

Abstract
The exocyst complex tethers vesicles at sites of fusion through interactions with small GTPases. The G-protein RalA resides on Glut4 vesicles, and binds to the exocyst after activation by insulin, but must then disengage to ensure continuous exocytosis. Here we report that after recognition of the exocyst by activated RalA, disengagement occurs through phosphorylation of its effector Sec5, rather than RalA inactivation. Sec5 undergoes phosphorylation in the G-protein binding domain, allosterically reducing RalA interaction. The phosphorylation event is catalyzed by protein kinase C and is reversed by an exocyst-associated phosphatase. Introduction of Sec5 bearing mutations of the phosphorylation site to either alanine or aspartate disrupts insulin-stimulated Glut4 exocytosis, as well as other trafficking processes in polarized epithelial cells and during development of zebrafish embryos. The exocyst thus serves as a “gatekeeper” for exocytic vesicles through a circuit of engagement, disengagement, and re-engagement with G proteins.
Introduction
Exocytotic vesicles rely on the exocyst complex for efficient delivery to the plasma membrane 1-3. Both the assembly and recognition of the exocyst require interactions with small GTPases that are spatially activated 4; G proteins such as Rho3 and Cdc42 in yeast 4 or TC10 in mammalian cells 5 direct exocyst assembly at the target membrane, whereas vesicular G proteins such as Sec4 in yeast 6 or RalA in mammalian cells 7-9 mediate its recognition by the vesicle.
Ral GTPases interact with the exocyst subunits Exo84 and Sec5 8,9. While the function of Exo84 is not well understood, Sec5 assembles as part of an exocyst sub-complex that marks the site of exocytosis at the plasma membrane 10,11. RalA binds to the exocyst after activation during cell growth and division 12,13, and functions in the delivery of receptors and transporters 14,15 to the cell surface, including the insulin-stimulated glucose transporter Glut4 7, and also acts as a cargo receptor for the motor complex Myo1c/Calmodulin 7. While exocyst recognition by RalA has received much attention, the mechanism underlying dissociation of the GTPase from the tethering complex prior to the fusion event remains unclear 1,16. Although G proteins usually disengage from effectors through the acceleration of their intrinsic GTPase activity by GAPs 17,18, it is not known whether RalA-exocyst dissociation results from RalA inactivation, or alternatively from modifications in its exocyst partners. We report here that upon delivery of cargo vesicles to the exocyst following the activation of RalA, its partner Sec5 undergoes hormone-stimulated phosphorylation within the Ral-binding domain, an event directly catalyzed by protein kinase C (PKC), resulting in the dissociation of the protein from active RalA. After disengaging from RalA, Sec5 undergoes dephosphorylation for another round of vesicle recognition. Thus, the RalA-exocyst interaction is dynamically regulated through a cycle of engagement, disengagement and re-engagement, crucial for the efficient delivery of exocytic vesicles.
Results
Active RalA promotes exocyst function independently of GTP hydrolysis
The disengagement of RalA from the exocyst could involve either inactivation of the G protein, or modifications on its binding partners. To test whether RalA inactivation is required for exocyst-mediated vesicle delivery, we introduced GTPase-deficient RalA into 3T3-L1 adipocytes, in which insulin stimulates glucose transport through the exocyst-dependent exocytosis of Glut4 5,7,19. Instead of inhibiting glucose transport, as observed with loss of exocyst function 5,7,20, expression of constitutively active RalA G23V produced a two-fold increase in basal glucose uptake, with an approximate 15% increase in insulin-stimulated levels (Figure 1a). Similar effects were seen upon expression of constitutively active RalA G23V after the endogenous protein was depleted by RNAi (Figure 1b, S1). The absence of alterations in proximal insulin signaling, or levels of the glucose transporters Glut4 and Glut1 (Figure 1c), suggested a selective effect on vesicle transport. To determine whether constitutively active RalA promotes glucose transport via the exocyst complex, we introduced point mutations into its effector domain to specifically uncouple active RalA from its effectors RalBP1 (RalA G23V/D49N) or the exocyst (RalA G23V/D49E) (Figure 1d). Interestingly, while RalAΔBP1 retained its ability to promote glucose transport, the exocyst-uncoupled RalAΔExocyst failed to do so (Figure 1e), despite harboring the constitutively active mutation G23V. Thus, while active RalA promotes glucose transport by binding to the exocyst, inactivation via GTP hydrolysis is not necessary for its displacement. Moreover, RalA G23V and RalA G23V/D49E maintained high GTP-bound ratios, comparable to wild type RalA loaded with GTPγS. RalA D49E, which cannot interact with the exocyst, displayed essentially the same GTP-bound ratio as did wild type RalA (Figure 1f, g), suggesting that the exocyst does not promote GTP hydrolysis of RalA, unlike the Sar1-COPII complex 17,18. Taken together, the data pointed to an alternative, GAP-independent mechanism for disengagement from the exocyst during vesicle transport.
Figure 1. RalA promotes exocyst function independently of GTP hydrolysis.

