

Abstract
Objective
To estimate the associations between neuronal homeostasis, neuroprotection, and oxidative stress candidate gene polymorphisms and neurodevelopmental disability.
Methods
This was a nested case-control analysis of a randomized trial of magnesium sulfate administered to women at imminent risk for early (prior to 32 weeks) preterm birth for the prevention of death or cerebral palsy in their offspring. We evaluated 21 single nucleotide polymorphisms (SNPs) in 17 genes associated with neuronal homeostasis, neuroprotection or oxidative stress in umbilical cord blood. Cases were infant deaths (n=43) and children diagnosed with cerebral palsy (n=24), mental delay (Bayley Mental Developmental Index <70; n=109) and/or psychomotor delay (Bayley Psychomotor Developmental Index <70; n=91). Controls were race- and gender-matched children with normal neurodevelopment. Associations between each SNP and each outcome were assessed in logistic regression models assuming an additive genetic pattern, conditional on maternal race and infant gender, and adjusting for study drug assignment, gestational age at birth, and maternal education.
Results
The odds of cerebral palsy were increased more than 2.5 times for each copy of the minor allele (A) of Vasoactive intestinal polypeptide (VIP) (rs17083008) (aOR 2.67 95% CI 1.09–6.55; p=0.03), and 4.5 times for each copy of the minor allele (T) of N-methyl-D-aspartate receptor subunit 3A (GRIN3A, rs3739722) (aOR 4.67 95% CI 1.36–16.01; p=0.01).The association between the receptor for advanced glycation endproducts (AGER, rs3134945) SNP and mental delay was modulated by study drug allocation (p=0.02).
Conclusions
VIP and NMDA receptor subunit 3A (GRIN3A) SNPs may be associated with cerebral palsy at age 2 in infants after preterm birth.
Introduction
Preterm delivery is a major risk factor for perinatal death and neurodevelopmental disability (NDD), including cerebral palsy (CP), in surviving infants. (1–3) It has been hypothesized that CP and other forms of NDD are the result of neuronal injury via inflammatory, hypoxic, excitatory, and/or oxidative mechanisms. Seventy percent of cases are now believed to be due to prenatal or perinatal factors, with birth asphyxia only playing a minor role in infants born preterm. (2, 4) Antenatal exposure to magnesium sulfate before anticipated early preterm delivery has been shown to reduce the risk of CP and motor dysfunction among survivors. (5–7) The final clinical outcome of at-risk neonates is probably affected by post-natal events and interactions with the postnatal environment. (1)
Genetic susceptibility may modify an individual’s risk for adverse outcomes both before and after delivery. (1, 8) There is evidence that certain genetic polymorphisms in inflammation pathways (e.g., cytokine polymorphisms) or thrombosis pathways (e.g., inherited thrombophilias) may be associated with a greater risk of CP and neurodevelopmental delay. (8, 9) There are limited data regarding the role of genetic polymorphisms in other pathways, particularly those involved with other mechanisms of neuronal injury. (8) Therefore, our objective was to evaluate the association between NDD at age 2 or death, and polymorphisms in candidate genes involved in neuronal homeostasis and protection, or oxidative stress, and to assess whether these genetic polymorphisms modulate the neuroprotective effects of magnesium sulfate.
Patients and Methods
This was a secondary analysis and a nested case-control study of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) Maternal-Fetal Medicine Units Network “Randomized Clinical Trial of the Beneficial Effects of Antenatal Magnesium Sulfate” (BEAM), in which women with singleton or twin gestations between 24–316/7 weeks of gestation at high risk for imminent preterm birth were randomized in a double-blind fashion to receive either magnesium sulfate infusion or identical-appearing placebo. (5) Group assignment in the original trial was made according to a computer-generated random sequence using the urn design, with stratification according to clinical center and, in twin pregnancies, weeks of gestation (<28 or ≥28). (10) Further details of the study, which was conducted at 20 institutions between December 1997 and May 2004, are described elsewhere. (5) The primary outcome of the trial was a composite of infant death or moderate to severe CP at the age of 2 years. Additionally, as secondary outcomes, the trial assessed other neurodevelopmental outcomes (psychomotor and mental delay) at age 2.
For the current analysis, we included infants who died by 1 year of corrected age or were diagnosed with CP, mental delay or psychomotor delay at or beyond 2 years of corrected age. DNA and two-year data on neurological outcomes were required for inclusion. Mental and psychomotor delay were defined by scores of <70 at age 2 years using the Bayley Scales of Infant Development II mental and psychomotor developmental indices (MDI and PDI, respectively) administered by a centrally certified psychologist or psychometrist. CP was determined by a centrally certified pediatrician or pediatric neurologist according to pre-specified criteria of gross motor delay, abnormality in muscle tone or movement, or reflexes. (5) The control group consisted of infants who survived until 2 years of age with normal neurodevelopment (Bayley MDI and PDI ≥ 85) and without CP, periventricular leukomalacia or intraventricular hemorrhage grade III or IV. We excluded cases of fetal demise and neonates with major congenital malformations or genetic syndromes. Randomly selected controls were matched to cases for ethnicity/race and infant gender, in order to minimize the chance of misidentifying a race or gender-based genotype as one associated with the outcome of interest. Controls were matched in a ratio of 4:1 to cases of CP, 2:1 to cases of CP or death, and 1:1 to cases of mental and psychomotor delay. This study was deemed exempt from institutional review board review since the data and samples were de-identified before the analysis was performed.
