

Abstract
Acetaminophen (ApAP) is an electron donor capable of reducing radicals generated by redox cycling of hemeproteins. It acts on the prostaglandin H synthases (cyclooxygenases; COXs) to reduce the protoporphyrin radical cation in the peroxidase site of the enzyme, thus preventing the intramolecular electron transfer that generates the Tyr385 radical required for abstraction of a hydrogen from arachidonic acid to initiate prostaglandin synthesis. Unrelated to this pharmacological action, metabolism of ApAP by CYPs yields an iminoquinone electrophile that is responsible for the hepatotoxicity, which results from high doses of the drug. We synthesized novel heterocyclic phenols predicted to be electron donors. Two of these inhibited the oxygenation of arachidonic acid by PGHS-1 and myoglobin and also were shown to be more metabolically stable and exhibited less direct cytotoxicity than acetaminophen. They are leading candidates for studies to determine whether they are free of the metabolism-based hepatotoxicity produced by acetaminophen.
Keywords: Acetaminophen, synthesis, heterocyclic phenols, toxicity, stability
Acetaminophen (ApAP; Paracetamol), with its analgesic and antipyretic properties, is one of the most widely used drugs. ApAP works by reducing the protoporphyrin radical cation in the peroxidase site of the prostaglandin H2 synthases (also know as cyclooxygenases, COX-1 and COX-2),1,2 thereby decreasing the production of the prostaglandins mediating fever and pain. We also have shown that acetaminophen can inhibit lipid peroxidation catalyzed by hemoglobin, myoglobin, and cytochrome c3,4 and that acetaminophen can protect the kidney from oxidative damage associated with rhabdomyolysis.3 This work provided the proof of concept that acetaminophen can be used in vivo to protect tissues from oxidative stress associated with hemeprotein redox cycling.
Unfortunately, acetaminophen is associated with hepatotoxicity, which may be severe,5−7 occurring both in association with intentional and accidental overdose and with administration to susceptible individuals. It has been implicated in nearly 50% of all acute liver failure in the United States alone.8 Acetaminophen’s hepatotoxicity is due to its 2-electron oxidation by the phase I cytochrome P450 (CYP) 2E1 and CYP3A4 in the liver.9−11 This leads to formation of the extremely reactive intermediate metabolite, N-acetyl-p-benzoquinone imine (NAPQI), which conjugates to glutathione, resulting in glutathione depletion in the liver.12 After GSH is depleted, NAPQI reacts with proteins13,14 and impairs mitochondrial function,15 leading to liver necrosis.
Taken together, the data indicate the need for a safer alternative to acetaminophen. In this letter, we describe the synthesis of acetaminophen analogues lacking the p-aminophenol structure that leads to formation of the toxic quinone imine electrophile metabolite. We also present the characterization of 2 lead compounds that are potent inhibitors of COX-1 and of myoglobin-induced lipid oxidation, but are metabolized by liver microsomes less rapidly than acetaminophen and do not exert direct cytotoxicity on cells in culture. Several nitrogen-containing highly potent phenolic antioxidants (1–11) have been prepared recently.16,17
For example, pyrimidinols (Figure 1, 1 and 2) as well as pyridinols (3 and 4) were proven to be highly effective inhibitors of COX-1.16
Figure 1.

Previously known pyrimidinols and pyridinols evaluated for their cytotoxicity and metabolic stability.
A number of trends can be inferred from previously published results for pyrimidinols (1 and 2) and pyridinols (3 and 4)16 as well as recently published vitamin B6-derivatives (5–11).17 In this letter, we evaluated efficiency to inhibit hemeproteins, cytotoxicity, and metabolic stability of those nitrogen-containing phenolic heterocycles seeking to identify the molecular structure motifs that correlate with higher activity and stability and lower levels of cytotoxicity. Our results provide information necessary for a rational design and synthesis of novel acetaminophen analogues with improved toxicity and stability profiles.
