′ 5′-ATGTGTGCAGAAGCTGCTGC-3′ |
60 | 35 | 204 |
| p21 | 5′-CTCAGAGGAGGCGCCATG-3′ 5′-GGGCGGATTAGGGCTTCC-3′ |
60 | 35 | 527 |
| Glyceraldehyde-3-phosphate dehydrogenase | 5′-TGAAGGTCGGAGTCAACGGATTTGGT-3′ 5′-AAATGAGCCCCAGCCTTCTCCATG-3′ |
55 | 40 | 325 |
Flow Cytometry
HEK293 cells were transfected with 20 μg of pSUPER vectors expressing 14-3-3τ or E2F siRNA by electroporation. Cells were then treated with 5 μM adriamycin for 48 h and apoptosis was quantified by propidium iodide flow cytometry (17). Alternatively, HEK293 cells were transfected with pcDNA3-GFP and pSUPER vectors and then treated with adriamycin. To assay apoptosis, cells were stained with annexin V-PE (Pharmigen) and 7-amino-actinomycin (7-AAD) (Pharmingen) as described (18). At least 5000 GFP-positive cells were gated for each sample, and the annexin/7-AAD profile of these cells was analyzed by flow cytometry. Annexin+, 7-AAD− and annexin +, 7-AAD+ cells were scored as apoptotic.
RESULTS
Interaction between 14-3-3τ and E2F1 Is Induced by Phosphorylation of E2F1 at Ser31 by ATM upon DNA Damage
The N terminus of E2F1 contains Ser 31 which is phosphorylated by ATM and is required for E2F1 induction during DNA damage (2). We used fusion proteins composed of the N-terminal portion of E2F1 and the GAL4 DNA binding domain to screen a HeLa cell cDNA library. Seventeen positive clones encoding nearly full-length 14-3-3τ (amino acids 39–247) were isolated of 12 million clones screened. The interaction was verified by selective growth of yeast cells expressing both N terminus of E2F1 and 14-3-3τ on plates lacking tryptophan, leucine, and histidine (Fig. 1A). Interestingly, the interaction was significantly impaired when Ser31 of E2F1 was mutated to alanine, suggesting that an efficient interaction required a serine residue on that position. To confirm their direct physical interaction, we examined in vitro binding between purified E2F1 and GST-14-3-3τ proteins. E2F1 protein was incubated with GST or GST-14-3-3τ, and the GST-14-3-3τ protein was pulled down with glutathione-Sepharose. GST-14-3-3τ but not GST pulled down E2F1 (Fig. 1B), demonstrating a direct interaction between 14-3-3τ and E2F1.
Fig. 1. Interaction of E2F1 and 14-3-3τ in vitro and in vivo.

A, E2F1 interacts with 14-3-3τ in yeast. The interaction between the E2F1 N terminus (amino acids 1–109; named E2F1N) and 14-3-3τ in Saccharomyces cerevisiae Y190 cells was verified by selective growth of transformants on a plate lacking tryptophan, leucine, and histidine and supplemented with 3-amino-1,2,4-triazole. Mutation at the Ser31 to alanine (S31A) inhibits the interaction. B, interaction of E2F1 and 14-3-3τ in vitro. Purified GST or GST-14-3-3τ was incubated with E2F1 protein, and a GST pull-down assay was performed. E2F1 protein was detected by immunoblotting. GST-14-3-3τ and GST were visualized by Ponceau S staining (bottom). C, interaction of endogenous E2F1 and 14-3-3τ during DNA damage. 293T cells were left untreated or treated with the radiomimetic agent NCS at 300 ng/ml for 3 h. Lysates were immunoprecipitated (IP) with normal rabbit IgG or rabbit anti-E2F1 antibody and immunoblotted (IB) as indicated. An aliquot of lysates was probed with anti-14-3-3τ antibody (bottom).
Next, we examined whether E2F1 interacted with 14-3-3τ in vivo. We detected an interaction between endogenous E2F1 and 14-3-3τ in 293T cells that had been treated with NCS, a radiomimetic chemical. The interaction between endogenous proteins in the absence of DNA damage was below detection in this experiment but was readily detectable upon NCS treatment (Fig. 1C). This result demonstrates that the interaction between endogenous E2F1 and 14-3-3τ is induced upon DNA damage.
NCS activates ATM kinase and leads to E2F1 phosphorylation at the Ser31 residue (2). This phosphorylation event might directly induce the binding between 14-3-3τ and E2F1. To test this hypothesis, we prepared cellular lysates from HEK293 cells, which were transfected with the plasmid expressing either wild-type E2F1 or S31A-E2F1 and treated with NCS. We measured the amounts of E2F1 in each lysate by E2F1 immunoblotting. Consistent with previous observation (2), the protein levels of wild-type E2F1, but not S31A mutant were induced by NCS (Fig. 2A). We then adjusted the amount of each input lysate, so that roughly equivalent amounts of E2F1 were incubated with GST or GST-14-3-3τ (Fig. 2B, bottom). GST-14-3-3τ pull-down was performed with glutathione-Sepharose. GST-14-3-3τ specifically interacted with wild-type E2F1, but not S31A mutant, and the interaction was enhanced upon NCS treatment (Fig. 2B, top). This result demonstrates that the interaction between E2F1 and 14-3-3τ is induced by Ser31 phosphorylation. Interestingly, there is weak interaction between 14-3-3τ and overexpressed HA-E2F1 even before NCS treatment (Fig. 2B, 0 h).