DNA extraction and SNP selection
DNA extraction, amplification and genotyping were done at Taueret Laboratories (Salt Lake City, Utah). Researchers and laboratory personnel were blinded to the case/control status of the biologic samples. DNA was extracted from umbilical cord serum that had been collected at the time of delivery using the PureGene DNA Purification System (Qiagen, Valencia, CA), per the manufacturer’s protocols and as previously described, and then amplified for better yield. (9) Serum samples were the only biologic material available for analysis. Due to DNA degradation and subsequent variation in size of PCR products, DNA extracted from serum is often of poorer quality compared to DNA extracted from whole blood. This can result in a larger number of missing genotype values when evaluating several SNPs, which can result in discrepant performance among SNP assays. We therefore, excluded children whose samples either failed to genotype or had >30% of their genotype results missing (n=47; 23 cases and 24 controls), and then excluded their corresponding case/control to maintain the matching design. We also randomly selected one twin from each pair of included twins (n=6) in order to avoid including related individuals in the analysis. Triplets and higher order multiple gestations were excluded from the primary trial. We selected 21 SNPs in 17 genes associated with oxidative stress and neuronal homeostasis and neuroprotection, based on the available literature and hypothesized causal pathways. (11–26) The SNPs that were included in our custom multiplex assay are shown in Table 1. SNP analysis was performed using the Taqman Single Nucleotide Polymorphism Allele Discrimination Assay for sample genotyping (Applied Biosystems, Inc., Foster City, CA). Predesigned TaqMan assays were used for genotyping, and genotypes were determined using Applied Biosystems automated Taqman genotyping software, SDS v2.1.
Table 1.
Single Nucleotide Polymorphisms of Selected Genes Included in the Analysis
| Gene | Pathway | Symbol | Polymorphism | RS Number |
|---|---|---|---|---|
| N-methyl-D-aspartate receptor 2B subunit gene | NHP | GRIN2B | −200 (T/G) | rs1019385 |
| N-methyl-D-aspartate receptor 3B subunit gene | NHP | GRIN3B | 1730 (C/T) | rs2240158 |
| GRIN3B | 1210 (C/T) | rs4807399 | ||
| N-methyl-D-aspartate receptor 3A subunit gene | NHP | GRIN3A | 3723 (G/A) | rs3739722 |
| Vasoactive intestinal peptide | NHP | VIP | (A/G) | rs17083008 |
| Vasoactive intestinal peptide receptor 2 | NHP | VPAC2 | 393 (C/A) | rs2098349 |
| 1760 (C/T) | rs885861 | |||
| Brain-derived neurotrophic factor | NHP | BDNF | Val66Met | rs6265 |
| Aquaporin-4 | NHP | AQP4 | Met278Thr (A/G) | rs3906956 |
| A/G | rs9951307 | |||
| Neuregulin-1 | NHP | NRG1 | C/T | rs35753505 |
| Transient receptor potential melastatin 7 | NHP | TRPM7 | Ile1482Thr | rs8042919 |
| Transthyretin | NHP | TTR | 76 (G/A) | rs1800458 |
| Reelin | NHP | RELN | V997L | rs362691 |
| −57 (T/C) | rs736707 | |||
| Receptor for advanced glycation endproducts | OX | AGER | 64 (C/A) | rs3134945 |
| Toll like receptor 4 | OX | TLR4 | Asp299Gly | rs4986790 |
| Kalirin | NHP | KALRN | −9759 (C/T) | rs11712039 |
| Inducible nitric oxide synthase | OX | NOS2 | −231 (C/T) | rs1137933 |
| Glutathione peroxidase 1 | OX | GPX1 | 35 (C/T) | rs1800668 |
| Superoxide dismutase 2 | OX | SOD2 | 24 (T/C) | rs4880 |
NHP, neuronal homeostasis and protection pathway; OX, oxidative stress pathway.
