

Abstract
cAMP is an important mediator of cystogenesis in polycystic kidney disease (PKD). Several adenylyl cyclase (AC) isoforms could mediate cAMP accumulation in PKD, and identification of a specific pathogenic AC isoform is of therapeutic interest. We investigated the role of AC6 in a mouse model of PKD that is homozygous for the loxP-flanked PKD1 gene and heterozygous for an aquaporin-2-Cre recombinase transgene to achieve collecting duct-specific gene targeting. Collecting duct-specific knockout of polycystin-1 caused massive renal cyst formation, kidney enlargement, and severe kidney failure, with a mean survival time of 2 months. In contrast, coincident collecting duct-specific knockout of polycystin-1 and AC6 (also homozygous for the floxed ADCY6 gene) markedly decreased kidney size and cystogenesis, improved renal function, reduced activation of the B-Raf/ERK/MEK pathway, and greatly increased survival. Absence of collecting duct AC6 did not alter urinary cAMP excretion or kidney cAMP concentration. In conclusion, AC6 is a key mediator of cyst formation and renal injury in a model of PKD.
Polycystic kidney disease (PKD) is the fourth leading cause of ESRD in the United States. Renal cyst development and expansion in PKD critically depend on vasopressin (AVP).1 Crossing Brattleboro rats, which have no AVP, with autosomal recessive PKD rats markedly inhibits cystogenesis, whereas an AVP analog restores the cystic phenotype.2 The recent Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes trial found that the AVP V2 receptor antagonist tolvaptan slowed the increase in kidney volume and the decline in kidney function in PKD patients over a 3-year period.3 The cystic effect of AVP is likely caused in large part by stimulation of cAMP.1 In cells from PKD kidneys, cAMP agonism stimulates cell growth, whereas in normal cells, cAMP inhibits cell growth.1 The mechanism of cAMP-stimulated cell proliferation has been largely ascribed to protein kinase A activation of the B-Raf/mitogen activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway.1,4 Intracellular Ca2+ may be important in determining the effect of cAMP on cell proliferation: under normal conditions, Akt, a serine-threonine protein kinase, inhibits B-Raf (another serine-threonine protein kinase); however, in PKD cells, intracellular Ca2+ is reduced, which decreases Akt activity, permitting cAMP activation of B-Raf.1 In addition, cyst fluid secretion is driven by chloride transport stimulated by cAMP.1,4 Thus, cAMP is a key factor in cyst formation and enlargement, and AVP is important in driving cAMP formation.
The renal collecting duct (CD) is the major source of cysts in humans and animal models of autosomal dominant PKD and autosomal recessive PKD.1 Given that cAMP plays a central role in the pathogenesis of PKD, it would be important to define which adenylyl cyclase (AC) isoforms are involved in AVP-mediated cyst formation in the CD. The CD contains intercalated and principal cells. Only principal cells give rise to cysts in mouse models of CD PKD1 deficiency, and only AC3, AC4, and AC6 are expressed in mouse principal cells.5 It is unknown which of these three AC isoforms is involved in AVP-stimulated cyst formation in the CD; however, AC3 and/or AC6 may be particularly important, because their expression has been localized to primary cilia (albeit in nonprincipal cells6,7), the cellular organelle found to be critically important in controlling cyst development.8
To begin to evaluate the role of individual AC isoforms in PKD renal disease, we have now studied mouse models of polycystin-1 deficiency with or without AC6 deficiency. Given that no specific AC isoform inhibitors exist (although this area represents an active area of drug development9), a genetic engineering approach was used. We previously reported a mouse model of PKD by selectively deleting the Pkd1 gene in CD principal cells.10,11 In this model, mice containing loxP sites within introns 1 and 4 of the Pkd1 gene are bred with mice transgenic for the aquaporin-2 promoter driving expression of Cre recombinase (AQP2-Cre); the AQP2 promoter is expressed in the kidney only within principal cells. In the current study, we found that mice with homozygous Pkd1 gene disruption in the CD (PKD knockout [KO]) have multiple large cysts and markedly enlarged kidneys at 33 days of age (Figure 1A). The mean survival was 59±6 days (Figure 1B), and BUN, used as an indicator of renal function, was greatly elevated (135±8 mg/dl) (Figure 1C). Thus, mice with homozygous Pkd1 gene disruption in the CD had rapid cyst progression, marked renal failure, and early mortality.
