

Abstract
Interaction of the chemokine CXCL12 with its receptor CXCR4 promotes neuronal function and survival during embryonic development and throughout adulthood. Previous studies indicated that μ-opioid agonists specifically elevate neuronal levels of the protein ferritin heavy chain (FHC), which negatively regulates CXCR4 signaling and affects the neuroprotective function of the CXCL12/CXCR4 axis. Here, we determined that CXCL12/CXCR4 activity increased dendritic spine density, and also examined FHC expression and CXCR4 status in opiate abusers and patients with HIV-associated neurocognitive disorders (HAND), which is typically exacerbated by illicit drug use. Drug abusers and HIV patients with HAND had increased levels of FHC, which correlated with reduced CXCR4 activation, within cortical neurons. We confirmed these findings in a nonhuman primate model of SIV infection with morphine administration. Transfection of a CXCR4-expressing human cell line with an iron-deficient FHC mutant confirmed that increased FHC expression deregulated CXCR4 signaling and that this function of FHC was independent of iron binding. Furthermore, examination of morphine-treated rodents and isolated neurons expressing FHC shRNA revealed that FHC contributed to morphine-induced dendritic spine loss. Together, these data implicate FHC-dependent deregulation of CXCL12/CXCR4 as a contributing factor to cognitive dysfunction in neuroAIDS.
Introduction
The chemokine CXCL12 and its cognate receptor CXCR4 perform multiple functions essential for CNS development and function, including guiding migration and differentiation of neuronal precursor cells (1), regulating cell cycle proteins (2–4), and modulating NMDA receptor signaling (5). These homeostatic functions take on added significance within the context of neuroinflammatory disease, as the CXCL12/CXCR4 signaling axis aids recovery by promoting survival of mature neurons, recruitment of neural and glial progenitor cells (6–8), and neuronal-glial communication (9).
Among the numerous etiologies of neuroinflammatory disease, CXCR4 dysfunction may be particularly relevant in the context of HIV-associated neurocognitive disorders (HAND). Due to its role as an HIV coreceptor and its expression in both neuronal and non-neuronal cells, CXCR4 has been directly implicated in HIV infection and neuropathogenesis (10). The neuropathology of HAND is complex, although consistent findings associated with severe forms include elevation of inflammatory cytokines (11), high levels of excitotoxins (12), neuronal loss (13), and decreased synaptic density (14–17). HAND is both accelerated and complicated by the frequent comorbidity of illicit drug abuse, primarily intravenous opiate abuse (18–20); compared with non–drug-abusing HIV patients, drug-abusing HIV patients exhibited increased HIV encephalitis frequency (21), enhanced microglia activation (22), promotion of giant cell formation (23), and increased blood-brain barrier disruption (21).
Several studies have shown that opiates, including morphine (the major metabolite of heroin, which easily reaches the brain), alter CXCR4 function in immune and neural cells (24–26). Recently, our group has shown that morphine (and selective μ-opioid receptor [MOR] agonists, such as DAMGO) specifically impairs CXCL12/CXCR4 signaling in neurons, both in vitro and in vivo, by increasing protein levels of ferritin heavy chain (FHC), a recently described CXCR4 regulator (25, 27–29). Morphine increases the level of FHC and its association with CXCR4, thereby inhibiting CXCR4 activation and downstream prosurvival signaling pathways (27). This novel CXCR4-regulatory function of FHC occurs seemingly independent from the protein’s primary role as a critical regulator of intracellular iron levels; in this context, FHC functions by binding, oxidizing, and sequestering free reactive iron, which is stably stored within the ferritin complex formed by FHC and its partner subunit, ferritin light chain (FLC). Ferritin’s ferroxidase activity is a critical property of FHC, responsible for oxidizing free elemental Fe2+ to the less-reactive Fe3+, and is essential in controlling the production of toxic free radicals (30).