Statistical Analysis
Demographic characteristics between cases with and without DNA samples available were compared using Chi-square, Fisher exact, or Wilcoxon Rank Sum test for maternal characteristics, and GEE (generalized estimating equations) for neonatal characteristics, in order to account for the correlation between siblings. (27) Demographic characteristics between cases and controls were compared using Chi-square, Fisher exact, or Wilcoxon Rank Sum test. Outcomes evaluated were CP, the combined outcome of CP or death, mental delay, and psychomotor delay. The combination of CP or death is necessary since CP and death are competing outcomes: the former can’t be assessed when the later occurs before a reliable diagnosis of CP can be made. Each SNP was tested for Hardy Weinberg equilibrium in the cases group (as controls were matched to cases in this study). Logistic regression modeling was used to analyze the association between each SNP and neonatal outcomes accounting for demographic and clinical variables significantly different between cases and controls or known a priori to be associated with neurodevelopmental outcomes. This included maternal education, which is associated with the results of the Bayley Scales of Infant Development (the source of our measure of psychomotor and mental delay), gestational age (GA) at delivery, which is associated with the various neurodevelopmental outcomes, and magnesium sulfate which has been shown to be associated with a decrease in CP. For each outcome-SNP combination, we first checked the interaction effect between the SNP and treatment assignment. If the interaction term was not significant, then we removed it from the model and focused on the SNP effect only. Genotypes were included as covariates in the regression model which assumed an additive genetic pattern. The additive genetic model assumes that each polymorphism confers additional risk and that having 2 copies of the minor allele has twice the effect of having 1 copy, and has been preferred for its robustness among the other genetic models. All the models were based on the logistic regression conditional on maternal race (African American, Caucasian or Hispanic) and neonatal gender (male or female). Because this was an exploratory study, no power calculation was conducted, and no adjustments were made for multiple comparisons. A two-sided P-value of less than 0.05 was considered to indicate statistical significance. Statistical analyses were performed using SAS statistical software (SAS Institute, Inc, Cary, NC).
The primary data collection was approved by the Institutional Review Boards of the Biostatistical Coordinating Center and the clinical sites at which patients were recruited, and all women provided written informed consent.
Results
The original trial (BEAM) included 2,444 infants, of whom 2,260 infants had long-term neurodevelopmental outcomes available. Out of those, 668 infants were diagnosed with CP, mental or psychomotor delay, or died; and satisfied our “cases” definition. However, out of those 668 infant “cases”, only 231 had DNA available to be analyzed. These were then matched to 231 infant “controls” by maternal ethnicity/race and infant gender. After excluding infant samples that failed to genotype or had >30% of their genotype missing (n=47; 23 cases and 24 controls), and after randomly selecting one twin from each pair (n=6; 5 cases and 1 control), the cohort remaining for analysis included 409 infants (203 cases and 206 controls). (Figure 1)
Figure 1.

Selection of cases and controls. The control group consisted of infants who survived until 2 years of age with normal neurodevelopment (Bayley Mental and Psychomotor Developmental Indices 85 or greater and without cerebral palsy, periventricular leukomalacia, or intraventricular hemorrhage grade 3 or 4).
Compared with the 425 infant “cases” from the parent cohort who satisfied the inclusion criteria but had no available DNA, the 203 infant “cases” with DNA samples available for testing were more likely to be born singleton and at a more advanced gestational age. (Table 2)
Table 2.
Characteristics of Cases With and Without DNA Samples Available
| Cases* n=628 |
|||
|---|---|---|---|
|
| |||
| DNA Samples† n=203 | No DNA Samples n=425 | P | |
|
| |||
| Ethnicity | 0.42 | ||
| African American | 91 (44.8) | 214 (50.4) | |
| Caucasian | 68 (33.5) | 131 (30.8) | |
| Hispanic | 44 (21.7) | 80 (18.8) | |
|
| |||
| Maternal education (years) | 12 [10,12] | 12 [11,13] | 0.10 |
|
| |||
| Allocated to magnesium sulfate | 97 (47.8) | 196 (46.1) | 0.70 |
|
| |||
| Gestational age at birth (weeks) | 29.6 [26.6, 31.6] | 27.4 [25.4, 29.9] | <0.001 |
|
| |||
| Singleton | 183 (90.1) | 352 (82.8) | 0.02 |
|
| |||
| Male gender‡ | 130 (64.0) | 264 (56.8) | 0.08 |
Cases were infants who died by age 1 or developed cerebral palsy, mental or psychomotor delay by age 2. Controls were infants who survived until 2 years of age with normal neurodevelopment and without cerebral palsy.
The sample size was based on the maternal patients with eligible neonatal outcomes.
Generalized estimating equation was used to adjusted for twins in the neonatal patients (DNA samples n=203 and no DNA samples n=465).
Data are median [interquartile range] or n (%) unless otherwise specified.
For this analysis, there were 43 infants who died, 24 children with CP, 109 with mental delay and 91 with psychomotor delay. There were eight children with CP and mental delay, 14 with CP and psychomotor delay, and 48 with mental and psychomotor delay. The maternal and neonatal characteristics of all cases and controls are summarized in Table 3. Compared with controls, cases were born at an earlier GA. Those with mental delay were born to mothers with less education than the matched controls, and those with CP were less likely to be from singleton gestations. There were no differences in ethnicity, study drug assignment, chorioamnionitis, or infant gender between cases and controls (Table 3).
Table 3.