Cytotoxicity was assessed by measuring total intracellular ATP levels in HepG2 cells. This represents a good marker of the overall cellular health18,19 as ATP levels are affected by mitochondrial integrity as well as by changes in cellular metabolism. Metabolic stability was assessed in vitro using rat liver microsomes.20
In addition to the hepatotoxicity caused by the toxic metabolite of acetaminophen, pyridine phenols (3–4) may produce direct cytotoxicity, especially in comparison with virtually nontoxic pyrimidine phenols (1–2) as we have previously shown.16 This can be explained by the formation of an unhindered cationic iminium ion (or quinone) and its subsequent reactivity toward nucleophiles.21,22 Extensive metabolism by liver microsomes may be further evidence of cationic iminium ion formation. In this regard, significantly lower toxicity of pyridinols 5 and 7 may be attributed to the vital role of C(5) methyl substituent,17 suggesting that a substituent in C(5) position can hinder this position for nucleophilic attack.
Excessively low ionization potential (IP), which leads to the formation of superoxide is the most likely cause of the instability and high direct cytotoxicity of compounds 10 and 11 as well as comparably elevated direct cytotoxicity of compound 6.17 Indeed, it has been shown that analogue 6 is a more potent antioxidant than 7 due to the higher degree of spin alignment between the aromatic ring and the adjacent nitrogen.17 High degree of toxicity shown by compounds 8 and 9 especially in comparison with analogues 6 and 7 can be partially attributed to their low IP value. This argument is supported by the observation of large amounts of oxygenation products in the microsome stability assay (Supporting Information) as well as by the higher electron donating effect of an alkyl chain vs a methyl substituent.23 Also, long alkyl chains of compounds 8 and 9 might result in a detergent-like cell membrane disruption, further elevating their toxicity.
The microsome stability assay showed that demethylation was a major metabolic path for almost all the NHMe or N(Me)2 containing analogues. It also showed rapid metabolic consumption of the highly potent COX-1 inhibitor 2 by both demethylation and oxidation pathways. Demethylation may lead to increased toxicity by conversion, for example, from analogue 7 to 6 and 5, both of which are highly cytotoxic.17
Three pyridinol analogues of acetaminophen (Figure 2) were designed based on the results described above. All compounds are related to acetaminophen. Their structures are designed to modulate the donor characteristics of the 6-amino group, similar to acetaminophen, and fix the geometry of ring substituents by the constraints of a fused ring. A similar structural motif can be found in uric acid, which is responsible for more than half of human blood plasma antioxidant capacity.24 Thus, a urea bridge has been chosen to protect the free amine. This structural feature was intended to modulate the IP value of these novel analogues to alleviate the metabolic instability and cytotoxicity shown by analogues 10 and 11. Since the presence or the absence of C(2) and C(4) substituents in the heterocyclic phenol ring did not drastically alter pyridines and pyrimidines toxicity profile, both analogues 12 (Scheme 1) and 13 have been prepared. The urethane analogue 14 of the urea analogue 13 was also prepared.
Figure 2.
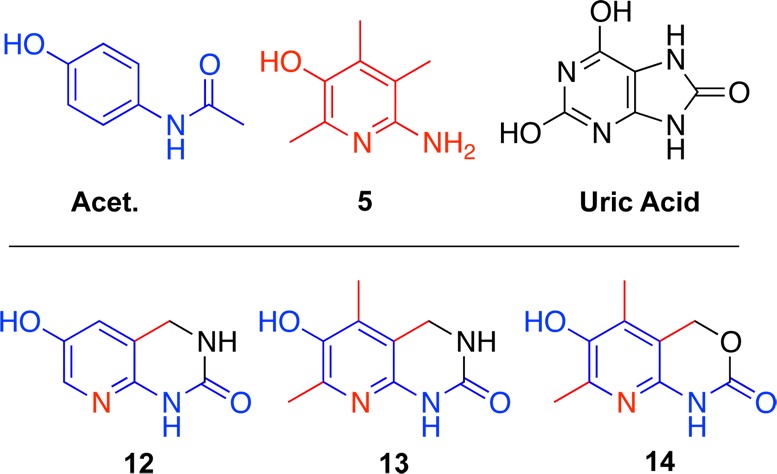
Analogue design.
Scheme 1. Synthesis of Pyridinol-Fused Ring Analogue 12.