(a) Constitutively active RalA enhances glucose uptake. 3T3-L1 Adipocytes were infected with lentivirus expressing GFP or RalA G23V and subjected to glucose uptake assay. Error represents SEM of triplicate samples. Asterisk: p<0.01. Experiment shown was representative of six independent repeats. (b) Constitutively active RalA up-regulated glucose transport after depletion of endogenous RalA. 3T3-L1 adipocytes were pre-treated for 72 hours with RalA UTR-siRNA by electroporation; GFP or constitutively active RalA (RalAG23V) were introduced into these cells by lentiviral expression. Cells were maintained in basal state and subjected to glucose uptake assay as in (a). Errors represent SEM. (c) Constitutively active RalA does not increase proximal insulin signaling or expression levels of glucose transporters. Cell lysates from (a) were subjected to western blotting (WB). The upper bands in RalA blotting represent exogenous forms. (d) Generation of active RalA mutants uncoupled from RalBP1 or the exocyst. Indicated FLAG-tagged RalA mutants were expressed in adipocytes via lentivirus, and subjected to immunoprecipitation (IP) and WB with indicated antibodies. (e) Active RalA promotes glucose uptake via the exocyst. Adipocytes were infected with lentivirus expressing indicated proteins and subjected to glucose uptake assay. Error represents SEM of triplicate samples. Asterisk: p<0.05, double asterisk: p<0.001.Experiment shown was representative of three independent repeats. (f) Uncoupling RalA from the exocyst does not affect GTP hydrolysis of the protein. Cells expressing the indicated RalA proteins were lysed and subjected to pull down assay; RalA bound to GST protein (active) and in cell lysates (total) were determined by WB. When indicated, cell lysates were loaded with 1mM GDP or 200μM GTPγS. In the case of active RalA or RalA loaded with GTPγS, only 10% of the pulldown in WB analysis to ensure linear range signals. (g) Quantification RalA GTP loading from four independent experiments as performed in (f). Errors represent SD.
The RalA-interacting exocyst subunit Sec5 is phosphorylated to disengage the G protein
We considered whether modifications on the RalA effector Sec5, the exocyst subunit connecting RalA to the holo-complex (Figure S2a) that marks exocytosis sites 9,10, might release RalA from the exocyst. The majority of the RalA-Sec5 interaction is initiated on intracellular membranes (Figure S2b), whereas the loss of plasma membrane-associated exocyst had little effect on RalA/exocyst interaction under basal conditions or in the presence of the RalGEF Rlf (Figure S2c, d), further indicating a disengagement event prior to vesicle fusion at the plasma membrane.
Treatment of cells with the phosphatase inhibitor Calyculin A led to slower migration of Sec5 on SDS-PAGE (Figure 2a), indicating reversible phosphorylation. A [32P] orthophosphate-labeled phospho-protein corresponding to the size of Sec5 (~105 Kd) was detected in exocyst immune complexes, and was reduced upon Sec5 knockdown (Figure S3a). HA-tagged Sec5 was also 32P-labeled in cells, in a manner dramatically enhanced by Calyculin A treatment (Figure S3b). Moreover, Serine/Threonine (Ser/Thr) phosphatase activity was found with the exocyst (Figure S3c), accounting for the stimulatory effect of Calyculin A on Sec5 phosphorylation.
Figure 2. The RalA-interacting exocyst subunit Sec5 undergoes hormone-stimulated phosphorylation in its effector domain.

(a) Inhibition of cellular phosphatase causes a mobility shift of Sec5. 3T3-L1 adipocytes were treated with 10nM Calyculin A for 30 minutes; cell lysates were subjected to SDS-PAGE and WB with indicated antibodies. (b) 32P metabolic labeling of Sec5. Cos-1 cells expressing the indicated constructs were labeled with 1mCi of ortho-phosphate (32P) for 2 hours. Cell lysates were subjected to immunoprecipitation (IP) with the indicated antibodies. After SDS-PAGE, phosphate incorporation was detected by autoradiography, whereas total protein was determined by WB with specific antibodies. When indicated, cells were treated with 10ng/μl EGF for 5 minutes. (c) EGF recruits Sec5 to plasma membrane ruffles. Cos-1 cells expressing HA-tagged Sec5 were treated with DMSO or 10ng/μl EGF for 5 minutes before subjected to immunostaining with the indicated antibodies. Images were at the same magnification; bar = 10μm. (d) Schematic of Sec5 truncation mutants. (e) Ral-Binding Domain (RBD) of Sec5 undergoes hormone-stimulated phosphorylation. Cos-1 cells expressing the indicated constructs were treated with EGF as in (b), and subjected to IP and WB with the indicated antibodies. (f) Mapping the phosphorylation site on Sec5 to Ser89. Wild type and Alanine-substituted RBDs were expressed in Cos-1 cells, and immunoprecipitated with HA antibody before being subjected to WB with the p-Motif antibody.
EGF enhanced phosphorylation of Sec5 in Cos-1 cells (Figure 2b), and also produced a concomitant recruitment of Sec5 to plasma membrane ruffles (Figure 2c), implying a functional role of the modification. Interestingly, Calyculin A, unlike EGF, did not translocate Sec5 to the plasma membrane, despite producing numerous membrane protrusions (Figure S3d), indicating that hormones might regulate Sec5 phosphorylation via kinase activation. EGF-stimulated Sec5 phosphorylation was also detected by an antibody that recognizes phosphorylated Ser/Thr following basic residues such as Lysine or Arginine (Figure S4a). Treatment of immunoprecipitated Sec5 with λ phosphatase eliminated reactivity of Sec5 with the p-Ser/Thr Motif antibody (Figure S4a). Insulin also stimulated Sec5 phosphorylation in 3T3-L1 adipocytes (Figure S4b).