Fig. 2. 14-3-3τ interacts with Ser31-phosphorylated E2F1.

A, Western blot analysis of E2F1 in HEK293 cells transfected with wild type (wt) E2F1 or S31A-E2F1 and treated with NCS (300 ng/ml) for the indicated time periods. β-actin immunoblotting (IB) was performed to ensure equal protein loading. B, 14-3-3τ interacts with ATM-phosphorylated E2F1. GST or GST-14-3-3τ was incubated with the cellular lysates from NCS-treated cells as described in A and pulled down with glutathione-Sepharose. The amounts of input lysates had been adjusted so that each input contained roughly equal levels of E2F1 (bottom). C, 14-3-3τ interacts with an E2F1 phosphopeptide. GST or GST-14-3-3τ was incubated with a biotin-labeled nonphosphorylated peptide (nP) derived from E2F1 sequences or a Ser31-phosphorylated peptide (pP) and pulled down with streptavidin-Sepharose (top) or glutathione-Sepharose (bottom) followed by GST immunoblotting. D, binding of 14-3-3τ with E2F1 peptides was tested by ELISA as described under “Experimental Procedures.” The data shown are the mean ± S.D. of triplicate experiments.
To further test whether 14-3-3τ could bind to the region around Ser31, we incubated GST or GST-14-3-3τ with a biotinylated E2F1 peptide (biotin-RLLDSS31QIVI) or phosphopeptide (biotin-RLLDSpS31QIVI) and pulled down the peptides with streptavidin-Sepharose. GST-14-3-3τ interacted with the phosphopeptide but not with the nonphosphorylated peptide (Fig. 2C). The interaction was dependent on 14-3-3τ, since GST did not interact with the phosphopeptide. The binding between 14-3-3τ and phosphopeptide was also confirmed by an independent assay. The nonphosphopeptide or phosphopeptide was coated to plates and then incubated with either purified GST or GST-14-3-3τ. The bound protein was measured using ELISA. 14-3-3τ also specifically interacted with the phosphopeptide in this assay (Fig. 2D). Together, we conclude that 14-3-3τ binds to Ser31-phosphorylated E2F1.
14-3-3τ Is Required for the Stabilization of E2F1 Protein during DNA Damage
The phosphorylation of E2F1 at Ser31 by ATM kinase stabilizes the E2F1 protein (2). Binding of 14-3-3τ to phosphorylated E2F1 suggests a role for 14-3-3τ in the stabilization of E2F1. To test that, we knocked down the expression of 14-3-3τ by pSUPER vector-based small interfering RNAs (siRNAs) (19). Two 14-3-3τ siRNAs significantly inhibited the expression of cotransfected FLAG-14-3-3τ in HEK293 cells without affecting the levels of coexpressed GFP (Fig. 3A). The induction of HA-E2F1 by NCS treatment was inhibited by both 14-3-3τ siRNAs (Fig. 3B). It is worth noting that the transfection in these experiments was performed by calcium phosphate method, and the transfection efficiency in HEK293 cells by this method in our hands is at most 30% as assayed by the percentage of GFP-positive cells in the pcDNA3-GFP-transfected cells. Therefore, the extent of 14-3-3τ siRNA-mediated knockdown of endogenous 14-3-3τ within the transfected cells would be much greater than it looks. We also took an independent approach to inhibit the function of 14-3-3τ. Prior study had shown that a 14-3-3τ mutant lacking the N-terminal dimerization domain (ΔN14-3-3τ) could function as a dominant negative inhibitor of endogenous 14-3-3τ (20). This dominant negative mutant also inhibited the induction of HA-E2F1 by NCS treatment (Fig. 3C). In the course of experiments, we found that the transfection efficiency in HEK293 cells by our electroporation protocol can reach greater than 90% as assayed by the percentage of GFP-positive cells in the pcDNA3-GFP-transfected cells. Therefore, we utilized electroporation to carry out further transfection experiments to examine the role of 14-3-3τ in the induction of endogenous E2F1. 14-3-3τ siRNA significantly blocked the induction of endogenous E2F1 by adriamycin treatment (Fig. 4A). To further investigate whether 14-3-3τ siRNA inhibited the stabilization of E2F1, the E2F1 protein half-life was measured in HEK293 cells treated with cycloheximide in the absence or presence of adriamycin (Fig. 4, B and C). The half-life of endogenous E2F1 in the absence of adriamycin was between 80 and 100 min but was extended to over 300 min upon adriamycin treatment. The half-life of E2F1 in 14-3-3τ siRNA-transfected cells was shortened to between 40 and 60 min. Importantly, 14-3-3τ siRNA prevented the stabilization of E2F1 by adriamycin treatment. Taken together, these results establish a role for 14-3-3τ in the control of E2F1 protein stability. It is worth noting that TopBP1, a BRCT domain-containing protein, also interacts with E2F1, but their interaction regulates E2F1 activity rather than stability (17). Moreover, TopBP1 siRNA does not inhibit the stabilization of E2F1 in response to DNA damage (18). The half-life of E2F1 has been examined by several investigators and falls in the range of 1–3 h (3, 15, 21–24). Therefore, our measurement (80–100 min) is within the general range reported in the literature. The difference between different reports is probably the result of different cell types, growing conditions, and methods used by each investigator.
