

Abstract
MicroRNA-122 (miR-122) plays a key role in hepatitis C virus (HCV) replication, but understanding exactly how it functions in the viral lifecycle has been elusive. HCV is a positive-strand virus with a messenger-sense RNA genome, to which miR-122 binds in a non-canonical fashion at two sites near the 5′ end. Recent studies show that miR-122 recruits Ago-2 to the genomic RNA, stabilizing it and slowing its decay in infected cells. This led us to investigate decay pathways that mediate degradation of the viral RNA. We found HCV RNA is degraded primarily by the cytoplasmic 5′ exonuclease Xrn1 in infected cells. miR-122 lost its stabilizing effect when cells were depleted of Xrn1 using an RNAi strategy, providing strong evidence that miR-122 acts to protect the viral RNA from Xrn1-mediated 5′ exonucleolytic decay. However, Xrn1 depletion did not rescue replication of a viral mutant defective in miR-122 binding, indicating that there is much more to miR-122’s actions than prevention of Xrn1 decay. Here, we consider the role of miR-122 in the viral lifecycle, and explore the possibility that it might function directly in viral RNA synthesis.
Keywords: micro-RNA, hepatitis C virus, translation, replication, RNA stability, Xrn1, RNA decay, PCBP2
Introduction
Hepatitis C virus (HCV) is a common cause of chronic liver disease and closely associated with the development of hepatocellular carcinoma. Classified within the Flaviviridae family of viruses, it has a single-stranded, messenger-sense RNA genome approximately 9.7 kb in length that contains a single, large open reading frame encoding a polyprotein under cap-independent translational control of an internal ribosome entry site (IRES).1 The replication of this viral RNA is uniquely dependent on a host-factor microRNA (miRNA), miR-122.2 miR-122 acts directly on the viral RNA, and not indirectly through its effects on hepatocellular metabolism.3 The critical role it plays in the HCV lifecycle is well documented by dose-dependent reductions in circulating HCV RNA following intravenous administration of a locked nucleic acid (LNA) antisense miR-122 “antagomir” to HCV-infected chimpanzees and humans.4,5
miRNAs are ~22 nt long RNA duplexes that regulate gene expression post-transcriptionally, typically by binding to the 3′ untranslated region (3′UTR) of mRNAs, repressing their translation and mediating their degradation.6,7 miR-122 is liver-specific and accounts for a large proportion of the mature miRNAs in hepatocytes,8 the cell targeted for infection by HCV. miR-122 binds to HCV RNA at two conserved sites (S1 and S2) within its 5′UTR, immediately upstream of the IRES (Fig. 1).9 Binding at both sites is important for fulfillment of the virus lifecycle and production of infectious virus,10 and involves both typical miRNA “seed” sequence interactions as well as supplemental base-pairing of 3′ miRNA bases.11-13 Similar to conventional miRNA action, miR-122 recruits Argonaute 2 protein (Ago2) to the viral RNA.14 However, in contrast to typical miRNA action, this stabilizes HCV RNA and slows its decay in infected cells.14 The stability conferred by the miR-122/Ago2 complex can be substituted functionally by addition of a 5′ cap, suggesting that it protects against 5′exonuclease-mediated decay.14

Figure 1. Two copies of miR-122 (red font) bind the extreme 5′ end of the positive-strand RNA genome of HCV (black font). The base-pair interactions shown have been established by genetic approaches and are important for viral replication. Seed sequence interactions are labeled S1 and S2. miR-122 recruits Ago2 to the RNA. PCPB2 binds to stem-loop 1 of the viral 5′NTR, formed by sequence within the most 5′ miR-122 binding site (S1).