A series of Sec5 truncation constructs were generated to map the phosphorylated residues (Figure 2d). Deletion of the N-terminal Ral-binding domain (RBD) of Sec5 largely decreased EGF-stimulated phosphorylation, whereas the RBD alone underwent EGF-stimulated phosphorylation (Figure 2e). An alanine scan indentified Ser89 as the phosphorylation site (Figure 2f), confirmed by a reciprocal immunoprecipitation with the p-Ser/Thr Motif antibody (Figure S4c) and metabolic labeling with 32P orthophosphate (Figure S4d). Interestingly, additional phosphorylation events on Sec5 were also observed.
Because the RBD of Sec5 is the RalA recognition site 11,21, we hypothesized that Ser89 phosphorylation may modulate RalA interaction. We generated a phospho-null version of Sec5 RBD by mutation of Ser89 to Alanine (S89A), as well as a phospho-mimetic Sec5 RBD by mutation of Ser89 to Aspartic acid (S89D). Wild type Sec5 RBD and the S89A mutant showed similar binding to active RalA, while the phospho-mimetic S89D mutant displayed little interaction (Figure 3a). Similar results were observed in reciprocal experiments (Figure 3b). Together, these data suggest that phosphorylation of Ser89 in the effector domain directly disrupts the Sec5/RalA interaction.
Figure 3. Sec5 effector domain phosphorylation disengages the G protein RalA during vesicle targeting.

(a) The phosphorylation mimetic substitution S89D inhibits Sec5 RBD binding to active RalA. Cos-1 cells expressing the indicated RBD constructs were lysed and subjected to pull down with immobilized GST-RalA G23V. RBD bound to RalA or present in total cell lysates were determined by WB. (b) Direct inhibition of RalA interaction by phospho-mimetic S89D RBD. Recombinant RBD of the indicated forms were purified from E.coli and immobilized on GSH beads before being used to pull down endogenous RalA from cell lysates. RalA bound to immobilized RBD or present in total cell lysates was determined by WB. (c) Replacement of endogenous Sec5 by exogenous copies of the protein. Adipocytes were infected with lentivirus expressing the indicated Myc-tagged Sec5 for 3 weeks before being subjected to cell lysis and IP with Myc antibody. IPs and total cell lysates were fractioned by SDS-PAGE and were subjected to WB analysis with indicated antibodies. Arrows indicated exogenous Sec5 (upper band) and endogenous Sec5 (lower band). (d) Perturbation of Sec5 Ser89 phosphorylation disrupts plasma membrane fusion of Glut4. Adipocytes were co-infected with lentivirus expressing the indicated Sec5 constructs and the Myc-Glut4-eGFP reporter at a 6:1 ratio for 3 weeks. Cells were starved for 4 hours before stimulation with vehicle or 100ng/μl insulin for 20 minutes, fixed with 10% formalin for 10 minutes without permeablization before being subjected to immunostaining with Myc (red) antibody and confocal microscopy. Images were at the same magnification; bar = 10μm. (e) Quantification of five different experiments (n=5) performed as in (d). A minimum of 400 cells in total were counted for each situation. The error bars represent SD. Asterisk: p<0.01.
The incorporation of Sec5 into the exocyst is independent of RalA, but controlled by other exocyst subunits including Sec8 (unpublished data and 22). Furthermore, the levels of Sec5 are tightly controlled by other exocyst subunits, as stably expressed wild type Sec5 caused a concomitant decrease of endogenous Sec5 (Figure 3c). Endogenous Sec5 was similarly replaced with both S89A and S89D mutants (Figure 3c). These results suggested that Sec5 Ser89 phosphorylation does not alter its assembly with other exocyst proteins, despite its inhibitory role in RalA binding, confirming that the exocyst provides docking sites for RalA-localized vesicles.
We expressed different versions of Sec5 into adipocytes along with the Myc-Glut4-eGFP, and assessed immunostaining of the Myc tag without permeabilization of the adipocytes (reflecting Glut4 fusion). While wild type Sec5 had little effect on insulin-stimulated Glut4 fusion with the plasma membrane, expression of Sec5 S89D markedly decreased Glut4 membrane fusion, consistent with the notion that RalA interaction is a prerequisite for exocyst function. Interestingly, expression of the S89A Sec5 also inhibited Glut4 fusion (Figure 3d, e), suggesting that disengagement of Sec5 from RalA by Ser89 phosphorylation is required for exocyst function.
Protein Kinase C (PKC) catalyzes Sec5 effector domain phosphorylation
We generated a polyclonal phospho-specific antibody that specifically reacted with phospho-Ser89 of Sec5, but not with S89A Sec5 (Figure S4e, f). Interestingly, the phospho-antibody recognizes S89D Sec5 RBD, but not the wild type or S89A form, thus confirming the phospho-mimetic property of the S89D substitution (Figure 4a). EGF-stimulated Ser89 phosphorylation of Sec5 in Cos-1 cells was abolished by two different inhibitors of protein kinase C (PKC), Gö6976 and Ro 31-8220, partially blocked by the PI 3-kinase/mTOR kinase inhibitor wortmannin, but was largely unaffected by other kinase inhibitors (Figure 4b). Addition to cells of the phorbol ester tetradecanoyl phorbol acetate (TPA), an activator of PKCs, stimulated Sec5 Ser89 phosphorylation (Figure 4c). The phosphorylation of Sec5 RBD was enhanced by PKCα expression, whereas the kinase-dead PKCα partially blocked TPA-stimulated phosphorylation of Ser89 (Figure 4c). Similar stimulation of phosphorylation by TPA was observed on endogenous Sec5, as determined by immunoprecipitation with the p-Ser89 antibody (Figure 4d). Inhibition of PKC activation by depletion of Rictor 23, or blockade of its catalytic activity with the PKC inhibitor Gö6850 or the cellular Ca2+ chelator BAPTA-AM all attenuated exocytosis of the Myc-Glut4-eGFP reporter after insulin stimulation (Figure 4e, f).