Fig. 3. 14-3-3τ is required for the induction of E2F1 protein accumulation upon DNA damage.

A, HEK293 cells were transfected with pFLAG-14-3-3τ, pGFP and pSUPER or pSUPER vectors expressing two different 14-3-3τ siR-NAs. Transfected 14-3-3τ was immunoprecipitated (IP) with FLAG-beads and immunoblotted (IB) with anti-14-3-3τ antibody. The expression of GFP protein in each transfectant serves as a control. B, knockdown of 14-3-3τ blocks the induction of E2F1 in response to NCS treatment. HEK293 cells, transiently transfected with 14-3-3τ siRNA and HA-E2F1, were treated with NCS (300 ng/ml) for 0, 1, 2, and 3 h. Western blot analysis was performed using antibodies as indicated. Proliferating cell nuclear antigen immunoblotting was performed to ensure equal protein loading. C, a dominant negative mutant ΔN14-3-3τ inhibited the induction of E2F1 by NCS. HEK293 cells were transfected with HA-E2F1 alone or with pFLAG-ΔN14-3-3τ and treated with NCS. Western blot analysis was performed using antibodies as indicated.
Fig. 4. 14-3-3τ is required for the stabilization of E2F1 protein upon DNA damage.

A, knockdown of 14-3-3τ blocks the induction of endogenous E2F1 by adriamycin treatment. HEK293 cells were transfected with 14-3-3τ siRNA and treated with adriamycin (5 μM) for 0–24 h. Western blot analysis (IB) was performed using antibodies as indicated. B, 14-3-3τ siRNA inhibits adriamycin-induced prolongation of E2F1 half-life. HEK293 cells were transfected with an empty vector (upper panels) or 14-3-3τ siRNA (lower panels) and were either left untreated (left panels) or treated with adriamycin (5 μM) for 24 h (right panels). The cells were then treated with cycloheximide (CHX; 20 μg/ml) for the time periods as indicated. The endogenous E2F1 protein was detected by Western blot analysis. C, The E2F1 levels in B were quantified by densitometry, and the relative abundance of E2F1 following CHX treatment was compared with that seen without CHX treatment and plotted.
14-3-3τ Inhibits E2F1 Ubiquitination
E2F1 is degraded through the ubiquitin-proteasome pathway (21, 23–27). To test whether 14-3-3τ inhibited E2F1 ubiquitination, we coexpressed HA-E2F1 and Myc-ubiquitin with increasing amounts of 14-3-3τ-expressing plasmids and measured E2F1 ubiquitination by immunoprecipitation/Western blotting assay in HEK293 cells. The effect of NCS on E2F1 ubiquitination was also tested. In order to visualize ubiquitinated E2F1, a proteasome inhibitor, MG-132, was added. Overexpression of 14-3-3τ resulted in inhibition of E2F1 ubiquitination in a dose-dependent manner (Fig. 5A). NCS treatment also inhibited E2F1 ubiquitination (Fig. 5A, right lane). We compared the S31A mutant E2F1 with wild-type E2F1 in the ubiquitination assay. Whereas 14-3-3τ greatly inhibited the ubiquitination of wild-type E2F1, it affected S31A-E2F1 to a much lesser degree (Fig. 5B), corresponding to the preferential binding of 14-3-3τ to wild-type E2F1 as compared with the S31A mutant (Fig. 2B).
Fig. 5. 14-3-3τ protects E2F1 from degradation by the ubiquitin-proteasome pathway.