Within the CNS, FHC and FLC proteins can be found in all cell types, but their relative ratio varies between neuronal and glial cells (31–35). FHC is predominant in oligodendrocytes, microglia, and neurons. FHC production is regulated in response to changing iron levels and inflammatory processes; of particular importance to HAND is the ability of inflammatory cytokines to regulate FHC levels. Specifically, the cytokines TNF-α and IL-1β, known to be increased within HAND patients (11), stimulate FHC protein expression (36, 37). These regulatory mechanisms support the possibility that the convergence of HIV and opiate exposure would increase FHC, which may contribute to neuronal dysfunction through effects on CXCR4 signaling. Here we propose that illicit opiate abuse contributes to the exacerbated neurological impairment seen in HIV-infected individuals through FHC-dependent disruption of neuronal CXCL12/CXCR4 signaling. As a measurement of neuronal damage, we focused on loss of dendritic spines, a highly consistent and likely causative event in HAND development (14, 15, 38, 39). Importantly, chronic treatment with morphine reduces spine density and excitatory synaptic activity (40), although the mechanisms involved are complex and not yet defined. Our present observations suggest that opiates and HIV negatively regulate neuronal CXCL12/CXCR4 signaling within human and nonhuman primates and identify decreased dendritic spine density as a consequence of FHC induction. Together, these data provide the first evidence of regulation of dendritic spine by CXCL12/CXCR4 (with consequent functional changes in neuronal activity) and disclose a novel mechanism in the neuropathogenesis and neurocognitive impairment seen in drug-abusing HIV patients.
Results
Expression of FHC protein in human brain cortical neurons is altered by disease state.
In order to assess alterations in FHC and CXCR4 signaling within human HIV-infected and drug-using individuals, we obtained postmortem samples of human frontal cortex from these patient populations. Our human cohort was divided into 4 disease groups: control (no history of drug use or HIV; n = 14), drug-using only (n = 7), HIV only (n = 19), and drug-using HIV (n = 11) (see also Methods and Supplemental Tables 1 and 2; supplemental material available online with this article; doi: 10.1172/JCI70090DS1).
As revealed by immunohistochemistry and multispectral image analysis (41), in which the OD of FHC was quantified specifically within cells staining positive for the neuronal marker MAP2 (Supplemental Figure 1), basal expression of FHC protein within human cortical neurons was normally low; within the control samples, the absolute OD of the FHC/chromogen complex was mainly distributed just above the level of detection (Figure 1A). In individuals with a history of drug use, mean FHC expression was significantly increased and also showed increased variability. HIV patients, both with and without a history of drug use, also showed significantly elevated mean neuronal FHC compared with controls (Figure 1A). Interestingly, FHC expression within neurons was skewed toward high levels, particularly in the drug-using only disease group, which suggests that the degree of FHC elevation within neurons is variable and may depend on the underlying pathological cause (i.e., drug use or HIV). These results are consistent with our previous studies in primary cultures of rat cortical neurons demonstrating a specific increase in neuronal FHC mediated by MOR activation, via either morphine or DAMGO (27). Additionally, these data demonstrated an association of HIV with elevated neuronal FHC in the absence of drug use. Combined HIV infection and drug use similarly associated with a significant elevation of FHC protein, with comparable mean levels among the drug-using only, HIV only, and drug-using HIV disease groups.
Figure 1. HIV infection and opiate drug use associate with increased FHC and decreased pCXCR4 within neurons of the prefrontal cortex in human HIV-infected and drug-using patients.

OD of FHC (A), pCXCR4 (active) (B), and total CXCR4 (C) within MAP2+ neurons was quantified among control (n = 7–14), drug-using only (DU; n = 5–7), HIV only (HIV; n = 5–16), and drug-using HIV (DU/HIV; n = 3–8) patients. Approximately 100 neurons were analyzed for each patient. Mean OD for each neuron and average neuronal OD for each patient group are shown. (D) Plotting each patient’s average FHC OD to that of pCXCR4 revealed a significant inverse correlation, indicative of a negative relationship between FHC expression and CXCR4 activation (n = 16; Pearson r = –0.724; P = 0.002). (E) No significant relationship was found between average neuronal FHC OD and age at death for any patient group. *P < 0.05, **P < 0.01, ***P < 0.001.