Figure 1.
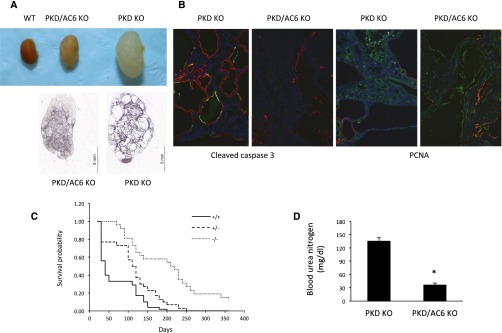
AC6 KO improves survival and lessens kidney disease in PKD KO mice. Coincident AC6 KO reduced kidney and cyst size. A shows representative images from 15 different kidneys of each genotype. B shows activated caspase 3 (apoptosis) or proliferating cell nuclear antigen (PCNA; proliferation), both of which are green, and AQP2 (red; to help show cyst walls, although not all cyst walls are red) staining in representative kidney sections from five different kidneys of each genotype. C shows mouse survival probability (all animals are PKD KO; legend shows AC6 genotype; n=26–52 per data point). D shows BUN at 33 days of age (n=15–27). *P<0.05 versus PKD KO alone.
To examine the role of AC6 in PKD, mice were generated with targeted homozygous disruption of both the Pkd1 and adenylyl cyclase 6 (ADCY6) genes. Mice with loxP-flanked exons 3–12 of the ADCY6 gene, which encode the first catalytic domain, were used (we previously reported CD-specific KO of AC6; these mice have a mild urinary concentrating defect and normal renal histology at 1 year of age12). A key feature of these double KO mice (PKD/AC6 KO) is that the Pkd1 and ADCY6 genes are deleted in the same cells (i.e., whenever Cre is expressed); regardless of whether 100% of principal cells are affected, both target genes have an extremely high likelihood of undergoing recombination. Notably, kidney polycystin-1 mRNA levels were similar between PKD KO and PKD/AC6 KO mice (Table 1), indicating comparable degrees of Pkd1 gene targeting, at least insofar as mRNA content is concerned. PKD/AC6 KO mice had enlarged kidneys compared with wild type but were significantly smaller and lighter, and they contained fewer cysts, with an average smaller cyst area than kidneys from PKD KO animals at 33 days of age (Figure 1, Table 1). Kidneys from PKD1 KO mice had readily detectable apoptosis (cleaved caspase 3) and proliferation (proliferating cell nuclear antigen) in cyst-lining cells, whereas apoptosis and proliferation were much less evident in kidneys from PKD/AC6 KO mice (Figure 1). PKD/AC6 KO mice manifested greatly increased survival compared with PKD KO mice (Figure 1). When PKD/AC6 KO mice died, the average age of death was 209±11 days; however, approximately one third of mice were alive at 1 year. The increased survival was associated with markedly reduced BUN (36±2 mg/dl) compared with PKD KO animals (Figure 1). Mice homozygous for PKD KO but heterozygous for AC6 KO had an intermediate survival time (Figure 1).
Table 1.
Effect of AC6 KO on kidney parameters in PKD KO mice
| Parameter | PKD KO | PKD/AC6 KO |
|---|---|---|
| Body weight (g) | 6.8±0.6 (n=27) | 11.0±0.9a (n=15) |
| Kidney weight (g) | 0.6±0.1 (n=27) | 0.3±0.1a (n=15) |
| Kidney/body weight (%) | 8±2 (n=27) | 3±1a (n=15) |
| Kidney area (mm3) | 791±76 (n=27) | 419±80a (n=15) |
| Cysts/kidney section | 161±35 (n=12) | 78±14a (n=12) |
| Cyst size (mm2) | 1.18±0.27 (n=12) | 0.46±0.10a (n=12) |
| Kidney polycystin-1 mRNA (ΔCT PKD1-GAPDH) | 8.24±0.10 (n=5) | 8.69±0.33 (n=5) |
All animals were euthanized at 33±2 days of age. ΔCT, threshold cycle; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
P<0.05 versus PKD KO alone.