A, increasing amounts of plasmids expressing 14-3-3τ were cotransfected with Myc-ubiquitin- and HA-E2F1-expressing constructs in HEK293 cells. The cells were then left untreated or treated with the proteasome inhibitor MG-132 (20 μM) and/or NCS (300 ng/ml) as indicated at the top. Whole cell extracts were subject to immunoprecipitation (IP) with HA beads followed by Western blot analysis to detect ubiquitinated E2F1. An aliquot of total lysates was immunoblotted (IB) with 14-3-3τ or β-actin antibody (lower panels). The numbers to the left of each panel indicate relative molecular mass markers (in thousands). B, 14-3-3τ preferentially protects wild-type E2F1, as compared with S31A mutant, from degradation by the ubiquitin-proteasome pathway. E2F1 ubiquitination assay in the presence of MG-132 was performed as in A. C, HEK293 cells were transfected with a Myc-ubiquitin expressing plasmid along with pSUPER or pSUPER-si14-3-3τ and left untreated or treated with NCS and/or MG-132. Lysates were immunoprecipitated with normal mouse IgG or a monoclonal E2F1 antibody, followed by immunoblotting to detect ubiquitinated E2F1. An aliquot of total lysates was immunoblotted with 14-3-3τ or β-actin antibody (lower panels).
The ubiquitination of endogenous E2F1 in the 14-3-3τ knockdown cells was also examined (Fig. 5C). Consistent with the results described above, depletion of 14-3-3τ led to an increase of E2F1 ubiquitination. E2F1 ubiquitination was inhibited by NCS treatment, but the levels of ubiquitination in 14-3-3τ knockdown cells remained elevated even during NCS treatment. Therefore, we conclude that 14-3-3τ regulates E2F1 ubiquitination to control its stability.
14-3-3τ and E2F1 Are Required for DNA Damage-induced Apoptosis
Given the proapoptotic activity of E2F1 and the role of 14-3-3τ in E2F1 stabilization, 14-3-3τ may be required for apoptosis during DNA damage. We again utilized RNA interference to test that. We transfected HEK293 cells with 14-3-3τ siRNA constructs using the electroporation method, by which we achieved greater than 90% of transfection efficiency. Both siRNAs effectively inhibited the expression of endogenous 14-3-3τ without altering the expression of 14-3-3τ (Fig. 6A). We treated mock-, empty vector-, or 14-3-3τ siRNA-transfected HEK293 cells with adriamycin and quantified the degree of apoptosis with propidium iodide/fluorescence-activated cell sorting. Whereas adriamycin induced extensive apoptosis in mock- and empty vector-transfected cells, apoptosis was significantly inhibited by both 14-3-3τ siRNAs (Fig. 6A). We also analyzed apoptosis by annexin V staining. In this experiment, pcDNA3-GFP expression vector was transiently cotransfected into HEK293 cells with pSUPER-si14-3-3τ, and the cells were treated with adriamycin. Apoptosis was analyzed by annexin V/7-AAD staining in GFP-positive cells. As shown in Fig. 6B, 14-3-3τ siRNA inhibited adriamycin-induced apoptosis. Together, these results clearly demonstrate a critical role for 14-3-3τ in DNA damage-induced apoptosis.
Fig. 6. A role for 14-3-3τ in adriamycin (Adr)-induced apoptosis.




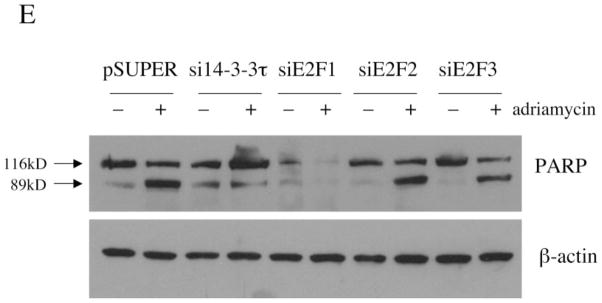
A, HEK293 cells were mock-transfected or transfected with pSUPER or pSUPER vectors expressing two different 14-3-3τ siRNAs and treated with adriamycin (5 μM) for 48 h. Apoptotic cells were counted by propidium iodide-flow cytometry. Inhibition of 14-3-3τ expression but not 14-3-3β expression in this experiment was confirmed by Western blot analysis (top). B, HEK293 cells were transfected with pSUPER or pSUPER-si14-3-3τ and pcDNA3-GFP, and treated with adriamycin as in A. Cells were then stained with annexin V-PE and 7-AAD. GFP-positive cells were gated and analyzed by flow cytometry. Top, the data shown are the mean ± S.E. of three independent experiments. *, p < 0.05 (t test) compared with the pSUPER vector group. Bottom, a representative flow cytometry profile. C, HEK293 cells were transfected with the plasmids expressing 14-3-3τ, E2F1, E2F2, or E2F3 siRNA and treated with adriamycin (5 μM) for 24 h. Apoptosis was scored by propidium iodide-flow cytometry. The data shown are the mean ± S.E. of three independent experiments. *, p < 0.05 (t test) compared with pSUPER vector group. Knockdown of 14-3-3τ or E2F by corresponding siRNA in these experiments was confirmed by immunoblotting (top). D, a representative propidium iodide-flow cytometry profile from the experiments described in C. E, the cell lysates from the experiments described in C were subject to Western blot analysis for PARP cleavage. β-actin immunoblot was performed to confirm equal protein loading.