Compared with cellular mRNA decay pathways, relatively little is known about the degradation of viral RNAs. As the positive-strand RNA of HCV serves directly as mRNA for viral protein translation, we considered HCV RNA likely to be subject to cellular mRNA decay pathways. The degradation of mRNAs typically initiates with removal of the 3′ poly-(A) tail and/or 5′ cap by deadenylation and decapping enzymes, followed by exonucleolytic degradation of the RNA body.15 The cytoplasmic 5′ exonuclease Xrn1 and 3′ exonuclease exosome complex are the two major exonucleases involved in this process.16,17 HCV RNA is generally considered to possess a free 5′ triphosphate, since it is sensed by the innate immune receptor RIG-I, and terminates in a 3′ stem-loop rather than a poly(A) tail. Therefore, neither deadenylation nor decapping would be required for HCV RNA decay, while either of the two exonuclease pathways could mediate its degradation.
Differential involvement of exonucleases in HCV RNA decay
We have used a relatively simple system to investigate HCV RNA decay in infected cells.14 Huh-7.5 cells are derived from a human hepatocellular carcinoma, express a moderate abundance of miR-122 and support the growth of selected strains of HCV. Huh-7.5 cells that were stably infected with a cell culture-adapted genotype 1a virus (H77S.3)18 were treated with a potent and selective small-molecule inhibitor of the viral NS5B RNA polymerase, PSI-6130 (β-D-2'-deoxy-2'-fluoro-2'-C-methylcytidine).19 This effectively arrests new viral RNA synthesis, and allowed us to assess the rate of decay of the viral RNA by real-time qRT-PCR. The rate of decay is much slower under these conditions than when synthetic RNA is electroporated into cells (t½ of ~11 h vs. 1.4 h),20 most likely because the replicating RNA is sequestered within membranous vesicles.21 Supplementing the cells with additional miR-122 by transfecting synthetic duplex miRNA significantly increased the half-life of the viral RNA (t½ ~18 h).14,20 siRNA-mediated depletion of the 5′ exonuclease Xrn1 slowed the decay rate to a similar extent, while depletion of PM/Scl-100, a component of the exosome complex, was without effect. Importantly, the stabilizing effects of miR-122 supplementation and Xrn1 depletion were not additive: miR-122 supplementation resulted in no additional stabilization of the viral RNA in Xrn1-depleted cells.20 This provides strong evidence that miR-122 stabilizes the viral RNA by antagonizing Xrn1-mediated degradation.
To confirm that 5′ degradation is the major pathway for decay of HCV RNA, we used circularization RT-PCR to identify RNAs that were partially degraded from either end. We found that intact HCV RNA could not be ligated (circularized) with T4 RNA ligase unless first treated with RNA polyphosphatase.20 This is consistent with the presence of a 5′ triphosphate, as this would prevent ligation.22 However, degradation intermediates were ligated without polyphosphatase treatment, allowing them to be amplified and cloned from cells persistently infected with HCV. Almost all of these intermediates contained 5′ truncations, while the 3′ ends were intact, suggesting that HCV RNA is predominantly degraded from its 5′end.20 Interestingly, the point of truncation within the 5′ end was not random, suggesting the influence of higher order structure or perhaps protein binding. These data provide further evidence that Xrn1 is responsible for HCV RNA decay within infected cells. Since the 5′ triphosphate must be removed before Xrn1 can degrade the RNA body, a cellular phosphatase/pyrophosphatase must also be involved. The miR-122/Ago2 complex could function to protect the 5′ triphosphate from this putative enzymatic activity, or from Xrn1 itself. Additional efforts will be needed to elucidate the detailed mechanism of HCV degradation.
Consistent with Xrn1 being primarily responsible for degradation of HCV RNA in infected cells, we found that Xrn1 depletion enhances both viral RNA abundance and production of infectious virus.20 This is in agreement with previous reports by Jones et al.23 and Ruggieri et al.24 that identified Xrn1 as an HCV restriction factor, and suggests that Xrn1-mediated degradation and HCV RNA synthesis compete with each other to determine HCV RNA abundance in cells. However, others have reported that Xrn1 depletion has no positive effects on HCV replication.25-27 This discrepancy could be due to different experimental systems, or possibly varied degrees of cellular toxicity accompanying Xrn1 depletion.