Figure 4. Protein Kinase C (PKC) catalyzes Sec5 effector domain phosphorylation that negatively regulates RalA interaction.

(a) Recognition of the phospho-mimetic S89D substitution by p-Ser89 antibody. Indicated GST fusion proteins purified from E.coli were blotted with p-Ser89 antibody after SDS-PAGE. (b) PKC inhibitors abolish Sec5 RBD Ser89 phosphorylation. Cos-1 cells expressing HA-tagged Sec5 RBD were treated with the indicated inhibitors. Phosphorylation was detected with the p-Ser89 antibody after anti-HA IP. (c) PKCα stimulates Sec5 Ser89 phosphorylation in vivo. Cos-1 cells expressing the indicated constructs were stimulated as described. Phosphorylation was detected with p-Ser89 antibody after anti-HA IP. (d) TPA induces phosphorylation of endogenous Sec5. Cos-1 cells were stimulated as in (b) before subjected IP and WB with the indiated antibody. (e) Loss of PKC activity inhibits Glut4 exocytosis. 3T3-L1 adipocytes expressing Myc-Glut4-eGFP reporter were transfected with control siRNA or Rictor siRNA, or treated with Gö6850 or BAPTA-AM to inhibit PKC activity. Cells were processed as in (2j) for confocal microscopy. Images were of the same magnification; bar = 10μm. (f) Quantification of four independent experiments (n=4) performed as in (e). A total of 400 cells were counted for each situation. Errors represent SD. Asterisk: p< 0.001. (g) Direct phosphorylation of Sec5 Ser89 by PKC isozymes. GST fragments containing Sec5 Ser89 (or S89A) were incubated with the indicated PKC isozymes. Phosphorylation was detected by 32P incorporation and p-Ser89 WB. (h) PKC phosphorylation directly inhibits RalA interaction with Sec5 RBD. Wild type or S89A GST-Sec5 RBD was incubated without or with recombinant PKCα before immobilized on GSH beads, then used to pull down endogenous RalA from cell lysates. (i) PKC phosphorylates Sec5 to inhibit RalA interaction in vivo. Cos-1 cells were transfected with HA-tagged Sec5 full length (FL) or RBD, FLAG-RalA G23VK47E, and wild type GST-PKCα. were subjected to anti-HA IP antibody after the indicated stimulation. (j) Ser89 is required for phosphorylation dependent disengagement of RalA-Sec5 interaction in vivo. Cos-1 cells were transfected with HA-tagged wild type Sec5 or S89A RBD, FLAG-RalA G23V, and GST-PKCα were stimulated as in (c), and subjected to IP and WB with the indicated antibodies.
Conventional PKCs (α and β) directly phosphorylated Ser89 on a GST-Sec5 RBD fusion protein, but did not phosphorylate the S89A mutant, whereas the novel PKC isoforms (δ and ε) produced only modest phosphorylation, and PKD/PKCμ was without effect (Figure 4g). GST-Sec5 RBD and its S89A mutant were incubated with recombinant PKCα plus ATP before immobilization on GSH beads. The immobilized proteins were then subjected to pull down to assess binding with active RalA. Phosphorylation of Sec5 on Ser89 led to decreased RalA binding, and this effect was not observed in the S89A mutant (Figure 4h). When tested in vivo, TPA-stimulated Ser89 phosphorylation in full length Sec5 or Sec5 RBD led to a coincident decrease in RalA binding (Figure 4i). This effect of TPA was not observed in the S89A mutant (Figure 4j), despite increased binding to RalA. Thus, protein kinase C-catalyzed phosphorylation of Sec5 at Ser89 in the effector domain reverses its interaction with RalA.
Allosteric phosphorylation on Sec5 contributes to cellular organization and organism development
We hypothesized that Sec5 effector domain phosphorylation may serve as a general mechanism for exocyst function. Consistent with the reported role of exocyst proteins in abscission during cell cycle 12,13, phosphorylated Sec5 is enriched in the abscission sites of the dividing daughter cells (Figure 5a). This phosphorylation may either take place within the interaction interface of RalA and Sec5, or at an allosteric site that causes a conformational change in the RBD. Examination of the Sec5 RBD structure 11,21 revealed that Ser89 does not occupy the RalA interaction interface 11, but rather makes contacts with the acidic side chain of an aspartic acid residue (Asp73) in a flexible loop region 21 (Figure 5b), suggesting that a negatively charged phospho-Ser89 might repel the side chain of Asp73, reducing interaction with activated RalA. Substitution of Asp73 in the RBD with a serine residue alleviated the inhibitory effect of the phospho-mimetic mutation S89D on RalA binding (Figure 5c), further indicating that Ser89 phosphorylation of Sec5 allosterically inhibits its interaction with RalA.
Figure 5. Sec5 phosphorylation contributes to a cyclic regulatory loop upon recognition by RalA.