We also examined the role of E2F in adriamycin-induced apoptosis in HEK293 cells utilizing the siRNA for E2F1, E2F2, and E2F3, respectively (18). The E2F siRNA specifically inhibited the expression of corresponding E2F without affecting the expression of other E2Fs (Fig. 6C, top). The 14-3-3τ siRNA inhibited the expression of 14-3-3τ as well as E2F1, due to its effect on E2F1 stability and ubiquitination. The apoptosis was analyzed by a propidium iodide/fluorescence-activated cell sorting assay (Fig. 6, C and D) or the proteolytic cleavage of PARP (Fig. 6E), which is commonly used as an apoptotic marker. Both 14-3-3τ siRNA and E2F1 siRNA significantly inhibited adriamycin-induced apoptosis. In contrast, E2F2 and E2F3 siRNAs did not have a significant effect on apoptosis. The unique role of E2F1 for apo-ptosis among the E2F family corresponds to the selective induction of E2F1 in response to DNA damage (2). Interestingly, the expression of PARP is reproducibly decreased in E2F1 siRNA-transfected cells (Fig. 6E), which is consistent with the observation that PARP is an E2F1-regulated gene (28). Together, we conclude that 14-3-3τ and E2F1 are required for DNA damage-induced apoptosis. The role of 14-3-3τ in apoptosis is probably mediated through the control of E2F1 stability.
14-3-3τ Regulates the Expression of E2F1 Target Genes
To further substantiate the regulation of E2F1 function by 14-3-3τ, we examined E2F1 target gene expression in 14-3-3τ siRNA-transfected cells. E2F1 target genes that are related to cell cycle, such as p107, cyclin E, and thymidine kinase, as well as target genes that are related to apoptosis, such as p73, Apaf-1, and caspase-3, -7, -8, and -9, were examined. HEK293 cells were transfected with 14-3-3τ siRNA and treated with adriamycin. The transcripts of several E2F1 target genes were analyzed by quantitative RT-PCR assay. To test the role of 14-3-3τ in other cells, we also performed similar experiments in NCS-treated U2OS cells. p73 has been identified as an authentic E2F1 apoptotic target gene in the response to DNA damage, and phosphorylation of Ser31 and acetylation of Lys117, Lys121, and Lys125 are required for efficient recruitment of E2F1 on the p73 promoter after DNA damage (4). Upon DNA damage, the transcript of p73 was induced in both HEK293 and U2OS cells (Fig. 7A). 14-3-3τ siRNA inhibited the expression and the induction of p73 transcript (Fig. 7A, top panel). 14-3-3τ siRNA also inhibited the expression of several other E2F1 targets, including Apaf-1, p107, and cyclin E in both HEK293 and U2OS cells (Fig. 7A). The expression of caspase-3, -7, -8, and -9 was reduced in 14-3-3τ siRNA-transfected HEK293 cells. In U2OS cells, caspase-3 was affected, but the effect on other caspases was less pronounced. 14-3-3τ siRNA did not affect the expression of p21, a p53 target gene. Interestingly, the expression of thymidine kinase (TK-1) was not affected by 14-3-3τ siRNA either. Since 14-3-3τ siRNA only affects the expression of E2F1, and not E2F3 (Fig. 6C), this result is consistent with recent reports that E2F3, but not E2F1, is responsible for the regulation of thymidine kinase (29). The effect of 14-3-3τ siRNA on E2F1 target gene expression correlated with that on E2F1 protein levels (Fig. 7B). Thus, we conclude that 14-3-3τ regulates E2F1 protein stability and consequently regulates its target gene expression.
Fig. 7. 14-3-3τ regulates the expression of E2F1 target genes.

A, HEK293 and U2OS cells were transfected with pSUPER or pSUPER-si14-3-3τ and were treated with adriamycin (5 μM) for 0, 6, or 12 h or NCS (300 ng/ml) for 0 or 5 h, respectively. RNA was extracted, and RT-PCR was performed using the primers specific for selected E2F1 target genes, p21, or glyceraldehyde-3-phosphate dehydrogenase. Mock RT-PCR represents a control reaction without the addition of RNA. B, a portion of cells in the experiments (A) was lysed, and cellular lysates were analyzed by Western blot analysis with indicated antibodies.