miR-122 functions beyond protecting HCV RNA
Viral RNAs with mutations in the S1 and S2 miR-122-binding sites fail to replicate,2,10 further demonstrating the importance of miR-122 in the viral lifecycle. If miR-122 acts only to protect the 5′ end of HCV RNA from exonucleolytic degradation, such RNAs might be expected to replicate in Xrn1-depleted cells. However, we found that near-complete depletion of Xrn1 in Huh-7.5 cells (conditions under which miR-122 shows no stabilizing effects on the viral RNA) failed to rescue the replication of HCV RNA containing single-base substitutions in both S1 and S2 that ablate miR-122 binding.20 Supplementing the cells with a miR-122 mutant containing complementary substitutions that restored base-pairing with the mutated viral RNA resulted in robust genome amplification. This key experiment shows that miR-122 has an additional, essential function in HCV replication beyond simply protecting the RNA genome from Xrn1-mediated degradation. Mortimer and Doudna28 reached a similar conclusion in a subsequent report describing the effects of miR-122 on Xrn1-mediated HCV RNA decay in a cell-free system.
What might that additional function of miR-122 be? Early data indicate that miR-122 promotes the amplification of subgenomic HCV replicons and, thus, functions independently of viral entry, assembly and release.2 On the other hand, miR-122 supplementation promotes protein expression from the HCV genome,10,29,30 and this has been interpreted as suggesting that it might enhance IRES-directed translation. The effect is about 2-fold, but we believe it is likely due to increased RNA stability.14 Using a cell-free system, Henke et al.29 found that single-stranded miR-122 promoted translation of a luciferase reporter RNA containing HCV 5′ and 3′UTRs.29,31 However, since duplex miR-122 is required for genome amplification,14 this is likely to be an artifact of the cell-free setting. Roberts et al.12 also reported that miR-122 enhanced translation of similar reporter RNAs in transfected cells. Although this was dependent upon the HCV IRES and not observed with similar reporter RNAs containing IRES sequences from other viruses, it is not clear that these observations can be extrapolated to genome-length HCV RNA. We found miR-122 binding is not required for ribosomes to load onto HCV RNA.20 We also observed no increase in viral protein expression when cells depleted of Xrn1 were treated with PSI-6130 (arresting viral RNA synthesis) and simultaneously supplemented with miR-122.20 Collectively, these data argue strongly against miR-122 enhancing IRES-directed translation initiation.
Could miR-122 function directly in viral RNA synthesis? Norman et al.3 demonstrated a decrease in 4-thiouridine-labeled HCV RNA synthesis in cells pulsed 6–7 h after transfection of an LNA anti-miR-122 antagomir. However, total HCV RNA was also decreased, and the amount of newly synthesized RNA was in fact slightly increased relative to total RNA. While elegant, this experiment is difficult to interpret due to the uncertain effects of miR-122 on stability of RNA pools within and outside of replication complexes. Other evidence comes from studies in which we found the addition of miR-122 or anti-miR-122 had no effect on ex vivo synthesis of HCV by isolated membrane-bound replicase complexes.32 This experiment reflects only the elongation phase of viral RNA synthesis, however, and unlike the situation in vivo, the isolated replicase complexes are likely to be sealed membrane-bound compartments allowing only limited access of miR-122. We do not think that either of these observations excludes a direct role for miR-122 in viral RNA synthesis, particularly during initiation.