HIV and drug use reduce CXCR4 phosphorylation in human brain cortical neurons.
Phosphorylation of the human chemokine receptor CXCR4 at Ser339 in the C terminus results from activation by the endogenous CXCR4 ligand CXCL12, and its presence indicates proper functioning of the CXCL12/CXCR4 signaling axis (27, 42, 43). In order to assess CXCR4 function in relation to our FHC findings, adjacent tissue sections were stained using an antibody that targets phosphorylated CXCR4-Ser339 (pCXCR4) (43). Comparison of pCXCR4 levels within MAP2+ neurons across disease groups revealed a significant reduction in individuals with a history of drug use or with HIV, as well as in the drug-using HIV group (Figure 1B).
Certain disease states have been shown to alter neuronal CXCR4 expression; therefore, low levels of pCXCR4 could be a consequence of low overall CXCR4 levels, rather than of inhibited receptor activation. We thus measured total neuronal CXCR4 levels, which revealed no reduction in total CXCR4 within any disease group (Figure 1C). These data indicate that the reduction of pCXCR4 seen in patients with HIV or a history of drug use is reflective of inhibited receptor activity, rather than regulated receptor expression. Interestingly, however, the HIV disease groups (both with and without a history of drug use) displayed significantly higher total CXCR4 levels (consistent with observations made in other models of HIV infection; refs. 44, 45), which suggests that the relative activation of CXCR4 in the 2 HIV groups is disproportionally lower than that in the drug-using only group. These findings suggest that an additive or synergistic disruption of neuronal CXCL12/CXCR4 signaling may exist between HIV and opiate use, despite the roughly comparable levels of pCXCR4 among the drug-using only, HIV only, and drug-using HIV disease groups.
FHC negatively correlates with pCXCR4 in vivo.
Our findings linking elevated neuronal FHC to both drug use and HIV infection, each independently, suggest that both MOR-dependent and -independent pathways regulate FHC during pathological conditions. In each of these states, however, an inverse relationship between the OD of FHC and the OD of pCXCR4 within individual patients can be expected, as a consequence of FHC’s ability to regulate CXCR4 signaling. In a parallel study of a subset of human patients from the larger cohort (i.e., the 4 best-preserved samples of each group uniformly analyzed in the same assay), neuronal expression of pCXCR4 and FHC within individual patients was plotted, which revealed a significant inverse relationship between FHC and pCXCR4 levels (n = 16; Pearson r = –0.724; P < 0.01; Figure 1D). This result was consistent with an important role of FHC in CXCR4 inhibition in vivo and supportive of our previous studies detailing this phenomenon in vitro (27). Collectively, these data showed that certain disease states, such as drug use (including opiate use) or CNS infection (including HIV), abnormally elevate neuronal FHC, resulting in disrupted function of the CXCL12/CXCR4 signaling axis.
Correlation of FHC with demographic and clinical data.
Expression of FHC protein can increase with age; therefore, age could be a potential confounding variable affecting these results. The age range of our patient cohort was 30–73 years (Figure 1E), with an average of 47.5 years. Correlations between age of death and average neuronal FHC expression levels for each patient were investigated among the 4 groups, and no significant correlation was found within any group (Figure 1E), which indicates that the elevated FHC expression is not caused by advanced age. Although this finding cannot necessarily be generalized across pediatric or geriatric patient populations, it suggests that the neurocognitive impairment seen in the largest HIV-infected age group within the United States (45–49 years old; ref. 46) is not due to age-related elevation of FHC within the CNS.