DISCUSSION
Recent studies have defined a pathway that links Rb/E2F cell cycle control with apoptosis. Particularly, the identification of ATM/Chk2 as a mediator that induces E2F1 in response to DNA damage provides us a conceptual framework to understand the interplay between growth control and stress response (2, 3). Understanding the mechanism of E2F1 induction is therefore important. Now we have identified 14-3-3τ as a critical regulator in the control of E2F1 protein stability. Through binding to ATM-phosphorylated E2F1, 14-3-3τ inhibits the ubiquitination of E2F1 and stabilizes E2F1. We also demonstrate a critical role of 14-3-3τ and E2F1 for adriamycin-induced apoptosis. Last, 14-3-3τ is shown to be required for the expression and induction of important E2F1 targets, including p73, Apaf-1, and caspases, and consequently is essential for DNA damage-induced apoptosis. The data presented in this paper allow us to propose a model in which association of 14-3-3τ with ATM-phosphorylated E2F1 inhibits the binding and function of an E3 ligase and leads to E2F1 stabilization. As such, 14-3-3τ regulates the expression of p73, Apaf-1, and some caspases for apoptosis (Fig. 8). In addition to ATM, Chk2 and p300/CREB-binding protein-associated factor (P/CAF) acetyl-transferase also contribute to E2F1 stabilization and activation (3, 4, 15). Whereas the dependence of these events is unclear, it is likely that these different modifications cooperate in the induction of E2F1, as seen in the activation of p53 in response to DNA damage (30).
Fig. 8. A model for the role of 14-3-3τ in the regulation of E2F1 stability during DNA damage.

Upon DNA damage, E2F1 is phosphorylated by ATM/ATR at Ser31 and by Chk2 at Ser364. E2F1 is also acetylated at Lys117, Lys120, and Lys125 by p300/CREB-binding protein-associated factor (P/CAF) acetyltransferase. Phosphorylation of Ser31 induces 14-3-3τ binding and protects E2F1 from ubiquitination and degradation.
14-3-3 binding increases the stability of other proteins, such as Wee1 (31), nicotinic receptor α4 subunit (32), ataxin-1 (33), p53 (34), and histone deacetylase 7 (35). 14-3-3σ interacts with p53 in response to adriamycin and blocks Mdm2-mediated p53 ubiquitination (34). Thus, the regulation of E2F1 ubiquitination by 14-3-3τ is analogous to the role of 14-3-3σ in the control of p53 ubiquitination. Several E3 ligases are proposed to target E2F1 for ubiquitination, including SCFSkp2 (23) and Mdm2 (22). Skp2 binds to the N terminus of E2F1, where the ATM phosphorylation site Ser31 resides (23). Thus, ATM-mediated phosphorylation might result in inhibition of Skp2 binding. However, binding of SCF complex is usually induced, rather than inhibited, by phosphorylation of its substrates (36). The expression of E2F1 protein in Skp2−/− mouse embryonic fibroblasts (MEFs) is not affected (37). Moreover, in an experiment involving primary MEFs prepared from four pairs of Skp2−/− and Skp2−/− sibling embryos, there was no significant difference in the induction of E2F1 accumulation upon cisplatin treatment between Skp2−/− and Skp2−/− MEFs.2 Therefore, the role of Skp2 in the regulation of E2F1 stability during DNA damage remains unclear. Likewise, the basal level and induction of E2F1 protein were indistinguishable between three pairs of p53−/−Mdm2−/− and p53−/−Mdm2−/− MEFs upon cisplatin treatment.3 Thus, the identity of E3 ligase(s) involved in E2F1 stability control during DNA damage remains elusive. The discovery of 14-3-3τ as a regulator for E2F1 ubiquitination would facilitate the identification of the relevant E3 ligase(s) for E2F1.
Two canonical 14-3-3 binding motifs have been defined as RSXpSXP and RXXXpSXP (38). However, 14-3-3 also binds to phosphoserines within different binding motifs, such as the Ser-rich motif (39), as well as motifs that do not conform to any of the above mentioned sequences or do not even require phosphorylation for binding (16, 40). The sequence of E2F1 phosphopeptide (RLLDSpS31QIVC) that binds to 14-3-3τ does not strictly conform to the classical binding motifs and may represent a new variation of the binding motif. It is interesting to note that p53 also contains an atypical 14-3-3 binding motif, and the interaction between p53 and 14-3-3 depends on ATM function as well (41). However, unlike E2F1, binding of 14-3-3 to p53 requires ATM-dependent dephosphorylation of pS376 to generate a binding motif at pS378, since double phosphorylation inhibits the 14-3-3 association (41).
Interestingly, the levels of E2F1 protein and ubiquitination are affected by 14-3-3τ siRNA even in the absence of DNA damage. 14-3-3τ interacts weakly with overexpressed E2F1 proteins (Fig. 2B). A weak interaction between endogenous 14-3-3τ and E2F1 in untreated HEK293 cells was seen in some co-immunoprecipitation experiments (data not shown). Thus, 14-3-3τ may be required for maintaining E2F1 protein stability even in normal growth conditions, and DNA damage simply augments their interaction.
The regulation of E2F1 stability by 14-3-3τ is important for E2F1 function. The examination of endogenous E2F1 target genes in the 14-3-3τ knockdown cells provides compelling evidence that 14-3-3τ regulates E2F1 function under physiologic conditions and genotoxic stress. Importantly, the E2F target genes that are affected by 14-3-3τ siRNA include those involved in apoptosis, such as p73, Apaf-1, and caspases. 14-3-3τ siRNA inhibits the expression and induction of p73 during DNA damage. Whereas the expression of Apaf-1 or caspases is either induced very mildly or not at all by adriamycin or NCS, their expression is decreased in the 14-3-3τ-depleted cells. Thus, the 14-3-3τ-depleted cells contain lower levels of proteins involved in apoptosis and therefore are less “proapoptotic” compared with the cells containing high E2F1 levels.