Current understanding of how HCV replicates its genome is incomplete, but miR-122 can be envisioned to contribute functionally to the initiation of RNA synthesis in at least two ways. During replication of the distantly related dengue virus, a complex stem-loop at the 5′ end of the positive-strand RNA acts as a promoter for the viral RNA-dependent RNA polymerase that binds it and, through a long-range interaction involving cyclization of the dengue genome, initiates minus-strand RNA synthesis at its 3′ end.33 The RNA structure formed by miR-122 interactions with the HCV 5′UTR could act in a similar fashion (Fig. 2A). Alternatively, miR-122 might function during a subsequent step in the replication cycle. Completion of negative-strand synthesis likely results in a genome-length duplex referred to as “replicative form” RNA (RF). The synthesis of new positive-strand RNA then initiates at the 3′ end of the negative-strand of the RF, which must first dissociate from the positive-strand to allow interactions with the polymerase. This is a thermodynamic problem common to all positive-strand RNA viruses. Despite absolute complementarity, it is likely that the RF termini “breath” to some extent, and that the strand separation required for positive-strand synthesis would be favored by stable secondary structure or protein binding at the end of either strand.34,35 miR-122, together with the protein factor(s) it recruits, could promote strand separation by binding the 5′ end of the RF positive-strand. This would free the 3′ end of the negative-strand, allowing for interactions with the polymerase and initiation of positive-strand synthesis (Fig. 2B). Poly(rC)-binding protein 2 (PCPB2) is thought to facilitate strand-separation during poliovirus replication by binding both ends of negative-strand RNA and promoting its circularization.34 Interestingly, PCPB2 binds to a stem-loop that lies within the S1 miR-122 binding site in the HCV positive-strand (Fig. 1).36,37 Thus, it could play a similar role, but on the opposing strand and in concert with miR-122, during HCV replication. These two hypothetical mechanisms are not mutually exclusive, and either or both could explain how miR-122 contributes to genome amplification.
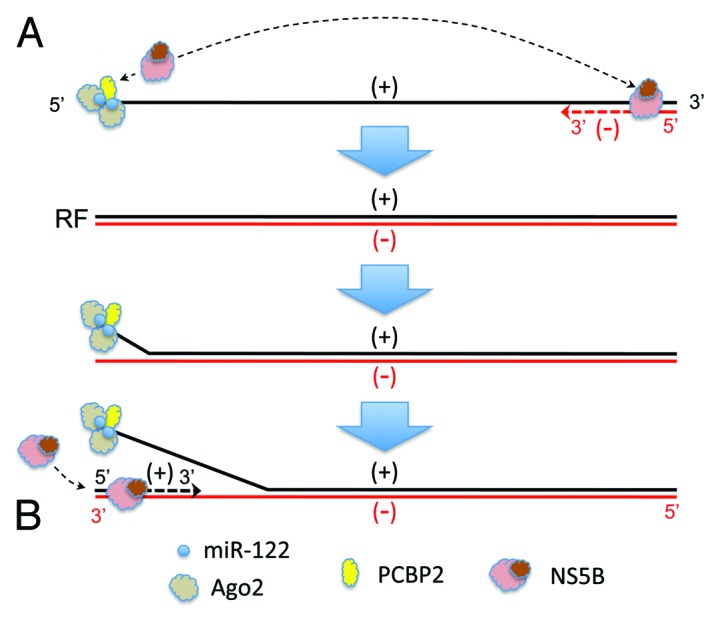
Figure 2. Potential mechanisms by which miR-122 could function to promote the initiation of (A) negative-strand or (B) positive-strand HCV RNA synthesis. Positive-strand (genome sense) RNA is shown in black, and negative-strand RNA in red. See text for details. RF, replicative form RNA.
These models raise a number of questions that have yet to be answered. First, does miR-122 remain bound to the viral RNA during later steps in the virus lifecycle, such as assembly and packaging of the genome? Is it present within extracellular virions? Also, are there proteins other than Ago2 that act as co-factors in miR-122 promotion of virus replication? Although Ago2 is essential for the actions of miR-122 on HCV RNA, this is not the case with Dicer, also a component of the RNA-induced silencing complex (RISC).14,30,38 Depleting cells of TRBP (transactivating response RNA-binding protein) or DDX6 (Rck/p54), both of which are involved in cellular miRNA function, may slow HCV replication, but neither is required for miR-122 to stimulate it.38,39 Thus, it seems that miR-122 requires only a subset of the host proteins required for cellular miRNA function, and perhaps only Ago2, in order to support HCV replication.
Apart from a specific function in RNA replication, miR-122 could help to mask the viral RNA from pathogen-recognition receptors, such as RIG-I (DDX58) and IFITs (interferon-induced protein with tetratricopeptide repeats) that sense RNAs with free 5′ triphosphates and are important anti-viral factors.40,41 In this way, miR-122 could function to diminish innate anti-viral defenses against HCV. There is no evidence for this as yet, however, and most of our studies have been performed in cells that lack interferon-mediated responses to HCV due to the absence of functional RIG-I and TLR3 (Toll-like receptor 3) expression.