Western analysis of cAMP-regulated signaling pathways involved in PKD disease progression was performed on PKD KO and PKD/AC6 KO mouse kidneys (Figure 2). PKD/AC6 KO mice at 33 days of age had significantly less expression of B-Raf, ERK1/2, phospho-ERK1/2, MEK1/2, and phopho-MEK1/2 compared with PKD KO mice, which is consistent with the notion that AC6-derived cAMP is involved in cell signaling processes involved in cyst formation and growth.
Figure 2.

AC6 KO reduces expression of B-Raf/ERK/MEK in PKD KO mice. A shows Western analysis for B-Raf, phospho–B-Raf, ERK1/2, phospho-ERK1/2, MEK1/2, phosphor-MEK1/2, and β-actin in whole kidneys from 33-day-old PKD KO and PKD/AC6 KO mice (n=6 per group). WT, one wild-type mouse lane. B shows densitometry analysis, normalizing to β-actin, for each blot. *P<0.05 versus PKD KO.
Urine volume was similar between PKD1 KO (0.14±0.2 ml/d) and PKD1/AC6 KO (0.15±0.2 ml/d) mice at 32 days of age. Urine cAMP concentration was similar between the two groups (37±8 pmol/ml in PKD KO and 39±7 pmol/ml in PKD/AC6 KO), whereas urinary cAMP excretion was also unchanged (0.51±0.18 pmol/d in PKD KO and 0.58±0.11 pmol/d in PKD/AC6 KO). Although it was not possible to isolate CD in appreciable amounts from diseased kidneys, total kidney cAMP concentration was determined. No difference was detected in renal cAMP concentration between PKD1 KO (222±37 pmol cAMP/mg total protein, n=5 mice, duplicate measurements in each mouse) and PKD1/AC6 KO (182±41 pmol cAMP/mg total protein, n=6 mice, duplicate measurements in each mouse) mice at 32 days of age. Thus, within our ability to detect changes, AC6 KO in the setting of PKD1 KO did not appreciably alter total urinary cAMP excretion or renal cAMP concentration.
Deficiency of a given adenylyl cyclase isoform being protective against PKD renal injury is not necessarily a given. Previous studies suggest that subcellular cAMP distribution within discrete microdomains is of importance.13,14 These microdomains typically contain, among other things, G protein–coupled receptors, adenylyl cyclases, cAMP-activated kinases, phosphodiesterases, and cytoskeletal anchoring proteins.13,14 Thus, a given adenylyl cyclase isoform has the potential to uniquely affect cell function, including potentially modulating cyst formation and growth. Another point is that a given agonist may activate more than one adenylyl cyclase isoform, resulting in differing biologic effects. For example, CD AC6 deficiency prevents AVP-stimulated epithelial Na channel activity in the CD15; however, CD water reabsorption is only mildly impaired by CD AC6 absence.12 Although these studies were conducted under different conditions (epithelial Na channel activity was studied in vitro, and water reabsorption was studied in vivo), they raise the possibility that AVP might exert different effects on the CD depending on which adenylyl cyclase isoforms are activated. It would seem most likely that AVP binding per se is not the determinant of which adenylyl cyclase isoforms are activated, but rather, other cellular modulatory systems are involved. The nature of such adenylyl cyclase regulatory systems is unknown; however, it does provide a basis for postulating that AVP activates more than one adenylyl cyclase isoform, but not all of these isoforms are necessarily involved in AVP-induced cyst formation. In a larger context, one must consider that other agonists that increase CD cAMP could also be involved in cyst formation in the setting of PKD and that one or more adenylyl cyclases may be specifically involved.