Maternal and Neonatal Characteristics of Cases and Controls
| Cerebral Palsy (n=24) | Controls (n=96) | P | Cerebral Palsy or Death (n=67) | Controls (n=134) | P | Psychomotor Delay (n=91) | Controls (n=91) | P | Mental Delay (n=109) | Controls (n=109) | P | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| Ethnicity | >0.99 | >0.99 | >0.99 | >0.99 | ||||||||
| African American | 11 (45.8) | 44 (45.8) | 35 (52.2) | 70 (52.2) | 35 (38.5) | 35 (38.5) | 50 (45.9) | 50 (45.9) | ||||
| Caucasian | 12 (50.0) | 48 (50.0) | 25 (37.3) | 50 (37.3) | 40 (44.0) | 40 (44.0) | 25 (22.9) | 25 (22.9) | ||||
| Hispanic | 1 (4.2) | 4 (4.2) | 7 (10.5) | 14 (10.5) | 16 (17.6) | 16 (17.6) | 34 (31.2) | 34 (31.2) | ||||
|
| ||||||||||||
| Maternal education (years) | 12 [11,14] | 12 [12,14.5] | 0.22 | 12 [11,13] | 12 [12,14] | 0.04 | 12 [10,13] | 12 [12,14] | 0.04 | 11 [10,12] | 12 [10,14] | <0.01 |
|
| ||||||||||||
| Allocated to magnesium sulfate | 12 (50.0) | 44 (45.8) | 0.71 | 34 (50.8) | 69 (51.5) | 0.92 | 41 (45.1) | 44 (48.4) | 0.66 | 52 (47.7) | 49 (45.0) | 0.68 |
|
| ||||||||||||
| Chorioamnionitis, clinical | 3 (12.5) | 13 (13.5) | >0.99 | 12 (17.9) | 16 (11.9) | 0.25 | 11 (12.1) | 11 (12.1) | >0.99 | 12 (11.0) | 12 (11.0) | >0.99 |
|
| ||||||||||||
| Gestational age at birth (wks) | 27.7 [26.4, 29.8] | 31.0 [29.1, 32.1] | <0.001 | 27.1 [25.7, 29.1] | 31.1 [29.1, 32.4] | <0.001 | 29.7 [26.7, 31.7] | 31.6 [29.6, 32.4] | <0.001 | 30.3 [27.7, 31.7] | 31.3 [29.7, 32.4] | <0.001 |
|
| ||||||||||||
| Preterm delivery prior to 37 weeks | 24 (100.0) | 95 (99.0) | >0.99 | 66 (98.5) | 131 (97.8) | >0.99 | 89 (97.8) | 88 (96.7) | >0.99 | 105 (96.3) | 107 (98.2) | 0.68 |
|
| ||||||||||||
| Preterm delivery prior to 28 weeks | 13 (54.2) | 11 (11.5) | <0.001 | 41 (61.2) | 15 (11.2) | <0.001 | 31 (34.1) | 10 (11.0) | <0.001 | 31 (28.4) | 7 (6.4) | <0.001 |
|
| ||||||||||||
| Cesarean delivery | 12 (50.0) | 34 (35.4) | 0.19 | 33 (49.3) | 43 (32.1) | 0.02 | 35 (38.5) | 38 (41.8) | 0.65 | 34 (31.2) | 42 (38.5) | 0.26 |
|
| ||||||||||||
| Singleton | 19 (79.2) | 90 (93.8) | 0.04 | 57 (85.1) | 128 (95.5) | 0.01 | 82 (90.1) | 84 (92.3) | 0.60 | 102 (93.6) | 104 (95.4) | 0.55 |
|
| ||||||||||||
| Male gender | 18 (75.0) | 72 (75.0) | >0.99 | 43 (64.2) | 86 (64.2) | >0.99 | 58 (63.7) | 58 (63.7) | >0.99 | 73 (67.0) | 73 (67.1) | >0.99 |
Out of 21 SNPs chosen, the assay for one SNP (NRG1, rs35753505) failed manufacturing, and another SNP (GRIN3B C/T, rs480739) failed to genotype in >30% of the samples, and these were excluded from further analysis. In the remainder, genotype determination was overall successful in 94% of samples (range 88 – 99 % for individual SNPs). Four SNPs (vasoactive intestinal polypeptide [VIP] rs17083008, GPX1 rs1800668, RELN rs362691, and VPAC2 rs885861) were in Hardy Weinberg disequilibrium (p<0.01).
Two SNPs (VIP rs17083008 and GRIN3A rs3739722) were associated with CP. The genotype frequency of these SNPs in cases and controls are summarized in Table 4. The VIP (A) allele and GRIN3A (T) allele were associated with increased risk of CP (adjusted OR (aOR) 2.67, 95% CI 1.09–6.55; p=0.03, and 4.67, 95% CI 1.36–16.01; p=0.01) respectively. We tried to add an additional interaction term but observed no significant interaction with magnesium sulfate treatment (p=0.86 and 0.89, respectively). The AGER rs3134945 SNP was differentially associated with mental delay, depending on exposure to magnesium sulfate. For this SNP there was an interaction with magnesium sulfate treatment (p=0.02), therefore we analyzed the association in the placebo and magnesium sulfate treated groups separately. Despite this significant interaction, however, there was no significant association between the minor allele (A) and mental delay either in the placebo group (aOR 0.51, 95% CI 0.24 – 1.07; p=0.08) or in the magnesium sulfate group (aOR 1.83, 95% CI 0.89 – 3.73; p=0.10). (Figure 2)
Table 4.