While the high degree of dependence of HCV RNA replication on miR-122 is incontrovertible and may well contribute to its hepatotropism, it is not absolute. HeLa cells that do not express miR-122 can be coaxed to support replication of subgenomic HCV RNA,42 and a recombinant RNA that replicates independently of miR-122 has been identified.43 The latter RNA contains a host U3 snoRNA sequence insertion that displaces the S1 miR-122-binding site near the 5′ end of the viral RNA. It replicates about 10-fold less efficiently than its wild-type parent and, importantly, is insensitive to miR122 sequestration. It is unclear how the U3 sequence substitutes for the native HCV sequence and supports HCV RNA replication. It is possible that it could provide binding sites for other miRNA(s), or possibly recruit proteins that facilitate HCV replication. Further study of this HCV variant could shed new light on the mechanism of HCV RNA synthesis as well as miR-122 function in the HCV lifecycle.
Future perspective
Since the discovery that miR-122 is required for efficient HCV replication, its role in this process has been extensively studied. Despite this, the exact mechanism underlying the dependence of HCV replication on miR-122 is still largely unknown. Our work has shown how miR-122 stabilizes the genomic RNA by protecting it against the 5′ exonuclease, Xrn1. This finding has revealed a novel action of a cellular miRNA, but the stabilizing action of miR-122 is insufficient to fully explain why miR-122 is required by the virus. Future efforts should dissect individual steps in the viral lifecycle impacted by miR-122, and identify the host and viral proteins involved. As the study of viruses often provides clues to underlying cellular processes, elucidating the molecular basis of the dependence of HCV replication on miR-122 could yield important insight into previously unrecognized miRNA actions44 and broaden our understanding of miRNA functions in eukaryotic cells.
Acknowledgment
This work was supported in part by NIH grants R01-AI095690 and R01-CA164029 and the University Cancer Research Fund.
Glossary
Abbreviations:
- HCV
hepatitis C virus
- IRES
internal ribosome entry site
- LNA
locked nucleic acid
- qRT-PCR
quantitative reverse transcription, polymerase chain reaction
- RF
replicative form RNA
- UTR
untranslated region
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Lukavsky PJ. Structure and function of HCV IRES domains. Virus Res. 2009;139:166–71. doi: 10.1016/j.virusres.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–81. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 3.Norman KL, Sarnow P. Modulation of hepatitis C virus RNA abundance and the isoprenoid biosynthesis pathway by microRNA miR-122 involves distinct mechanisms. J Virol. 2010;84:666–70. doi: 10.1128/JVI.01156-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–94. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 5.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol. 2012;19:586–93. doi: 10.1038/nsmb.2296. [DOI] [PubMed] [Google Scholar]
- 7.Meijer HA, Kong YW, Lu WT, Wilczynska A, Spriggs RV, Robinson SW, et al. Translational repression and eIF4A2 activity are critical for microRNA-mediated gene regulation. Science. 2013;340:82–5. doi: 10.1126/science.1231197. [DOI] [PubMed] [Google Scholar]
- 8.Chang J, Nicolas E, Marks D, Sander C, Lerro A, Buendia MA, et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004;1:106–13. doi: 10.4161/rna.1.2.1066. [DOI] [PubMed] [Google Scholar]