Regulation of E2F1 stability by 14-3-3τ is important for apoptosis during DNA damage. In line with the selective accumulation of E2F1 protein in response to DNA damage (2), adriamycin-induced apoptosis is selectively inhibited by the siRNA of E2F1 but not E2F2 or E2F3 (Fig. 6, C–E). Previously, we showed that etoposide-induced apoptosis is attenuated in E2F1−/− murine thymocytes (2). The results obtained from siRNA experiments further support a critical role of E2F1 for DNA damage-induced apoptosis. The role of E2F1 and 14-3-3τ in apoptosis may depend on cellular contexts. For example, the difference in the response of primary MEFs with different E2F1 genotypes to various chemotherapeutic drugs, as measured by the extent and kinetics of G1/S arrest and apoptosis or by colony formation assay, is either not significant (2) or very small (42). In this case, the response to genotoxic stress may be primarily dictated by p53. However, in some transformed cells, such as HEK293 cells in which p53 is disabled by E1B, the levels of E2F1 become important in determining the occurrence of apoptosis. Considering that p53 is mutated in roughly half of human cancers and most cancer cells contain excessive E2F activities because of deregulation in the Rb pathway, 14-3-3τ may be an important determinant for chemosensitivity in cancer therapy.
Acknowledgments
We thank Marion Spell (University of Alabama at Birmingham Flow Cytometry Core Facility) for the fluorescence-activated cell sorting analysis and Hua Fan for technical assistance.
Footnotes
The work was supported by the General Motors Cancer Research Scholar Award (to W.-C. L.) and National Institutes of Health Grants CA100857 (to W.-C. L.) and CA100848 (to F.-T. L.).
The abbreviations used are: E3, ubiquitin-protein isopeptide ligase; PARP, poly(ADP-ribose) polymerase; GST, glutathione S-transferase; RT, reverse transcription; 7-AAD, 7-amino-actinomycin; GFP, green fluorescent protein; NCS, neocarzinostatin; siRNA, small interfering RNA; HA, hemagglutinin; MEF, mouse embryo fibroblast.
W.-C. Lin and K. Nakayama, unpublished results.
W.-C. Lin, unpublished results.
References
- 1.Cam H, Dynlacht BD. Cancer Cell. 2003;3:311–316. doi: 10.1016/s1535-6108(03)00080-1. [DOI] [PubMed] [Google Scholar]
- 2.Lin WC, Lin FT, Nevins JR. Genes Dev. 2001;15:1833–1844. [PMC free article] [PubMed] [Google Scholar]
- 3.Stevens C, Smith L, La Thangue NB. Nat Cell Biol. 2003;5:401–409. doi: 10.1038/ncb974. [DOI] [PubMed] [Google Scholar]
- 4.Pediconi N, Ianari A, Costanzo A, Belloni L, Gallo R, Cimino L, Porcellini A, Screpanti I, Balsano C, Alesse E, Gulino A, Levrero M. Nat Cell Biol. 2003;5:552–558. doi: 10.1038/ncb998. [DOI] [PubMed] [Google Scholar]
- 5.Stevens C, La Thangue NB. DNA Repair (Amst) 2004;3:1071–1079. doi: 10.1016/j.dnarep.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 6.Dyson N. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 7.Nevins JR. Cell Growth Differ. 1998;9:585–593. [PubMed] [Google Scholar]
- 8.Trimarchi JM, Lees JA. Nat Rev Mol Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 9.Hallstrom TC, Nevins JR. Proc Natl Acad Sci U S A. 2003;100:10848–10853. doi: 10.1073/pnas.1831408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Proc Natl Acad Sci U S A. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kowalik TF, DeGregori J, Leone G, Jakoi L, Nevins JR. Cell Growth Differ. 1998;9:113–118. [PubMed] [Google Scholar]
- 12.Moroni MC, Hickman ES, Denchi EL, Caprara G, Colli E, Cecconi F, Muller H, Helin K. Nat Cell Biol. 2001;3:552–558. doi: 10.1038/35078527. [DOI] [PubMed] [Google Scholar]
- 13.Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, Vousden KH. Nature. 1998;395:124–125. doi: 10.1038/25867. [DOI] [PubMed] [Google Scholar]
- 14.Nahle Z, Polakoff J, Davuluri RV, McCurrach ME, Jacobson MD, Narita M, Zhang MQ, Lazebnik Y, Bar-Sagi D, Lowe SW. Nat Cell Biol. 2002;4:859–864. doi: 10.1038/ncb868. [DOI] [PubMed] [Google Scholar]