Genotype in Children With Cerebral Palsy (Cases) Compared With Controls in the Absence of Magnesium Sulfate Interaction
| SNP | Outcome | Genotype | Cases | Controls | aOR (95% CI) | P* |
|---|---|---|---|---|---|---|
| VIP rs17083008 |
Cerebral palsy | AA | 8 (40.0) | 16 (19.1) | 2.67 | 0.03 |
| AG | 4 (20.0) | 21 (25.0) | (1.09, | |||
| GG | 8 (40.0) | 47 (56.0) | 6.55) | |||
| GRIN3A rs3739722 |
Cerebral palsy | CC | 9 (47.4) | 63 (71.6) | 4.67 | 0.01 |
| CT | 10 (52.6) | 21 (23.9) | (1.36, | |||
| TT | 0 (0) | 4 (4.6) | 16.01) |
Data reported as n (%)
SNP, single nucleotide polymorphism; aOR, adjusted odds ratio VIP, vasoactive intestinal polypeptide; GRIN3A, N-methyl-D-aspartate receptor 3A subunit gene.
All the regression models were adjusted for treatment assignment, maternal education, and gestational age. The interaction term between the SNP and the treatment assignment was not significant; therefore it was removed from the model. All the models were based on the logistic regression conditional on maternal race (African American, Caucasian, or Hispanic) and neonatal gender (male or female).
Figure 2.
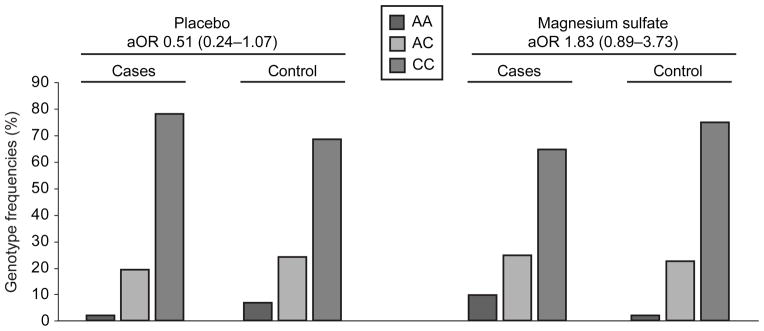
Genotype frequencies and adjusted odds ratios for advanced glycation endproducts specific receptor (AGER; rs3134945) polymorphism and association with mental delay. The Y axis is the percentage of various genotypes. aOR, adjusted odds ratio; CI, confidence interval.
Discussion
In this study, we found that polymorphism in the vasoactive intestinal polypeptide gene (VIP, rs17083008) and GRIN3A gene (which encodes the N-methyl-D-aspartate receptor 3A subunit (NR3A) were associated with an increased risk of cerebral palsy. The AGER, which encodes the receptor for advanced glycation endproducts (RAGE), rs3134945 SNP was differentially associated with mental delay, depending on exposure to magnesium sulfate.
VIP is an endogenous neuromodulator and neuroprotective peptide in the CNS. (28) Its neuroprotective role against ischemic and glutamate induced excitatory brain injury is mediated by VIP receptors (VPAC 1 and 2 subtypes). (14, 15) VIP stimulates astrocytes to produce neuroprotective agents in particular the potent activity-dependent neurotrophic factor (ADNF) and activity-dependent neuroprotective protein (ADNP), and inhibits the production of inflammatory cytokines by activated microglia. (28) Functional genetic polymorphisms in both VIP and VPAC2 genes have been described. (14–16) In particular, the VIP (rs17083008), located in the promoter/regulatory region, is predicted to have a potential functional role since it alters a transcription factor-binding site, may be associated with lower VIP levels, and is thought to be associated with bipolar and other neurobehavioral disorders. (16)
N-methyl-D-aspartate (NMDA) receptors are tetrameric glutamate/calcium ion channels expressed on astrocytes and oligodendrocytes, and essential in excitatory synaptic transmission in the CNS. (29, 30) It has been postulated that NMDA receptors play an essential role in the pathogenesis of CP. (31, 32) Briefly, in the setting of hypoxic injury, cells shift to anaerobic metabolism, increase lactate and glutamate production. The latter then binds and coactivates NMDA receptors, leading to influx of Ca++ intracellularly, lipid peroxidation, free radical production and cell death/injury. Magnesium is a non-competitive inhibitor of these NMDA receptors blocking the influx of Ca++ into the cells and thus protects against cell damage/death. (31, 32) There are 3 major subunits of the NMDA tetramer (NR1, NR2 and NR3). (29) NR3A is one of the two types of the NR3 subunit and has been shown to be highly expressed in oligodendrocytes where glutamate toxicity leads to damage in the myelin sheath. Its expression in the dendirtic spines implies a role in synaptic stability and neuronal activity. (29, 30) The NMDA NR3A are abundantly expressed on the myelin sheath, and are only weakly blocked by extracellular Mg2+, making them more vulnerable to glutamate toxicity damage. (29) The currently described SNP is an exonic polymorphism at position 3723 in the GRIN3A gene that encodes the NR3A, and has been found to be associated with Alzheimer disease. (22)
Last, animal and human studies suggest that inflammation and oxidative stress in the developing fetal brain are associated with brain injury and subsequent development of CP (31–33). The host’s defense to an inflammatory insult involves a group of intracellular proteins called damage-associated molecular pattern molecules or DAMPs, which when released into the extracellular milieu after an insult, activate the receptor for advanced glycation end-products (RAGE) (33). This will then lead to activation of multiple cellular signaling cascades mediated by NFκB, and amplification of the inflammatory and oxidative stress signaling, potentiating and accelerating cellular damage and dysfunction. (33). We have previously shown in a nested case control study from the same cohort that a naturally occurring soluble truncated variant of RAGE (sRAGE) was decreased in the cord blood of neonates who ultimately died after preterm birth (34), indicating a possible role of RAGE activation in neurodevelopmental disability or death. In this study, we have found that the AGER SNP (rs3134945), which is located in the 3′ untranslated region of the AGER gene and its function is still unknown, was differentially associated with mental delay, depending on exposure to magnesium sulfate. However, despite this significant interaction, there was no significant association between the minor allele (A) and mental delay in either group.