Reagents and conditions: (a) (i) NBS, NH4Ac, MeCN, 0°C to RT, 12 h; (ii) 5% HCl, NaOH, pH > 10, H2O, 0 °C, 20 min 91%; (b) (i) BH3-THF, THF, 0 °C to reflux, 12 h; (ii) 15% HCl in H2O, 0 °C to reflux, 3 h; (iii) NaOH, to pH > 12, 71%; (c) (i) CDI, DMF, RT; (ii) H2O, RT to reflux to 0 °C, 20 min, 86%. (d) Boc2O, DMAP, THF, RT, 30 min, 97%; (e) (i) KOAc, Pd(OAc)2, bis-(pinacolato)diboron, DMF; (ii) NaBO3·2H2O, THF/H2O; (f) (i) HCl/MeOH, RT to 60 °C, 1.5 h; (ii) NaHCO3, H2O, 0 °C, 5 h, 75%.
The previously known 2-amino-5-bromo-nicotinonitrile (15)25 has been prepared from commercially available 2-amino-nicotinonitrile via an efficient nuclear monobromination, by treatment with N-bromosuccinimide (NBS) in the presence of a catalytic amount of NH4OAc.26 Reduction by the borane–tetrahydrofuran complex (BH3-THF) followed by acidic deprotection of the resulting borane furnished the diamino compound (16), which was converted to the cyclic urea (17) by action of 1,1′-carbonyldiimidazole (CDI). tert-Butyloxycarbonyl (Boc) protection of exchangeable amide protons is important for success of the Pd catalyzed borylation reaction–oxidation two-step sequence.27 Therefore, the Boc protected cyclic urea (18) was reacted with bis-(pinacolato)diboron, and the crude product was oxidized giving rise to a separable mixture of mono- and di-protected phenols (19 and 20). Either mono- or di-protected phenols (or their mixture) can be deprotected by the reaction with methanolic solution of hydrochloric acid followed by recrystallization.
The synthesis of analogues 13 (Scheme 2) and 14 originated from commercially available vitamin B6, which was converted to compound 21 by a highly efficient two-step sequence.17 Bromination with concentrated hydrobromic acid28 afforded crude 22, which was converted to compound 23 by reaction with sodium azide. Diazo-substitution furnished compound 24. Both diazo and azido groups were simultaneously reduced by palladium-on-carbon catalyzed hydrogenation. Diamine 25 was converted to pyridinol-fused ring analogue 13 via CDI induced cyclic urea formation. Analogue 14 was produced from compound 21, which was converted to 6-amino-5-(hydroxymethyl)-2,4-dimethylpyridin-3-ol by a known procedure29 followed by protection with benzyl chloroformate (CbzCl) and subsequent cyclization under the basic conditions.
Scheme 2. Synthesis of Pyridinol-Fused Ring Analogue 13.

Reagents and conditions: (a) Zn, AcOH (Ac = acetyl), reflux, 3 h, 93% over two steps; (b) 2 M HCl in ether, MeOH, reflux, 1 h; (c) HBr reflux, 20 min; (d) NaN3, DMF, RT, 12 h, 66% over two steps; (e) PhNH2, NaNO2, HCl then NaOH; (f) H2, Pd/C, HCl, MeOH, 12 h, 57% over two steps; (g) (i) CDI, DMF, RT, 12 h; (ii) AcOH, H2O, RT, 8 h; (iii) NaHCO3, 20% MeOH/H2O, 3 h, 66%; (h) PhNH2, HCl, NaNO2, NaOH, H2O, 0 °C, 1 h, 95%; (i) H2,Pd/C, MeOH, RT, 6 h, 90%; (j) CBzCl, NaHCO3; (k) K2CO3, MeOH/H2O.
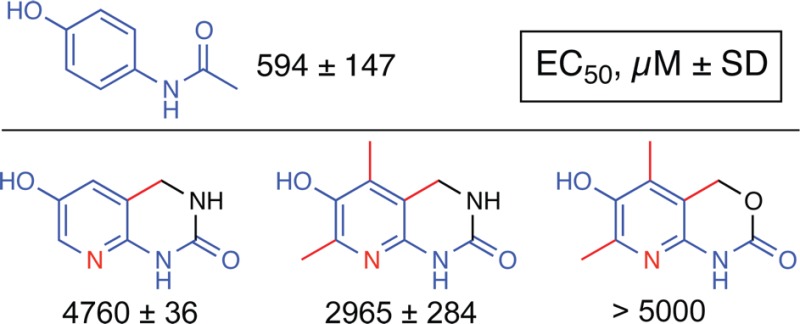
These three analogues have been evaluated for their microsomal stability and cellular toxicity as described in the experimental section. Analogues 12, 13, and 14 have displayed a toxicity and stability profile, which is consistent with our structure-to-biological property relationship analysis described above. All three compounds have no direct cytotoxicity at concentrations below the millimolar range. Their EC50 values for cytotoxicity are 5 to more than 10 times higher than that of acetaminophen (Table 1). All three compounds were metabolized by human liver microsomes to a lesser extent than acetaminophen, suggesting that in vivo metabolism by microsomes would be less as well.