(a) Sec5 Ser89 phosphorylation is concentrated at the abscission site during cytokinesis. Cos-1 cells were stained with antibodies against Sec8 (green) and p-Ser89 Sec5 (red), and subjected to confocal microscopy. When indicated, cells were stained with equal amount of antibodies plus p-Ser89 antigenic peptide. Images were at the same magnification; bar = 10μm. (b) Structural illustration of spatially positioning of Ser89 in Sec5 RBD. Figures were generated from the structure of Sec5 Ral binding domain (PDB ID: 1HK6) using Pymol software (DeLano Scientific LLC). (c) D73S substitution alleviates the inhibitory effects on RalA interaction observed with S89D RBD. Lysates of Cos-1 cells expressing indicated constructs were incubated with immobilized GST-RalA G23V. RBD bound to RalA or present in total cell lysates was determined by WB. (d) Reconstitution of phosphorylation-dependent disengagement between RalA and Sec5. Upper: Schematic of the hypothetic sequential events during phosphorylation-induced disengagement. Lower: A complex between recombinant His-RalA loaded with GTPγS and GST-Sec5 RBD was incubated with PKC, and was immobilized by GSH column. RalA bound to Sec5, or released into supernatant, was determined by WB. (e) Loss of Sec5 phosphorylation leads to accumulation of binding with endogenous RalA. Cos-1 cells expressing the indicated constructs were stimulated as indicated before being subjected to anti-HA IP. Bound RalA were detected by WB using protein-A conjugated secondary antibody. (f) Total internal reflection fluorescent microscope (TIRFM) to evaluate the role of Sec5 phosphorylation. Representative images from Cos-1 cell over-expressing pHluorin-Transferrin receptor (TfR) and wild type Sec5 (upper left) or Sec5 S/A (lower left). Scale bar = 10μm. Representative fusion events marked by red boxes 1 & 2, or red boxes 3 & 4. Upper right, time series images of sample fusion events (1 & 2) for Sec5 WT and (3 & 4) for Sec5 S/A. Each panel = 5μm. Lower right, pHluorin-TfR fusion events per area (μm2) per time (min) observed in Sec5 WT and Sec5 S/A Cos cells. Over 1500 fusion events from three different experiments (n=3) were quantified. Errors represent SD. Asterisk: p<0.05.
We also performed a reconstitution experiment to define the sequential order of the regulatory events. GST-Sec5 RBD was incubated with GTPγS-loaded His-RalA. The complex was separated from unbound RalA via GSH columns, and then incubated with activated PKC in the presence of ATP/Magnesium. PKC catalyzed Sec5 phosphorylation when bound to RalA, subsequently disengaging the two proteins (Figure 5d). Importantly, dissociated RalA underwent a second round of interaction with fresh, unphosphorylated Sec5 RBD, thus allowing the process to continue.
To determine whether RalA might potentiate Sec5 phosphorylation after its membrane recruitment, we generated a chimeric protein by fusing the C-terminal membrane-targeting sequence of RalA with Sec5 RBD (RBD-CAAX, Figure S5a); this chimera localized to RalA-residing membranes. Compared with cytosolic RBD, phosphorylation of the chimeric protein was enhanced (Figure S5b). Moreover, while Sec5 RBD S89D failed to co-immunoprecipitate with endogenous RalA, the S89A version of the Sec5 RBD displayed increased binding in cells (Figure 5e), despite possessing a similar binding capacity in vitro (Figure 3a, b). A similar increase was observed with FLAG-tagged, constitutively active RalAG23V (Figure S5c), ruling out potential effects on RalA GTP loading by RBDs, and further suggesting that the loss of Ser89 phosphorylation leads to accumulation of RalA-bound Sec5. We then examined active RalA-residing vesicles that were directed to the regions of plasma membrane expansion, a process that involves the exocyst and cytoskeletal proteins 22,24. Wild type Sec5 localized to vesicles bearing activated RalA G23V; however, a portion of the vesicles displayed little or decreased Sec5 localization when approaching the cell edge. In contrast, Sec5 S89A showed persistent co-localization to RalA-containing vesicles, even when these vesicles are targeted to long narrow protrusions of plasma membrane (Figure S5d).
These data prompted us to further investigate the impact of these mutants on vesicle fusion by total internal reflective fluorescent microscopy (TIRFM). We introduced into Cos-1 cells pHfluorin-tagged transferrin receptor (TfR), an exocyst-dependent cargo that also undergoes endocytic recycling 25, with S89A Sec5. The kinetics of exocytic fusion of pHfluorin-TfR-containing vesicles were not significantly different from those in cells expressing wild type Sec5 and the reporter (Figure 5f), consistent with the notion that tethering is distinct from fusion. Quantitative analysis revealed that S89A Sec5 expression decreased plasma membrane fusion events averaged by surface area and time (Figure 5f, supplementary movie 1, 2, and supplementary table S1). These data further support the hypothesis that Sec5 phosphorylation disengages the interacting G protein to regulate the dynamics of the exocytic program.
A survey of the Sec5 effector domain sequence revealed more than 90% identity between humans and zebrafish, whereas the full length proteins are ~75% identical between the two organisms (Figure S6a). Furthermore, Ser89 and its flanking sequences are highly conserved in the several vertebrate organisms (Figure S6b). MDCK cells expressing wild type Sec5 displayed morphology and distribution of the tight junction marker ZO-1 identical to that of the neighboring cells without the exogenous protein (Figure 6a, upper panel). However, replacement of endogenous Sec5 with either S89A or S89D Sec5 led to a disruption of cellular morphology and accumulation of multi-nucleated cells (Figure 6b; middle and lower panel, Figure 6a), suggesting that perturbation of the phosphorylation cycle of Sec5 disrupts exocyst function.
Figure 6. Sec5 phosphorylation contributes to cellular organization and organism development.