The major strength of this study is that it was nested in a multicenter trial where the outcomes were clearly defined and the data and specimen carefully collected in a standard fashion. Umbilical cord serum samples were the only biologic material available for analysis. Because of the poorer quality of DNA extracted from serum, we eliminated individual serum samples and specific SNP assays that genotyped poorly compared to other samples and SNPs. We then excluded their corresponding case/control to maintain the matching the status. We used an elimination cut-off of >30% missing results for serum samples and SNP assays. Given the relatively small number of candidate SNPs, a more rigorous cut-off would have resulted in exclusion of a large number of high-quality serum samples with as few as one missing genotype. After excluding serum samples that entirely failed to genotype (n=26) and samples with >30% of genotypes missing (n=21), the average individual sample genotype call rate over the 19 markers was 98%. Using the same exclusions, the 19 SNPs had an average genotype call rate of 97%. This elimination method for both individual serum samples and specific SNP assays resulted in a robust genetic analysis without bias towards inclusion of erroneous genotypes. Our study was limited by the sample size which did not permit us to evaluate gene-environment interactions on subsequent survival and neurodevelopmental outcomes.
Table 2 compares infant “cases” with or without DNA available, and it was intended to see whether the remaining sample is representative of the entire study cohort. We speculated that the difference in GA and twin gestations seen between infants who had DNA samples available, compared with those who did not, may be related to difficulty in obtaining sufficient cord blood from the smallest neonates and in multiple gestations. These differences are not a significant source of systematic selection bias for the case-control secondary analysis reported, although they may limit ability to extrapolate to the primary study population. We also performed an analysis with a covariate for twin vs. singleton gestation, and all results have remained similar. In addition, data were not available to assess the influence of other variables that may be associated with adverse neurodevelopmental outcomes such as histologic chorioamionitis, funisitis, and acidosis at birth. As this was an exploratory analysis, no corrections were made for multiple comparisons and all associations were reported despite the possibility that some associations may be statistically significant but not clinically meaningful, and some statistically significant associations may have been due to chance (type I error). Our findings need to be confirmed in other cohorts. The possibility of false negatives cannot be excluded as well. Given the limited number of SNPs analyzed, the genetic variation within the selected genes was not fully captured.
In conclusion, survival and risk of adverse neurodevelopmental outcomes are determined by complex interactions between genes and the intrauterine and postnatal environment. VIP and NMDA receptor subunit 3A (GRIN3A) SNPs may be associated with cerebral palsy at age 2 in infants after preterm birth. This work supports the potential role of genetic predisposition to death and neurodevelopmental disability in preterm infants, and suggests that genetic predisposition may alter responses to antenatal magnesium sulfate treatment and risk of death and adverse neurodevelopmental outcomes. Knowledge of individual fetal genetic risk may ultimately lead to new preventative and therapeutic strategies that optimize neurodevelopment after preterm birth.
Supplementary Material
Acknowledgments
The project described was supported by grants from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) [HD27869, HD34208, HD34116, HD40544, HD27915, HD34136, HD21414, HD27917, HD27860, HD40560, HD40545, HD40485, HD40500, HD27905, HD27861, HD34122, HD40512, HD53907, HD34210, HD21410, HD36801, HD19897]MO1-RR-000080, and by the National Institute of Neurological Disorders and Stroke (NINDS) and does not necessarily represent the official views of the NICHD, NINDS, or the National Institutes of Health.
The authors thank Allison T. Northen, M.S.N, R.N., for protocol development and coordination between clinical research centers; Elizabeth Thom, Ph.D. and Steven Weiner, M.S., for protocol and data management and statistical analysis; and Michael W. Varner, M.D., Deborah G. Hirtz, M.D., and Karin Nelson, M.D. for protocol development and oversight.