Table 1. Stability and Direct Cytotoxicity of Novel Generation of Pyridinolsa.
| N | microsomal stability % left | cellular toxicity (EC50, μM ± SEM) |
|---|---|---|
| ApAP | 65 ± 12 | 594 ± 147 |
| 12 | 108 ± 12 | 4760 ± 36 |
| 13 | 87 ± 7 | 2965 ± 284 |
| 14 | 97 ± 3 | >5000 |
Microsomal stability is expressed as % of unmodified compound remaining after reaction. Microsomal stability values represent means ± SD. Cellular toxicity is expressed as the concentration causing a 50% decrease in total cellular ATP levels in HepG2 cells. Cellular toxicity values represent means ± SEM.
Next, the efficiency of these ApAP analogues to inhibit hemeprotein-catalyzed lipid oxidation was tested using COX-1 and myoglobin as reported previously.2,3 As shown in Table 2, all analogues are able to inhibit myoglobin-induced arachidonic oxidation within the same order of magnitude, with analogue 14 being the least potent. In contrary, analogue 14 is most potent in inhibiting COX-1 activity followed by ApAP and analogue 12. Interestingly, analogue 13 was not able to significantly inhibit COX-1 at concentrations up to 1 mM.
Table 2. Inhibition by ApAP of Hemeprotein-Catalyzed Oxidation of Arachidonic Acida.
The difference in the myoglobin-induced arachidonic oxidation assay values for analogues 13 and 14 can be explained by their electronic properties. Indeed, more electron-withdrawing oxygen would somewhat diminish analogue 14’s ability to reduce myoglobin. While the reverse trend in COX-1 inhibition is a subject of further studies, we can speculate that it is related to the constrains of the COX-1 binding pocket; interactions with the heme pocket has been shown to affect bioflavonoid activities.30 This possible mechanism is supported by the evidence showing that analogue 12 is more active that the dimethyl analogue 13; the methyl group positive inductive effect would have predicted the opposite effect. Further computational studies, which are outside of this letter’s scope, may shed some light on this phenomenon.
In conclusion, we investigated a number of potent phenolic heterocycles with respect to their efficiency in inhibiting hemeprotein-catalyzed lipid oxidation and also their metabolic stability and toxicity. Several important structure–stability and structure–toxicity relationship studies were used for the design of novel heterocyclic acetaminophen analogue series. Our results indicate that two analogues (12 and 14) may represent a good alternative to acetaminophen with a similar efficiency and better cytotoxicity profile. These analogues are promising candidates for studies in animal models of hepatotoxicity to determine whether they represent lead compounds for development of drugs that could replace acetaminophen.
Acknowledgments
We are thankful to Dr. Donald F. Stec for his help with 2D NMR characterization of the compounds.
Supporting Information Available
Experimental procedures and NMR spectra of key compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ (R.V.S.) Institute of Imaging Science, Vanderbilt University, 1161 21st Avenue South, MCN, AA-1105, Nashville, Tennessee 37232-2310, United States.
Author Present Address
⊥ (B.-S.J.) College of Pharmacy, Yeungnam University, Gyeongsan 712-749, Republic of Korea.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by NIGMS Center for Clinical Pharmacology and Drug Toxicology (GM-15431). J.A.O. is the Thomas F. Frist, Sr. Professor of Medicine.
The authors declare no competing financial interest.