(a) Perturbation of the Sec5 phosphorylation cycle leads to defects in epithelial cells. MDCK cells stably expressing the indicated HA-Sec5 proteins were fixed in formalin and stained with ZO-1(green), HA (red), and DAPI (blue). Images were at the same magnification; bar = 10μm. (b) Quantification of multi-nucleated cells expressing the indicated form of Sec5 from 3 different experiments (n=3) performed as in (a). A minimum of 600 cells in total were counted. Errors represent SEM. Left, WB to confirm replacement of endogenous Sec5 with cells lysates prepared from the same experiments. (c) Perturbation of the Sec5 phosphorylation cycle in zebrafish embryos causes severe deformation and lethality. Zebrafish embryos were injected with 0.5 mM MO oligos alone or in combination with indicated mRNAs encoding different versions of mouse Sec5 around 4-8 cell stages. Embryos were monitored up to 96 hpf. (d) Quantification embryos with normal morphology or only minimal degeneration as in (c). A minimum of ~150 embryos in total were phenotyped in a double-blind manner from 3-4 independent experiments (n=4 for MO alone and WT, n=3 for mutants). The error bars represent SD.
Loss of exocyst function causes embryonic lethality in different organisms, including Drosophila and mouse 26,27, suggesting an evolutionarily conserved role of the complex during development. We thus hypothesized that loss of Sec5 may produce embryonic defects during the development of zebrafish, in which a single, non-duplicated ortholog of Sec5 was found. Injection of morpholino (MO) oligonucleotides that bind the 5’ UTR of Sec5 mRNA into zebrafish embryos caused embryonic degeneration and lethality in a dose-dependent fashion (Figure S6c), with most embryos dead or severely deformed at MO concentrations greater than 0.5mM. The defects could be attributed to loss of Sec5, as re-introducing the wild type mouse Sec5 mRNA largely reversed the embryonic lethality caused by MO oligos (Figure 6c, d). Unlike the wild type protein, neither S89A nor S89D Sec5 rescued the lethality resulting from loss of Sec5 (Figure 6c, d), further confirming the necessity of the Ser89 phosphorylation cycle for exocyst function. Thus, we propose that phosphorylation of Sec5 drives its disengagement from RalA following vesicle recognition in a variety of physiological systems (Figure 7).
Figure 7. Proposed model describing the role of the cyclic phosphorylation of Sec5 in an engagement-disengagement cycle between the exocyst and its upstream G protein.
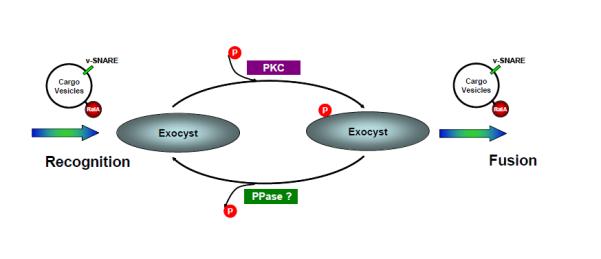
After its activation, the vesicular GTPase RalA interacts with the exocyst protein Sec5, in the process catalyzing the engagement between cargo vesicles and the exocyst. Sec5 subsequently undergoes PKC-dependent phosphorylation within the Ral-binding domain. This phosphorylation allosterically reduces the affinity of its interaction with RalA, resulting in the release of the vesicle from the exocyst so that it can fuse with the plasma membrane. Sec5 then undergoes dephosphorylation by a resident phosphatase, recycling the exocyst for the next round of vesicle recognition.
Discussion
Regulated exocytosis requires the actions of distinct machineries to coordinate the sorting, trafficking, tethering, docking and fusion of exocytotic vesicles 28,29. The exocyst complex integrates the later stages of this process by tethering exocytic vesicles prior to their membrane fusion 1, through interactions with several small GTPases, including RalA that directs the vesicle to the exocyst 4. However, little is known about the mechanisms governing the disengagement of the exocyst from the G proteins. Here we present evidence that phosphorylation of the exocyst subunit Sec5 by PKC disassociates the effector protein from RalA, in the process ensuring the proper function of the exocyst.
The exocyst targets vesicles to specific domains of cells where the fusion apparatus is concentrated 1,30. While studies from yeast suggest that vesicle tethering by the exocyst may be closely coupled with the final membrane fusion 1,31,32, the nature of this cooperative processes is poorly understood in higher organisms, particularly regarding the transition from long-range tethering factors to the assembly of relatively small SNARE fusion apparatus 1,2. In this regard, freeing vesicles from the tethering factor likely would maximize the chances of v-SNAREs to make contact with their cognate t-SNAREs on the plasma membrane.
While freeing the vesicles may offer advantages for fusion, disengagement of the G protein and the effector complex also free the exocyst for the continuous flow of vesicles to the plasma membrane and maximize the output of key transport machineries for rapid and efficient delivery of vesicles. Moreover, the exocyst complex is unlikely to be in excess over cargo molecules, especially those that undergo continuous recycling for sustained exocytosis like Glut4 33. Thus, Sec5 phosphorylation allows active recycling of the complex to continuously provide targeting signals for incoming vesicles. Hence, the exocyst may represent a limiting factor during regulated exocytosis, raising the possibility that the complex not only serves to target vesicles 32, but also as a “gatekeeper” to control their access to the plasma membrane.