Footnotes
Financial Disclosure: The authors did not report any potential conflicts of interest.
Presented in part at the 31st Annual Meeting of the Society for Maternal Fetal Medicine 2011, in San Francisco, CA.
References
- 1.American College of Obstetricians and Gynecologists and the American Academy of Pediatrics. Neonatal Encephalopathy and Cerebral Palsy: Defining the Pathogenesis and Pathophysiology. Washington, DC: ACOG; Sep, 2003. [Google Scholar]
- 2.Paneth N, Hong T, Korzeniewski S. The descriptive epidemiology of cerebral palsy. Clin Perinatol. 2006;33:251–67. doi: 10.1016/j.clp.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 3.Saigal S, Doyle LW. An overview of mortality and sequelae of preterm birth from infancy to adulthood. Lancet. 2008;371:261–69. doi: 10.1016/S0140-6736(08)60136-1. [DOI] [PubMed] [Google Scholar]
- 4.Hagberg B, Hagberg G, Beckung E, Uvebrandt P. Changing panorama of cerebral palsy in Sweden: VIII. Prevalence and origin in the birth year period 1991–1994. Acta Paediatr. 2001;90:271–7. [PubMed] [Google Scholar]
- 5.Rouse DJ, Hirtz DG, Thom E, et al. for the Eunice Kennedy Shriver NICHD Maternal-Fetal Medicine Units Network. A randomized trial of magnesium sulfate for the prevention of cerebral palsy. NEJM. 2008;359:895–905. doi: 10.1056/NEJMoa0801187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doyle LW, Crowther CA, Middleton P, Marret S, Rouse D. Magnesium sulphate for women at risk of preterm birth for neuroprotection of the fetus. The Cochrane Database of Systematic Reviews. 2009;(1):Art. No.: CD004661. doi: 10.1002/14651858/CD004661.pub3. [DOI] [PubMed] [Google Scholar]
- 7.Costantine MM, Weiner SJ. Effects of antenatal exposure to magnesium sulfate on neuroproetction and mortality in preterm infants. A meta-analysis. Obstet Gynecol. 2009;114:354–364. doi: 10.1097/AOG.0b013e3181ae98c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Callaghan ME, MacLennan AH, Haan EA, Dekker G South Australian Cerebral Palsy Research Group. The genomic basis of cerebral palsy: a HuGE systematic literature review. Hum Genet. 2009;126:149–72. doi: 10.1007/s00439-009-0638-5. [DOI] [PubMed] [Google Scholar]
- 9.Clark EA, Mele L, Wapner RJ, Spong CY, Sorokin Y, Peacman A, et al. Association of fetal inflammation and coagulation pathway gene polymorphisms with neurodevelopmental delay at age 2 years. Am J Obstet Gynecol. 2010;203:83.e1–10. doi: 10.1016/j.ajog.2010.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei LF, Lachin JM. Properties of urn randomization in clinical trials. Controlled Clin Trials. 1988;9:345–64. doi: 10.1016/0197-2456(88)90048-7. [DOI] [PubMed] [Google Scholar]
- 11.Kleffner I, Bungeroth M, Schiffbauer H, et al. The role of aquaporin 4 polymorphism in the development of brain edema after MCA occlusion. Stroke. 2008;39:1333–35. doi: 10.1161/STROKEAHA.107.500785. [DOI] [PubMed] [Google Scholar]
- 12.Sorani MD, Manley GT, Giacomini KM. Genetic variation in human aquaporins and effects on phenotypes of water homeostasis. Human Mutation. 2008;29:1108–1117. doi: 10.1002/humu.20762. [DOI] [PubMed] [Google Scholar]
- 13.Dai Q, Shrubsole MJ, Ness RM, Schlundt D, Cai Q, Smalley WE, et al. The relation of magnesium and calcium intakes and a genetic polymorphism in the magnesium transporter to colorectal neoplasia risk. Am J Clin Nutr. 2007;86:743–51. doi: 10.1093/ajcn/86.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delgado M, Robledo G, Rueda B, Varela N, O’Valle F, Hernandez-Cortes P, Caro M, Orozco G, et al. Genetic Association of Vasoactive Intestinal Peptide Receptor With Rheumatoid Arthritis Altered Expression and Signal in Immune Cells. Arthritis Rheumatism. 2008;58:1010–9. doi: 10.1002/art.23482. [DOI] [PubMed] [Google Scholar]
- 15.Sun W, Hong J, Zang YCQ, Liu X, Zhang JZ. Altered expression of vasoactive intestinal peptide receptors in T lymphocytes and aberrant Th1 immunity in multiple sclerosis. Int Immunol. 2006;18:1691–700. doi: 10.1093/intimm/dxl103. [DOI] [PubMed] [Google Scholar]