The findings in the current study may well be relevant to human PKD. A recent study found that, although cAMP was elevated in PKD cells obtained from human kidneys (compared with normal human kidney [NHK] cells), AVP causes a greater increase in NHK cell cAMP,16 suggesting that the total amount of cellular cAMP is less important than its localized effects within the cell. Inhibition of calmodulin, which reduces AC6 and AC3 activity, decreased AVP-induced cAMP in PKD but not NHK cells. Targeting AC3 might provide insights, although it should be noted that mice with global AC3 deletion have markedly reduced GFR.17 In contrast, mice with global AC6 deletion seem to develop and grow normally, albeit with some decrease in urine concentrating ability.18
These findings raise the possibility of targeting AC6 in PKD as a therapeutic option. Although AVP receptor antagonism slows PKD progression, it is associated with a significant discontinuation rate because of substantially increased urine output.3 In addition, AC6 is very proximal in the cystogenesis signaling system; hence, its blockade could have multiple effects on downstream mediators. Currently, inhibitors of specific AC isoforms do not exist; however, this area remains an area of active drug development.9,19,20 Even if targeting AC6 per se is ultimately not a viable therapeutic option, continued studies examining the signaling pathways that AC6 specifically regulates (which could be done with our mice) may lead to new insights into potential therapeutic targets in PKD.
Concise Methods
Animal Study Approval
All experiments were carried out in accordance with the guidelines of the animal care and use policy of the University of Utah Health Sciences Center and the Department of Veterans Affairs.
Transgenic Mouse Lines
Principal cell-specific deletion of the Pkd1 gene in mice was achieved as previously described.11 Two mouse lines were used: (1) mice containing a transgene expressing Cre recombinase under the control of AQP2-Cre that express Cre selectively in renal collecting duct principal cells and (2) mice with loxP-flanked exons 1 and 4 of the Pkd1 gene (Pkd1cond). AQP2-Cre transgenic female mice hemizygous for the Pkd1cond allele were bred with male mice homozygous for the Pkd1cond allele. Offspring heterozygous for AQP2-Cre and homozygous for the Pkd1cond allele have principal cell-specific KO of the Pkd1 gene (CD-PKD1-KO).
Mice with both Pkd1 and ADCY6 gene principal cell-specific KO (CD PKD/AC6 KO) were achieved by breeding mice with loxP-flanked (floxed) exons 3–12 of the ADCY6 gene with mice with loxP-flanked exons 1 and 4 of the Pkd1 gene (Pkd1cond). Mice were bred to be doubly homozygous for the floxed Pkd1 and ADCY6 genes (the latter as previously reported12). These mice were crossed with AQP2-Cre mice through a series of breedings to obtain mice that were homozygous for the floxed Pkd1 gene, heterozygous for AQP2-Cre, and either homozygous or heterozygous for the floxed ADCY6 gene.
Genotyping
Tail DNA was PCR-amplified to screen for the floxed Adcy6 alleles using primers AC6-Ex12F 5′-GGA AAG TAG ATC CTC GTC TCG G-3′ and AC6-Ex13R2 5′-CCT ACT TAC AAG AAC CGC AGG AG-3′, which amplify the region containing the 3′ loxP site. DNA was PCR-amplified for the Pkd1cond and Pkd1wt alleles using primers GerF2 (5′-GGC TAT AGG ACA GGG ATG ACA T-3′) and GerR6 (5′-CAT ATT CCT CAC CTG GGA ACA G-3′). The amplified product of the Pkd1cond allele is 34 bp longer than the wild-type allele, corresponding to the loxP site in intron 4. Screening was also performed for the presence of the AQP2-Cre transgene using primers mAQP2 F 5′-GAG ACG TCA ATC CTT ATC TGG AG-3′and creTag R 5′-GCG AAC ATC TTC AGG TTC TGC GG-3′. These primers amplify the junction between the mouse AQP2 promoter and the Cre gene.
Quantitation of Polycystin-1 mRNA
Reverse transcription was performed on 2 μg total kidney RNA from PKD1 and PKD1/AC6 KO mice with oligo(dt) and Superscript III reverse transcription according to the manufacturer's protocol (Invitrogen, Grand Island, NY). The resulting cDNA was then assayed for relative expression of polycystin-1 mRNA using the Taqman Gene Expression Assay (polycystin-1 probe: catalog number Mm00465434_m1; glyceraldehyde-3-phosphate dehydrogenase probe: catalog number Mm03302249_g1; Applied Biosystems, Carlsbad, CA).
Kidney Phenotyping
PKD KO and PKD/AC6 KO mice of both sexes were euthanized at 33 days of age after being weighed. The kidneys were removed, weighed, and measured in three dimensions. One kidney was fixed in formalin, stored in 70% ethanol, imbedded in paraffin, and sectioned (4 µM). Kidney sections (three per kidney) were measured for cyst number and cyst sizes by a blinded individual. Some kidney sections were used for immunostaining. The other kidney was used for Western analysis.