- 15.Ianari A, Gallo R, Palma M, Alesse E, Gulino A. J Biol Chem. 2004;279:30830–30835. doi: 10.1074/jbc.M402403200. [DOI] [PubMed] [Google Scholar]
- 16.Fu H, Subramanian RR, Masters SC. Annu Rev Pharmacol Toxicol. 2000;40:617–647. doi: 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- 17.Liu K, Lin FT, Ruppert JM, Lin WC. Mol Cell Biol. 2003;23:3287–3304. doi: 10.1128/MCB.23.9.3287-3304.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu K, Luo Y, Lin FT, Lin WC. Genes Dev. 2004;18:673–686. doi: 10.1101/gad.1180204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brummelkamp TR, Bernards R, Agami R. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 20.Seimiya H, Sawada H, Muramatsu Y, Shimizu M, Ohko K, Yamane K, Tsuruo T. EMBO J. 2000;19:2652–2661. doi: 10.1093/emboj/19.11.2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martelli F, Livingston DM. Proc Natl Acad Sci U S A. 1999;96:2858–2863. doi: 10.1073/pnas.96.6.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blattner C, Sparks A, Lane D. Mol Cell Biol. 1999;19:3704–3713. doi: 10.1128/mcb.19.5.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marti A, Wirbelauer C, Scheffner M, Krek W. Nat Cell Biol. 1999;1:14–19. doi: 10.1038/8984. [DOI] [PubMed] [Google Scholar]
- 24.Hofmann F, Martelli F, Livingston DM, Wang Z. Genes Dev. 1996;10:2949–2959. doi: 10.1101/gad.10.23.2949. [DOI] [PubMed] [Google Scholar]
- 25.Hateboer G, Kerkhoven RM, Shvarts A, Bernards R, Beijersbergen RL. Genes Dev. 1996;10:2960–2970. doi: 10.1101/gad.10.23.2960. [DOI] [PubMed] [Google Scholar]
- 26.Campanero MR, Flemington EK. Proc Natl Acad Sci U S A. 1997;94:2221–2226. doi: 10.1073/pnas.94.6.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohta T, Xiong Y. Cancer Res. 2001;61:1347–1353. [PubMed] [Google Scholar]
- 28.Ma Y, Croxton R, Moorer RL, Jr, Cress WD. Arch Biochem Biophys. 2002;399:212–224. doi: 10.1006/abbi.2002.2761. [DOI] [PubMed] [Google Scholar]
- 29.Giangrande PH, Zhu W, Rempel RE, Laakso N, Nevins JR. EMBO J. 2004;23:1336–1347. doi: 10.1038/sj.emboj.7600134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E. Genes Dev. 1998;12:2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Jacobs C, Hook KE, Duan H, Booher RN, Sun Y. Cell Growth Differ. 2000;11:211–219. [PubMed] [Google Scholar]
- 32.Jeanclos EM, Lin L, Treuil MW, Rao J, DeCoster MA, Anand R. J Biol Chem. 2001;276:28281–28290. doi: 10.1074/jbc.M011549200. [DOI] [PubMed] [Google Scholar]
- 33.Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, Aitken A, Skoulakis EM, Orr HT, Botas J, Zoghbi HY. Cell. 2003;113:457–468. doi: 10.1016/s0092-8674(03)00349-0. [DOI] [PubMed] [Google Scholar]
- 34.Yang HY, Wen YY, Chen CH, Lozano G, Lee MH. Mol Cell Biol. 2003;23:7096–7107. doi: 10.1128/MCB.23.20.7096-7107.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Song S, Liu Y, Ko SH, Kao HY. J Biol Chem. 2004;279:34201–34208. doi: 10.1074/jbc.M405179200. [DOI] [PubMed] [Google Scholar]
- 36.Harper JW. Trends Cell Biol. 2002;12:104–107. doi: 10.1016/s0962-8924(01)02238-3. [DOI] [PubMed] [Google Scholar]
- 37.Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, Shirane M, Tsunematsu R, Tsukiyama T, Ishida N, Kitagawa M, Hatakeyama S. EMBO J. 2000;19:2069–2081. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 39.Liu YC, Liu Y, Elly C, Yoshida H, Lipkowitz S, Altman A. J Biol Chem. 1997;272:9979–9985. doi: 10.1074/jbc.272.15.9979. [DOI] [PubMed] [Google Scholar]
- 40.Yaffe MB. FEBS Lett. 2002;513:53–57. doi: 10.1016/s0014-5793(01)03288-4. [DOI] [PubMed] [Google Scholar]
- 41.Waterman MJ, Stavridi ES, Waterman JL, Halazonetis TD. Nat Genet. 1998;19:175–178. doi: 10.1038/542. [DOI] [PubMed] [Google Scholar]
- 42.Meng RD, Phillips P, El-Deiry WS. Int J Oncol. 1999;14:5–14. [PubMed] [Google Scholar]