- 16.Soria V, Martinez-Amoros E, Escaramis G, Valero J, Perez-Egea R, Garcia C, et al. Differential Association of Circadian Genes with Mood Disorders: CRY1 and NPAS2 are Associated with Unipolar Major Depression and CLOCK and VIP with Bipolar Disorder. Neuropscychopharmacology. 2010;35:1279–1289. doi: 10.1038/npp.2009.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheeran B, Talelli P, Mori F, et al. A common polymorphism in the brain-derived neurotrophic factor gene (BDNF) modulates human cortical plasticity and the response to rTMS. J Physiol. 2008;586:5717–5725. doi: 10.1113/jphysiol.2008.159905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serajee AJ, Zhong H, Huq AHM. Association of Reelin gene polymorphism with autism. Genomics. 2006;87:75–83. doi: 10.1016/j.ygeno.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 19.Krug T, Manso H, Gouveia L, Sobral J, Xavier JM, Albergaria I, et al. Kalirin: a novel genetic risk factor for ischemic stroke. Hum Genet. 2010;127:513–23. doi: 10.1007/s00439-010-0790-y. [DOI] [PubMed] [Google Scholar]
- 20.Hoffmann I, Bueter W, Zscheppang K, Brinkhaus MJ, Liese A, Riemke S, Dörkc T, Dammannb O, Dammanna CEL. Neuregulin-1, the fetal endothelium, and brain damage in preterm newborns. Brain Behav Immun. 2010;24:784–91. doi: 10.1016/j.bbi.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li A, He L. Association study between the NMDA receptor 2Bsubunit gene (GRIN2B) and schizophrenia: A HuGEreview and meta-analysis. Genet Med. 2007;9(1):4–8. doi: 10.1097/01.gim.0000250507.96760.4b. [DOI] [PubMed] [Google Scholar]
- 22.Liu HP, Lin WY, Liu SH, Wang WF, Tsai CH, Wu BT, et al. Genetic Variation in N-Methyl- D –Aspartate Receptor Subunit NR3A but Not NR3B Influences Susceptibility to Alzheimer’s Disease. Dement Geriatr Cogn Disord. 2009;28 :521–7. doi: 10.1159/000254757. [DOI] [PubMed] [Google Scholar]
- 23.Gibson CS, MacLennan AH, Dekker GA, et al. Candidate genes and cerebral palsy: a population based study. Pediatrics. 2008;122:1079–1085. doi: 10.1542/peds.2007-3758. [DOI] [PubMed] [Google Scholar]
- 24.Rajaraman P, Hutchinson A, Rothman N, Black PM, Fine HA, Loeffler JS, et al. Oxidative response gene polymorphisms and risk of adult brain tumors. Neuro-Oncology. 2008;10:709–715. doi: 10.1215/15228517-2008-037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaens KHJ, Van Der Kallen CJH, Van Greenvenbroek MMJ, et al. Receptor for advanced glycation end product polymorphisms and type 2 diabetes. The CODAM study. Ann NY Acad Sci. 2008;1126:162–165. doi: 10.1196/annals.1433.013. [DOI] [PubMed] [Google Scholar]
- 26.Arbour NC, Lorenz E, Schutte BC, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 27.Liang KY, Zeger SL. Longitudinal Data Analysis Using Generalized Linear Models. Biometrika. 1986;73:13–22. [Google Scholar]
- 28.Dejda A, Sokotowska P, Nowak JZ. Neuroprotective potential of three neuropeptides PACAP, VIP and PHI. Pharmacol rep. 2005;57:307–320. [PubMed] [Google Scholar]
- 29.Mayer M. Glutamate receptors at atomic resolution. Nature. 2006;440:456–462. doi: 10.1038/nature04709. [DOI] [PubMed] [Google Scholar]
- 30.Karadottir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischemia. Nature. 2005;438:1162–6. doi: 10.1038/nature04302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvarez-Diaz A, Hilario E, Goni de Cerio F, Valls-i-soler A, Alvarez-Diaz FJ. Hypoxic ischemic injury in the immature brain – key vascular and cellular changes. Neonatology. 2007;92:227–235. doi: 10.1159/000103741. [DOI] [PubMed] [Google Scholar]
- 32.Costantine MM, Drever N. Antenatal exposure to magnesium sulfate and neuroprotection in preterm infants. Obstet Gynecol Clin North Am. 2011;38:351–366. doi: 10.1016/j.ogc.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 33.Buhimschi CS, Baumbusch MA, Dulay AT, Oliver EA, Lee S, Zhao G, et al. Characterization of RAGE, HMGB1, and S100 beta in inflammation-induced preterm birth and fetal tissue injury. Am J Pathol. 2009;175:958–75. doi: 10.2353/ajpath.2009.090156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Costantine MM, Weiner SJ, Rouse DJ, Hirtz DG, Varner MW, Spong CY, Mercer BM, Iams JD, Wapner RJ, Sorokin Y, Thorp JM, Ramin SM, O’Sullivan MJ, Peaceman AM, Simhan HN for the Eunice Kennedy Shriver National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network. Umbilical Cord Blood Biomarkers of Neurologic Injury and the Risk of Cerebral Palsy or Infant Death. Int J Dev Neurosci. 2011;29:917–22. doi: 10.1016/j.ijdevneu.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.