Immunostaining
Kidney sections were subjected to heat-induced antigen retrieval, equilibrated in phosphate-buffered saline (PBS), and blocked with 1% BSA in PBS (BSA-PBS). Sections were incubated with 1:100 goat anti-AQP2 (C-17; Santa Cruz Biotechnology, Santa Cruz, CA) and 1:50 rabbit anticaspase-3 (cleaved form; Chemicon-Millipore, Billerica, MA) or 1:50 rabbit antiproliferating cell nuclear antigen (AbCam, Cambridge, MA) overnight at 4–8°C, washed with BSA-PBS, incubated for 1 hour with 1:50 Alexa Fluor 488 donkey anti-rabbit (Invitrogen) and 1:400 Alexa Fluor 555 donkey anti-goat (Invitrogen) in BSA-PBS, washed with PBS, and mounted in mounting medium with 4′,6-diamidino-2-phenylindole (H-1500; Vector Laboratories, Burlingame, CA). Sections were inspected with a DM-2500 Olympus Microscope, and the red, green, and blue channels were recorded and merged using a computer interface.
Western Analysis
Whole kidneys were isolated from mice and immediately homogenized in HBSS containing 15 mM Hepes (pH 7.4). An aliquot was removed, and protein concentration was determined by the Bradford method. The remainder was solubilized (1:1) in Laemmli buffer containing 0.5% lithium dodecyl sulfate, boiled for 10 minutes, and stored at −20°C. Samples were run on a denaturing NUPAGE 4%–12% Bis⋅Tris Minigel using a MOPS Buffer System (Invitrogen, Carlsbad, CA). Proteins were transferred to a polyvinylidene difluoride plus nylon membrane by electroelution and visualized with the Advance ECL System (GE Healthcare, Piscataway, NJ). Densitometry was performed with a Bio-Rad Gel Documentation System (Hercules, CA).
Antibodies used for immunoblotting were incubated with membranes in blocking buffer (5% milk and 1% BSA⋅Tris-buffered saline–0.1% Tween 20) overnight at 4°C at concentrations from 1:25,000 to 1:100,000. The primary antibodies used were rabbit polyclonal antibodies to β-actin, phospho–B-Raf, B-Raf, ERK1/2, phospho-ERK1/2, MEK1/2, or phospho-MEK1/2 (Cell Signaling Technology, Danvers, MA). Secondary horseradish peroxidase-conjugated antibodies (goat anti-rabbit; Santa Cruz Biotechnology) were incubated at room temperature in blocking buffer. After visualization, blots were stripped and incubated with anti–β-actin antibody at 1:5000 (to control for loading) and then exposed to secondary antibody as above.
Blood, Whole-Kidney, and Urine Assays
A 24-hour sample was collected at 33 days of age. Urine volume was measured, and urine was assayed for cAMP using a commercially available immunoassay according to the manufacturer’s protocol (Assay Design, Ann Arbor, MI). On the next day at the time of euthanasia, blood was collected, and plasma analyzed for BUN using a commercially available assay (Quantichrom DIUR-500; Bioassay Systems, Hayward, CA). Some kidneys from each genotype were snap frozen and homogenized while frozen in cold 5% trichloroacetic acid; an aliquot was taken for determination of total protein (Bradford method), and the remainder was centrifuged at 600×g for 10 minutes at 4°C. The supernatant was extracted with water-saturated ether, the aqueous extract was dried and then reconstituted in assay buffer, and it was analyzed for cAMP content as above.
Statistical Analyses
Data are presented as mean ± SEM. Data were compared using the two-sided unpaired t test. The exception was data examining survival in three mouse genotypes, where one-way ANOVA was used. The criterion for significance was P<0.05.
Disclosures
None.
Acknowledgments
The authors thank Dr. Gregory Germino at the National Institutes of Health for providing the floxed Polycystic Kidney Disease 1 mice.