Conventional PKCs have long been recognized as key regulators of exocytosis, particularly in neuronal systems 34-36. PKC activation increases the size of the Ready Release Pool (RRP) of synaptic vesicles 37,38, a process that also involves RalA and perhaps the exocyst 37. Furthermore, the mammalian exocyst interacts with atypical PKC in regulating cell migration 39 , suggesting that other members of this kinase family might fine tune exocyst function via phosphorylation of Sec5 or other exocyst subunits. The ability of kinases to directly modulate the partner of the RalA GTPase bypasses the need for GAP proteins to disengage the tethering complex by inactivating RalA, thus reserving RalGAPs for regulating the G protein by upstream signals 40.
The precise roles of the various steps in insulin-stimulated glucose transport remain uncertain and controversial 33,41,42. Insulin regulates exocyst assembly via activation of TC10 5, and induces its recognition of the Glut4 vesicle via stimulation of RalA 7. We propose that after activation of RalA by insulin 7 via the PI 3-kinase pathway that inactivates the Ral GAP complex 40, the G protein binds the exocyst complex via Sec5, thus directing the vesicle to sites on the plasma membrane where SNARE proteins assemble. After binding to Sec5 and localizing at or near the plasma membrane, this exocyst subunit undergoes phosphorylation on Ser89, an event that allosterically disassociates Sec5 from RalA, likely facilitating the release of the vesicle from the exocyst for interaction with the t-SNARE proteins at the site of fusion. Once released from RalA, Sec5 is dephosphorylated by resident phosphatases, enabling the recognition of another vesicle via RalA. It is tempting to speculate that Sec5 phosphorylation represents the last regulated step in insulin-stimulated glucose transport, before an irreversible fusion process at the plasma membrane.
Supplementary Material
Acknowledgements
The work is supported by NIH RO1 DK061618 and DK076906 to ARS, and P60 DK020572 to the Diabetes Research and Training Center at University of Michigan. DG is an investigator of Howard Hughes Medical Institute.
References
- 1.Munson M, Novick P. The exocyst defrocked, a framework of rods revealed. Nat Struct Mol Biol. 2006;13:577–81. doi: 10.1038/nsmb1097. [DOI] [PubMed] [Google Scholar]
- 2.Cai H, Reinisch K, Ferro-Novick S. Coats, tethers, Rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Dev Cell. 2007;12:671–82. doi: 10.1016/j.devcel.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Grote E, Carr CM, Novick PJ. Ordering the final events in yeast exocytosis. J Cell Biol. 2000;151:439–52. doi: 10.1083/jcb.151.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Novick P, Guo W. Ras family therapy: Rab, Rho and Ral talk to the exocyst. Trends Cell Biol. 2002;12:247–9. doi: 10.1016/s0962-8924(02)02293-6. [DOI] [PubMed] [Google Scholar]
- 5.Inoue M, Chang L, Hwang J, Chiang SH, Saltiel AR. The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature. 2003;422:629–33. doi: 10.1038/nature01533. [DOI] [PubMed] [Google Scholar]
- 6.Guo W, Roth D, Walch-Solimena C, Novick P. The exocyst is an effector for Sec4p, targeting secretory vesicles to sites of exocytosis. Embo J. 1999;18:1071–80. doi: 10.1093/emboj/18.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen XW, Leto D, Chiang SH, Wang Q, Saltiel AR. Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c. Dev Cell. 2007;13:391–404. doi: 10.1016/j.devcel.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 8.Moskalenko S, et al. The exocyst is a Ral effector complex. Nat Cell Biol. 2002;4:66–72. doi: 10.1038/ncb728. [DOI] [PubMed] [Google Scholar]
- 9.Moskalenko S, et al. Ral GTPases regulate exocyst assembly through dual subunit interactions. J Biol Chem. 2003;278:51743–8. doi: 10.1074/jbc.M308702200. [DOI] [PubMed] [Google Scholar]
- 10.Jin R, et al. Exo84 and Sec5 are competitive regulatory Sec6/8 effectors to the RalA GTPase. Embo J. 2005;24:2064–74. doi: 10.1038/sj.emboj.7600699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukai S, Matern HT, Jagath JR, Scheller RH, Brunger AT. Structural basis of the interaction between RalA and Sec5, a subunit of the sec6/8 complex. Embo J. 2003;22:3267–78. doi: 10.1093/emboj/cdg329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen XW, Inoue M, Hsu SC, Saltiel AR. RalA-exocyst-dependent recycling endosome trafficking is required for the completion of cytokinesis. J Biol Chem. 2006;281:38609–16. doi: 10.1074/jbc.M512847200. [DOI] [PubMed] [Google Scholar]
- 13.Gromley A, et al. Centriolin anchoring of exocyst and SNARE complexes at the midbody is required for secretory-vesicle-mediated abscission. Cell. 2005;123:75–87. doi: 10.1016/j.cell.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 14.Feig LA. Ral-GTPases: approaching their 15 minutes of fame. Trends Cell Biol. 2003;13:419–25. doi: 10.1016/s0962-8924(03)00152-1. [DOI] [PubMed] [Google Scholar]