Supplementary Material
References
- Ouellet M.; Percival M. D. Mechanism of acetaminophen inhibition of cyclooxygenase isoforms. Arch. Biochem. Biophys. 2001, 387, 273–280. [DOI] [PubMed] [Google Scholar]
- Boutaud O.; Aronoff D. M.; Richardson J. H.; Marnett L. J.; Oates J. A. Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H(2) synthases. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 7130–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutaud O.; Moore K. P.; Reeder B. J.; Harry D.; Howie A. J.; Wang S.; Carney C. K.; Masterson T. S.; Amin T.; Wright D. W.; Wilson M. T.; Oates J. A.; Roberts L. J. II. Acetaminophen inhibits hemoprotein-catalyzed lipid peroxidation and attenuates rhabdomyolysis-induced renal failure. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 2699–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Vergeade A.; Shi Q.; Zackert W. E.; Gruenberg K. C.; Bokiej M.; Amin T.; Ying W.; Masterson T. S.; Zinkel S. S.; Oates J. A.; Boutaud O.; Roberts L. J. Acetaminophen inhibits cytochrome c redox cycling induced lipid peroxidation. Biochem. Biophys. Res. Commun. 2012, 423, 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson R.; Ross D.; Berlin T.; Olsson L. I.; Moldeus P. Prostaglandin synthase catalyzed metabolic activation of p-phenetidine and acetaminophen by microsomes isolated from rabbit and human kidney. J. Pharmacol. Exp. Ther. 1985, 235, 475–480. [PubMed] [Google Scholar]
- Larson A. M.; Polson J.; Fontana R. J.; Davern T. J.; Lalani E.; Hynan L. S.; Reisch J. S.; Schiødt F. V.; Ostapowicz G.; Shakil A. O.; Lee W. M. Acetaminophen-induced acute liver failure: Results of a United States multicenter, prospective study. Hepatology 2005, 42, 1364–1372. [DOI] [PubMed] [Google Scholar]
- Makin A. J.; Wendon J.; Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987–1993). Gastroenterology 1995, 109, 1907–1916. [DOI] [PubMed] [Google Scholar]
- Lee W. M. Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. Hepatology 2004, 40, 6–9. [DOI] [PubMed] [Google Scholar]
- Nelson S. D.; Dahlin D. C.; Rauckman E. J.; Rosen G. M. Peroxidase-mediated formation of reactive metabolites of acetaminophen. Mol. Pharmacol. 1981, 20, 195–199. [PubMed] [Google Scholar]
- Nelson S. D. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin. Liver. Dis. 1990, 10, 267–278. [DOI] [PubMed] [Google Scholar]
- Mitchell J. R.; Jollow D. J.; Potter W. Z.; Davis D. C.; Gillette J. R.; Brodie B. B. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J. Pharmacol. Exp. Ther. 1973, 187, 185–194. [PubMed] [Google Scholar]
- Mitchell J. R.; Jollow D. J.; Potter W. Z.; Gillette J. R.; Brodie B. B. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J. Pharmacol. Exp. Ther. 1973, 187, 211–217. [PubMed] [Google Scholar]
- Jollow D. J.; Mitchell J. R.; Potter W. Z.; Davis D. C.; Gillette J. R.; Brodie B. B. Acetaminophen-induced hepatic necrosis. II. Role of covalent binding in vivo. J. Pharmacol. Exp. Ther. 1973, 187, 195–202. [PubMed] [Google Scholar]
- James L. P.; Letzig L.; Simpson P. M.; Capparelli E.; Roberts D. W.; Hinson J. A.; Davern T. J.; Lee W. M. Pharmacokinetics of acetaminophen–protein adducts in adults with acetaminophen overdose and acute liver failure. Drug Metab. Dispos. 