This research was supported by a Merit Review Award from the Department of Veterans Affairs.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
References
- 1.Wallace DP: Cyclic AMP-mediated cyst expansion. Biochim Biophys Acta 1812: 1291–1300, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Wu Y, Ward CJ, Harris PC, Torres VE: Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 19: 102–108, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS, TEMPO 3:4 Trial Investigators : Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 367: 2407–2418, 2012. 23121377 [Google Scholar]
- 4.Torres VE, Harris PC, Pirson Y: Autosomal dominant polycystic kidney disease. Lancet 369: 1287–1301, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Strait KA, Stricklett PK, Chapman M, Kohan DE: Characterization of vasopressin-responsive collecting duct adenylyl cyclases in the mouse. Am J Physiol Renal Physiol 298: F859–F867, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raychowdhury MK, Ramos AJ, Zhang P, McLaughin M, Dai XQ, Chen XZ, Montalbetti N, Del Rocío Cantero M, Ausiello DA, Cantiello HF: Vasopressin receptor-mediated functional signaling pathway in primary cilia of renal epithelial cells. Am J Physiol Renal Physiol 296: F87–F97, 2009 [DOI] [PubMed] [Google Scholar]
- 7.Bishop GA, Berbari NF, Lewis J, Mykytyn K: Type III adenylyl cyclase localizes to primary cilia throughout the adult mouse brain. J Comp Neurol 505: 562–571, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Torres VE, Harris PC: Polycystic kidney disease: Genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J Intern Med 261: 17–31, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Pierre S, Eschenhagen T, Geisslinger G, Scholich K: Capturing adenylyl cyclases as potential drug targets. Nat Rev Drug Discov 8: 321–335, 2009 [DOI] [PubMed] [Google Scholar]
- 10.Raphael KL, Strait KA, Stricklett PK, Baird BC, Piontek K, Germino GG, Kohan DE: Effect of pioglitazone on survival and renal function in a mouse model of polycystic kidney disease. Am J Nephrol 30: 468–473, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raphael KL, Strait KA, Stricklett PK, Miller RL, Nelson RD, Piontek KB, Germino GG, Kohan DE: Inactivation of Pkd1 in principal cells causes a more severe cystic kidney disease than in intercalated cells. Kidney Int 75: 626–633, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roos KP, Strait KA, Raphael KL, Blount MA, Kohan DE: Collecting duct-specific knockout of adenylyl cyclase type VI causes a urinary concentration defect in mice. Am J Physiol Renal Physiol 302: F78–F84, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooper DM: Compartmentalization of adenylate cyclase and cAMP signalling. Biochem Soc Trans 33: 1319–1322, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Edwards HV, Christian F, Baillie GS: cAMP: Novel concepts in compartmentalised signalling. Semin Cell Dev Biol 23: 181–190, 2012 [DOI] [PubMed] [Google Scholar]
- 15.Roos KP, Bugaj V, Mironova E, Stockand JD, Ramkumar N, Rees S, Kohan DE: Adenylyl cyclase VI mediates vasopressin-stimulated ENaC activity. J Am Soc Nephrol 24: 218–227, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinto CS, Reif GA, Nivens E, White C, Wallace DP: Calmodulin-sensitive adenylyl cyclases mediate AVP-dependent cAMP production and Cl- secretion by human autosomal dominant polycystic kidney cells. Am J Physiol Renal Physiol 303: F1412–F1424, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pluznick JL, Zou DJ, Zhang X, Yan Q, Rodriguez-Gil DJ, Eisner C, Wells E, Greer CA, Wang T, Firestein S, Schnermann J, Caplan MJ: Functional expression of the olfactory signaling system in the kidney. Proc Natl Acad Sci U S A 106: 2059–2064, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rieg T, Tang T, Murray F, Schroth J, Insel PA, Fenton RA, Hammond HK, Vallon V: Adenylate cyclase 6 determines cAMP formation and aquaporin-2 phosphorylation and trafficking in inner medulla. J Am Soc Nephrol 21: 2059–2068, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamenetsky M, Middelhaufe S, Bank EM, Levin LR, Buck J, Steegborn C: Molecular details of cAMP generation in mammalian cells: A tale of two systems. J Mol Biol 362: 623–639, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beazely MA, Watts VJ: Regulatory properties of adenylate cyclases type 5 and 6: A progress report. Eur J Pharmacol 535: 1–12, 2006 [DOI] [PubMed] [Google Scholar]