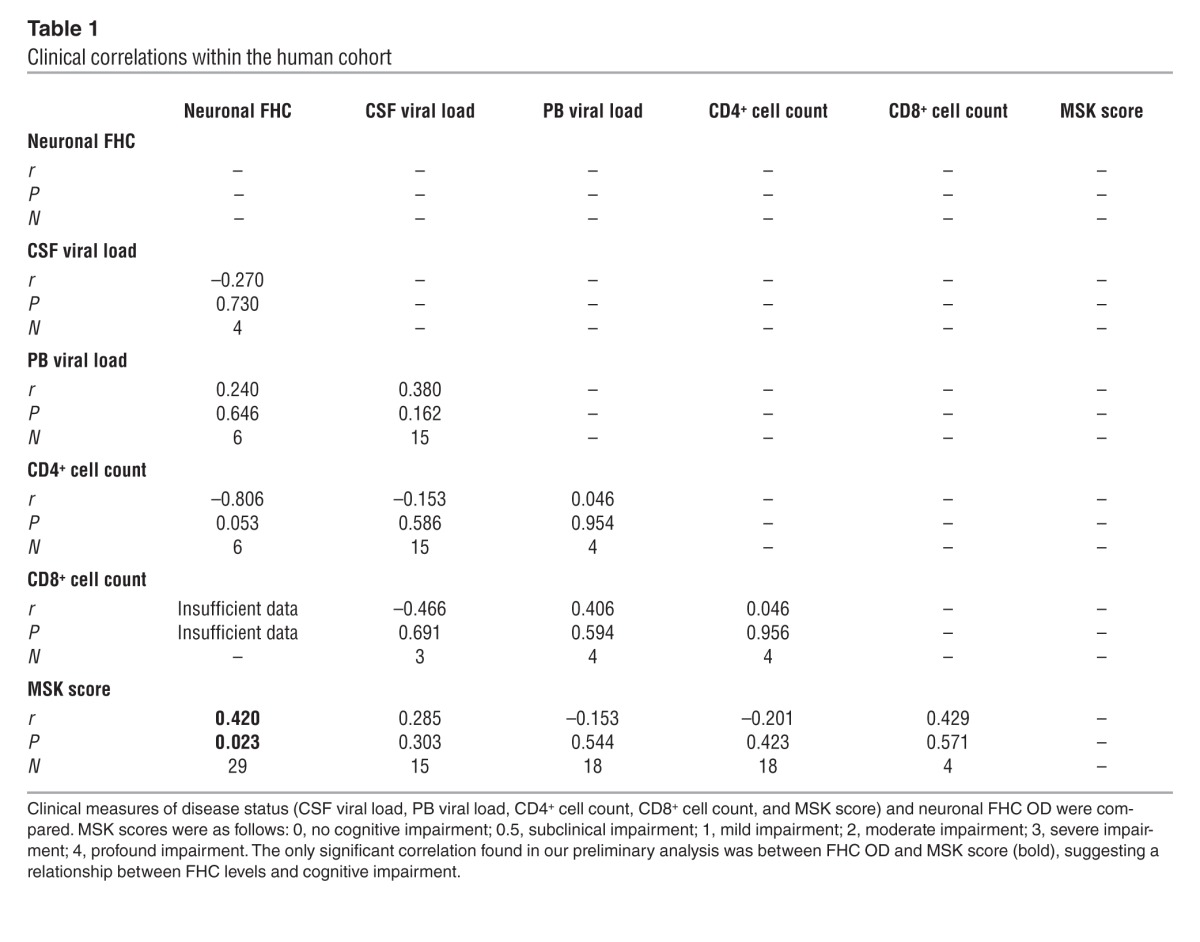
In order to determine clinical correlations between neuronal FHC and other important HIV biomarkers more broadly, a correlational matrix was generated for the patient cohort, including each patient’s neuronal FHC OD, viral load within the cerebrospinal fluid (CSF) and peripheral blood (PB), CD4+ and CD8+ cell counts, and Memorial Sloan Kettering (MSK) score, a clinical measure of cognitive impairment (Table 1). Interestingly, cognitive impairment correlated significantly with neuronal FHC, but not with any other measurement. Although these results were preliminary (due to the small number of patients) and correlational, they suggest that FHC alterations may be a sensitive indicator of HAND neuropathogenesis.