- 9.Jopling CL, Schütz S, Sarnow P. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe. 2008;4:77–85. doi: 10.1016/j.chom.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jangra RK, Yi M, Lemon SM. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J Virol. 2010;84:6615–25. doi: 10.1128/JVI.00417-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Machlin ES, Sarnow P, Sagan SM. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc Natl Acad Sci USA. 2011;108:3193–8. doi: 10.1073/pnas.1012464108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts AP, Lewis AP, Jopling CL. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res. 2011;39:7716–29. doi: 10.1093/nar/gkr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimakami T, Yamane D, Welsch C, Hensley L, Jangra RK, Lemon SM. Base pairing between hepatitis C virus RNA and microRNA 122 3′ of its seed sequence is essential for genome stabilization and production of infectious virus. J Virol. 2012;86:7372–83. doi: 10.1128/JVI.00513-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimakami T, Yamane D, Jangra RK, Kempf BJ, Spaniel C, Barton DJ, et al. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc Natl Acad Sci USA. 2012;109:941–6. doi: 10.1073/pnas.1112263109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–26. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 16.Jones CI, Zabolotskaya MV, Newbury SF. The 5′ → 3′ exoribonuclease XRN1/Pacman and its functions in cellular processes and development. Wiley Interdiscip Rev RNA. 2012;3:455–68. doi: 10.1002/wrna.1109. [DOI] [PubMed] [Google Scholar]
- 17.Liu Q, Greimann JC, Lima CD. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell. 2006;127:1223–37. doi: 10.1016/j.cell.2006.10.037. [DOI] [PubMed] [Google Scholar]
- 18.Shimakami T, Welsch C, Yamane D, McGivern DR, Yi M, Zeuzem S, et al. Protease inhibitor-resistant hepatitis C virus mutants with reduced fitness from impaired production of infectious virus. Gastroenterology. 2011;140:667–75. doi: 10.1053/j.gastro.2010.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stuyver LJ, McBrayer TR, Tharnish PM, Clark J, Hollecker L, Lostia S, et al. Inhibition of hepatitis C replicon RNA synthesis by beta-D-2′-deoxy-2′-fluoro-2′-C-methylcytidine: a specific inhibitor of hepatitis C virus replication. Antivir Chem Chemother. 2006;17:79–87. doi: 10.1177/095632020601700203. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Masaki T, Yamane D, McGivern DR, Lemon SM. Competing and noncompeting activities of miR-122 and the 5′ exonuclease Xrn1 in regulation of hepatitis C virus replication. Proc Natl Acad Sci USA. 2013;110:1881–6. doi: 10.1073/pnas.1213515110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, et al. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol. 2003;77:5487–92. doi: 10.1128/JVI.77.9.5487-5492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevens A. Purification and characterization of a Saccharomyces cerevisiae exoribonuclease which yields 5′-mononucleotides by a 5′ leads to 3′ mode of hydrolysis. J Biol Chem. 1980;255:3080–5. [PubMed] [Google Scholar]
- 23.Jones DM, Domingues P, Targett-Adams P, McLauchlan J. Comparison of U2OS and Huh-7 cells for identifying host factors that affect hepatitis C virus RNA replication. J Gen Virol. 2010;91:2238–48. doi: 10.1099/vir.0.022210-0. [DOI] [PubMed] [Google Scholar]
- 24.Ruggieri A, Dazert E, Metz P, Hofmann S, Bergeest JP, Mazur J, et al. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe. 2012;12:71–85. doi: 10.1016/j.chom.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ariumi Y, Kuroki M, Kushima Y, Osugi K, Hijikata M, Maki M, et al. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. J Virol. 2011;85:6882–92. doi: 10.1128/JVI.02418-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pager CT, Schütz S, Abraham TM, Luo G, Sarnow P. Modulation of hepatitis C virus RNA abundance and virus release by dispersion of processing bodies and enrichment of stress granules. Virology. 2013;435:472–84. doi: 10.1016/j.virol.2012.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scheller N, Mina LB, Galão RP, Chari A, Giménez-Barcons M, Noueiry A, et al. Translation and replication of hepatitis C virus genomic RNA depends on ancient cellular proteins that control mRNA fates. Proc Natl Acad Sci USA. 2009;106:13517–22. doi: 10.1073/pnas.0906413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mortimer SA, Doudna JA. Unconventional miR-122 binding stabilizes the HCV genome by forming a trimolecular RNA structure. Nucleic Acids Res. 2013;41:4230–40. doi: 10.1093/nar/gkt075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henke JI, Goergen D, Zheng J, Song Y, Schüttler CG, Fehr C, et al. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008;27:3300–10. doi: 10.1038/emboj.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson JA, Zhang C, Huys A, Richardson CD. Human Ago2 is required for efficient miR-122 regulation of HCV RNA accumulation and translation. J Virol. 2011;85:2342–50. doi: 10.1128/JVI.02046-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goergen D, Niepmann M. Stimulation of Hepatitis C Virus RNA translation by microRNA-122 occurs under different conditions in vivo and in vitro. Virus Res. 2012;167:343–52. doi: 10.1016/j.virusres.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 32.Villanueva RA, Jangra RK, Yi M, Pyles R, Bourne N, Lemon SM. miR-122 does not modulate the elongation phase of hepatitis C virus RNA synthesis in isolated replicase complexes. Antiviral Res. 2010;88:119–23. doi: 10.1016/j.antiviral.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Filomatori CV, Lodeiro MF, Alvarez DE, Samsa MM, Pietrasanta L, Gamarnik AV. A 5′ RNA element promotes dengue virus RNA synthesis on a circular genome. Genes Dev. 2006;20:2238–49. doi: 10.1101/gad.1444206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perera R, Daijogo S, Walter BL, Nguyen JH, Semler BL. Cellular protein modification by poliovirus: the two faces of poly(rC)-binding protein. J Virol. 2007;81:8919–32. doi: 10.1128/JVI.01013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andino R, Rieckhof GE, Baltimore D. A functional ribonucleoprotein complex forms around the 5′ end of poliovirus RNA. Cell. 1990;63:369–80. doi: 10.1016/0092-8674(90)90170-J. [DOI] [PubMed] [Google Scholar]
- 36.Fukushi S, Okada M, Kageyama T, Hoshino FB, Nagai K, Katayama K. Interaction of poly(rC)-binding protein 2 with the 5′-terminal stem loop of the hepatitis C-virus genome. Virus Res. 2001;73:67–79. doi: 10.1016/S0168-1702(00)00228-8. [DOI] [PubMed] [Google Scholar]
- 37.Wang L, Jeng KS, Lai MM. Poly(C)-binding protein 2 interacts with sequences required for viral replication in the hepatitis C virus (HCV) 5′ untranslated region and directs HCV RNA replication through circularizing the viral genome. J Virol. 2011;85:7954–64. doi: 10.1128/JVI.00339-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang C, Huys A, Thibault PA, Wilson JA. Requirements for human Dicer and TRBP in microRNA-122 regulation of HCV translation and RNA abundance. Virology. 2012;433:479–88. doi: 10.1016/j.virol.2012.08.039. [DOI] [PubMed] [Google Scholar]
- 39.Jangra RK, Yi M, Lemon SM. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site-directed translation. J Virol. 2010;84:6810–24. doi: 10.1128/JVI.00397-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abbas YM, Pichlmair A, Górna MW, Superti-Furga G, Nagar B. Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature. 2013;494:60–4. doi: 10.1038/nature11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang F, Ramanathan A, Miller MT, Tang GQ, Gale M, Jr., Patel SS, et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479:423–7. doi: 10.1038/nature10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu Q, Guo JT, Seeger C. Replication of hepatitis C virus subgenomes in nonhepatic epithelial and mouse hepatoma cells. J Virol. 2003;77:9204–10. doi: 10.1128/JVI.77.17.9204-9210.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li YP, Gottwein JM, Scheel TK, Jensen TB, Bukh J. MicroRNA-122 antagonism against hepatitis C virus genotypes 1-6 and reduced efficacy by host RNA insertion or mutations in the HCV 5′ UTR. Proc Natl Acad Sci USA. 2011;108:4991–6. doi: 10.1073/pnas.1016606108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.García-Sastre A, Evans MJ. miR-122 is more than a shield for the hepatitis C virus genome. Proc Natl Acad Sci USA. 2013;110:1571–2. doi: 10.1073/pnas.1220841110. [DOI] [PMC free article] [PubMed] [Google Scholar]