- 15.Shipitsin M, Feig LA. RalA but not RalB enhances polarized delivery of membrane proteins to the basolateral surface of epithelial cells. Mol Cell Biol. 2004;24:5746–56. doi: 10.1128/MCB.24.13.5746-5756.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boyd C, Hughes T, Pypaert M, Novick P. Vesicles carry most exocyst subunits to exocytic sites marked by the remaining two subunits, Sec3p and Exo70p. J Cell Biol. 2004;167:889–901. doi: 10.1083/jcb.200408124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antonny B, Schekman R. ER export: public transportation by the COPII coach. Curr Opin Cell Biol. 2001;13:438–43. doi: 10.1016/s0955-0674(00)00234-9. [DOI] [PubMed] [Google Scholar]
- 18.Antonny B, Madden D, Hamamoto S, Orci L, Schekman R. Dynamics of the COPII coat with GTP and stable analogues. Nat Cell Biol. 2001;3:531–7. doi: 10.1038/35078500. [DOI] [PubMed] [Google Scholar]
- 19.Ewart MA, Clarke M, Kane S, Chamberlain LH, Gould GW. Evidence for a role of the exocyst in insulin-stimulated Glut4 trafficking in 3T3-L1 adipocytes. J Biol Chem. 2005;280:3812–6. doi: 10.1074/jbc.M409928200. [DOI] [PubMed] [Google Scholar]
- 20.Inoue M, Chiang SH, Chang L, Chen XW, Saltiel AR. Compartmentalization of the exocyst complex in lipid rafts controls Glut4 vesicle tethering. Mol Biol Cell. 2006;17:2303–11. doi: 10.1091/mbc.E06-01-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mott HR, et al. Structure of the GTPase-binding domain of Sec5 and elucidation of its Ral binding site. J Biol Chem. 2003;278:17053–9. doi: 10.1074/jbc.M300155200. [DOI] [PubMed] [Google Scholar]
- 22.Spiczka KS, Yeaman C. Ral-regulated interaction between Sec5 and paxillin targets Exocyst to focal complexes during cell migration. J Cell Sci. 2008;121:2880–91. doi: 10.1242/jcs.031641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. Embo J. 2008;27:1919–31. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zuo X, et al. Exo70 interacts with the Arp2/3 complex and regulates cell migration. Nat Cell Biol. 2006;8:1383–8. doi: 10.1038/ncb1505. [DOI] [PubMed] [Google Scholar]
- 25.Lim JE, et al. A mutation in Sec15l1 causes anemia in hemoglobin deficit (hbd) mice. Nat Genet. 2005;37:1270–3. doi: 10.1038/ng1659. [DOI] [PubMed] [Google Scholar]
- 26.Friedrich GA, Hildebrand JD, Soriano P. The secretory protein Sec8 is required for paraxial mesoderm formation in the mouse. Dev Biol. 1997;192:364–74. doi: 10.1006/dbio.1997.8727. [DOI] [PubMed] [Google Scholar]
- 27.Murthy M, Garza D, Scheller RH, Schwarz TL. Mutations in the exocyst component Sec5 disrupt neuronal membrane traffic, but neurotransmitter release persists. Neuron. 2003;37:433–47. doi: 10.1016/s0896-6273(03)00031-x. [DOI] [PubMed] [Google Scholar]
- 28.Chieregatti E, Meldolesi J. Regulated exocytosis: new organelles for non-secretory purposes. Nat Rev Mol Cell Biol. 2005;6:181–7. doi: 10.1038/nrm1572. [DOI] [PubMed] [Google Scholar]
- 29.Jahn R, Sudhof TC. Membrane fusion and exocytosis. Annu Rev Biochem. 1999;68:863–911. doi: 10.1146/annurev.biochem.68.1.863. [DOI] [PubMed] [Google Scholar]
- 30.Hsu SC, TerBush D, Abraham M, Guo W. The exocyst complex in polarized exocytosis. Int Rev Cytol. 2004;233:243–65. doi: 10.1016/S0074-7696(04)33006-8. [DOI] [PubMed] [Google Scholar]
- 31.Wiederkehr A, De Craene JO, Ferro-Novick S, Novick P. Functional specialization within a vesicle tethering complex: bypass of a subset of exocyst deletion mutants by Sec1p or Sec4p. J Cell Biol. 2004;167:875–87. doi: 10.1083/jcb.200408001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Novick P, et al. Interactions between Rabs, tethers, SNAREs and their regulators in exocytosis. Biochem Soc Trans. 2006;34:683–6. doi: 10.1042/BST0340683. [DOI] [PubMed] [Google Scholar]
- 33.Chen XW, Saltiel AR. TIRFing out studies on Glut4 trafficking. Dev Cell. 2007;12:4–5. doi: 10.1016/j.devcel.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 34.Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7:281–94. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 35.Morgan A, et al. Regulation of exocytosis by protein kinase C. Biochem Soc Trans. 2005;33:1341–4. doi: 10.1042/BST0331341. [DOI] [PubMed] [Google Scholar]
- 36.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–71. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Polzin A, Shipitsin M, Goi T, Feig LA, Turner TJ. Ral-GTPase influences the regulation of the readily releasable pool of synaptic vesicles. Mol Cell Biol. 2002;22:1714–22. doi: 10.1128/MCB.22.6.1714-1722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gillis KD, Mossner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–20. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 39.Rosse C, et al. An aPKC-exocyst complex controls paxillin phosphorylation and migration through localised JNK1 activation. PLoS Biol. 2009;7:e1000235. doi: 10.1371/journal.pbio.1000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen XW, et al. A Ral GAP complex links PI 3-kinase/Akt signaling to RalA activation in insulin action. Mol Biol Cell. 22:141–52. doi: 10.1091/mbc.E10-08-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–52. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 42.Saltiel AR, Pessin JE. Insulin signaling pathways in time and space. Trends Cell Biol. 2002;12:65–71. doi: 10.1016/s0962-8924(01)02207-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.