Table 1.
Clinical correlations within the human cohort
Morphine and SIV independently regulate neuronal FHC within nonhuman primates.
HAND is a highly complex pathogenic process, further complicated in drug-using patient populations by widespread polydrug abuse. Several factors that could influence the regulation of neuronal FHC, such as opiate dosage, frequency of use, and time between the last administration and death, may have additionally increased variability within our human cohort. In order to control for these inherent variables, the above-described methods were applied to a nonhuman primate model of SIV- and opiate-induced neuropathology. 4 groups of Indian rhesus macaques were studied: morphine-treated only, SIV-infected only, morphine-treated and SIV-infected, and saline-delivered control (Supplemental Table 3). Treatments began with i.v. infection using the SIV strain SIVMac251 (or vehicle), followed by a 3-month regimen of morphine (see Methods). Animals were then sacrificed, and tissue sections were processed and stained using protocols and reagents identical to those used for the human tissue.
Consistent with our human data, neuronal FHC (measured as the OD of FHC quantified specifically within MAP2+ neurons) was significantly elevated in animals that received morphine only (Figure 2A). However, the variation among the morphine-treated macaques was reduced compared with that of the human drug-using only group, likely a reflection of both greater homogeneity and strictly controlled monodrug treatment in the animal population. Importantly, the significant elevation of neuronal FHC after administration of morphine alone, without other drugs of abuse, argues strongly for a mechanistic role of opioid receptors in neuronal FHC regulation (namely, MOR, as we previously demonstrated; refs. 25, 27). SIV-infected animals also showed significant elevation of FHC within cortical neurons; however, in this group, the variability of FHC within neurons was greater than in the morphine-treated group (Figure 2A). The combined morphine-treated, SIV-infected group also showed a trend for elevated neuronal FHC, comparable to the morphine and SIV groups. Overall, the macaque model was consistent with respect to the human population and supported our hypothesis that morphine and HIV/SIV elevate neuronal FHC in vivo.
Figure 2. SIV infection and morphine treatment associate with increased FHC and decreased pCXCR4 within neurons of the prefrontal cortex in rhesus macaques.

The OD of FHC (A), pCXCR4 (active) (B), and total CXCR4 (C) within MAP2+ neurons was quantified among control (n = 7), morphine-treated only (Mor; n = 3), SIV-infected only (SIV; n = 4), and morphine-treated, SIV-infected (SIV/Mor; n = 2) macaques. 100 neurons were analyzed per animal. Mean OD for each neuron and average neuronal OD for each treatment group are shown. Note that statistical analysis excluded morphine-treated SIV animals (n = 2). (D) Plotting each animal’s average FHC OD to that of pCXCR4 revealed a significant inverse correlation, indicative of a negative relationship between FHC expression and CXCR4 activation (n = 15; Pearson r = –0.547; P = 0.035). (E) Among the 4 SIV-infected macaques, FHC and pCXCR4 levels were apparently stratified: 2 animals expressed higher levels of FHC and lower levels of pCXCR4 (red outline), and 2 animals expressed lower levels of FHC and higher levels of pCXCR4 (blue outline). Clinical data identified the animals with greater FHC as rapid progressors that retained an elevated viral load after infection, suggesting a relationship between FHC induction and disease severity. *P < 0.05, **P < 0.01.
FHC correlates with reduced pCXCR4 within the nonhuman primate model.