2009, 37, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fannin R. D.; Russo M.; O’Connell T. M.; Gerrish K.; Winnike J. H.; Macdonald J.; Newton J.; Malik S.; Sieber S. O.; Parker J.; Shah R.; Zhou T.; Watkins P. B.; Paules R. S. Acetaminophen dosing of humans results in blood transcriptome and metabolome changes consistent with impaired oxidative phosphorylation. Hepatology 2010, 51, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam T. G.; Nara S. J.; Zagol-Ikapitte I.; Cooper T.; Valgimigli L.; Oates J. A.; Porter N. A.; Boutaud O.; Pratt D. A. Pyridine and pyrimidine analogs of acetaminophen as inhibitors of lipid peroxidation and cyclooxygenase and lipoxygenase catalysis. Org. Biomol. Chem. 2009, 7, 5103–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serwa R.; Nam T. G.; Valgimigli L.; Culbertson S.; Rector C. L.; Jeong B. S.; Pratt D. A.; Porter N. A. Preparation and investigation of vitamin B6-derived aminopyridinol antioxidants. Chemistry 2010, 16, 14106–14114. [DOI] [PubMed] [Google Scholar]
- Storer R. D.; McKelvey T. W.; Kraynak A. R.; Elia M. C.; Barnum J. E.; Harmon L. S.; Nichols W. W.; DeLuca J. G. Revalidation of the in vitro alkaline elution/rat hepatocyte assay for DNA damage: improved criteria for assessment of cytotoxicity and genotoxicity and results for 81 compounds. Mutat. Res., Genet. Toxicol. 1996, 368, 59–101. [DOI] [PubMed] [Google Scholar]
- Cree I. A.; Andreotti P. E. Measurement of cytotoxicity by ATP-based luminescence assay in primary cell cultures and cell lines. Toxicol. In Vitro 1997, 11, 553–556. [DOI] [PubMed] [Google Scholar]
- Mondal S. K.; Mazumder U. K.; Mondal N. B.; Banerjee S. Optimization of rat liver microsomal stability assay using HPLC. Int. J. Biol. Sci. 2008, 8, 1110–1114. [Google Scholar]
- Dahlin D. C.; Miwa G. T.; Lu A. Y.; Nelson S. D. N-Acetyl-p-benzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc. Natl. Acad. Sci. U.S.A. 1984, 81, 1327–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchepin R.; Moller M. N.; Kim H.-Y. H.; Hatch D. M.; Bartesaghi S.; Kalyanaraman B.; Radi R.; Porter N. A. Tyrosine-lipid peroxide adducts from radical termination: Para coupling and intramolecular Diels–Alder cyclization. J. Am. Chem. Soc. 2010, 132, 17490–17500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey F. A.; Sundberg R. J.. Advanced Organic Chemistry, Part A: Structure and Mechanisms, 5th ed.; Springer-Verlag: New York, 2007. [Google Scholar]
- Maxwell S. R. J.; Thomason H.; Sandler D.; Leguen C.; Baxter M. A.; Thorpe G. H. G.; Jones A. F.; Barnett A. H. Antioxidant status in patients with uncomplicated insulin-dependent and non-insulin-dependent diabetes mellitus. Eur. J. Clin. Invest. 1997, 27, 484–490. [DOI] [PubMed] [Google Scholar]
- Cai L. S.; Cuevas J.; Temme S.; Herman M. M.; Dagostin C.; Widdowson D. A.; Innis R. B.; Pike V. W. Synthesis and structure–affinity relationships of new 4-(6-iodo-H-imidazo[1,2-a]pyridin-2-yl)-N-dimethylbenzeneamine derivatives as ligands for human beta-amyloid plaques. J. Med. Chem. 2007, 50, 4746–4758. [DOI] [PubMed] [Google Scholar]
- Das B.; Venkateswarlu K.; Majhi A.; Siddaiah V.; Reddy K. R. A facile nuclear bromination of phenols and anilines using NBS in the presence of ammonium acetate as a catalyst. J. Mol. Catal. A: Chem. 2007, 267, 30–33. [Google Scholar]
- Medina J. R.; Henry T. A.; Axten J. M. A mild and general method for the synthesis of 2-substituted-5-hydroxypyrimidines. Tetrahedron Lett. 2006, 47, 7363–7365. [Google Scholar]
- Sakuragi T.; Kummerow F. A. The vitamin B6 derivatives structurally analogous to thiamine and their biological activity. Arch. Biochem. Biophys. 1957, 71, 303–310. [DOI] [PubMed] [Google Scholar]
- Nam T.-G.; Ku J.-M.; Rector C. L.; Choi H.; Porter N. A.; Jeong B.-S. Pyridoxine-derived bicyclic aminopyridinol antioxidants: synthesis and their antioxidant activities. Org. Biomol. Chem. 2011, 9, 8475–8482. [DOI] [PubMed] [Google Scholar]
- Wang P.; Bai H. W.; Zhu B. T.. Structural basis for certain naturally occurring bioflavonoids to function as reducing co-substrates of cyclooxygenase I and II. PLoS One 2010, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.