Also consistent with our human data, macaques treated with morphine displayed a significant reduction in pCXCR4 levels, within a comparatively small range (Figure 2B). SIV-infected animals additionally showed a significant decrease in pCXCR4; however, as with FHC, the range of pCXCR4 expression was greater than that in the morphine-treated group. Neuronal pCXCR4 also trended toward a decrease in the morphine-treated, SIV-infected group.
We also sought to measure the overall expression of neuronal CXCR4 to confirm that the decreased expression of pCXCR4 was not due to a decrease in overall neuronal CXCR4 within these animals. Levels of total neuronal CXCR4 were unchanged in the morphine-treated group, but elevated in SIV-infected animals (Figure 2C); thus, reduced pCXCR4 levels cannot be explained by a reduction in overall receptor levels. Interestingly, both human HIV infection and macaque SIV infection resulted in a significant increase in neuronal CXCR4 (Figure 1C and Figure 2C), which suggests that SIV, like HIV, causes dysregulation of neuronal CXCR4 expression independent of morphine treatment. As in the human groups, comparing macaque mean neuronal FHC expression with mean neuronal pCXCR4 expression revealed a significant negative correlation (n = 15; r = –0.547, P = 0.035; Figure 2D), further supporting the notion that pathologic alterations in pCXCR4 are associated with changes in FHC.
Correlation of neuronal FHC with disease status.
Notably, the 4 animals of the SIV-infected group were easily stratified by FHC expression: 2 showed substantially higher FHC levels than the others, suggesting the presence of 2 distinct populations within this group. Consistent with our previous data, the 2 SIV-infected animals with higher FHC levels showed lower pCXCR4 levels (Figure 2E), again supporting a correlation between neuronal FHC and pCXCR4 levels, even within a single group. Interestingly, after multispectral image analysis, the corresponding clinical data was unblinded, which revealed that the higher FHC expressers retained an elevated viral load after inoculation, whereas lower FHC expressers did not (T. Fischer-Smith, unpublished observations). Additionally, at the time of sacrifice, histopathological evidence of encephalitis was not yet present in the SIV-infected animals, which suggests that FHC elevation may precede pathological changes associated with HIV/SIV encephalitis. Together, these findings revealed a consistent association of neuronal FHC induction and concomitant CXCR4 inhibition with HIV/SIV disease progression, which may play an important role in HIV neuropathogenesis.
Role of iron-binding activity in FHC regulation of CXCR4.
The ability of FHC to regulate CXCR4 signaling, an activity that has been observed in multiple cell types, including neurons, may occur independent of the protein’s well-characterized iron-sequestering functions. We sought to determine whether this effect of FHC is a consequence of its iron-binding activity, or a truly novel function of the protein. This was addressed by comparing the CXCR4-regulatory effects of mutant FHC 222, a mutated form of FHC unable to bind or oxidize iron (47), to those of WT FHC. Human osteosarcoma (HOS) cells expressing high levels of CXCR4 were transfected with either FHC plasmid and selected to generate stable cell lines. First, in order to confirm the differential effects of these proteins on iron sequestration, free intracellular iron levels were compared using the fluorescent probe calcein. Calcein is delivered as a calcein-AM ester, which passes across the plasma membrane into cells and is cleaved by cytosolic esterases to produce membrane-impermeable calcein. The fluorescence of this trapped calcein is quenched after chelation with labile iron; therefore, the degree of quenching is a commonly used estimation of free intracellular iron levels (47). As expected, cells overexpressing WT FHC displayed greater calcein fluorescence than control cells expressing empty vector (EV), similar to cells treated with the iron chelator deferoxamine (DFO). However, calcein fluorescence was not altered in cells expressing mutant FHC 222 (Figure 3A), consistent with its inability to bind iron. The effects of the WT FHC and mutant FHC 222 proteins on CXCR4 signaling were then compared by treating each cell line with CXCL12 and measuring the activation of downstream signaling pathways. Phosphorylation of CXCR4 and activation of its downstream target Akt in response to CXCL12 were reduced both in cells expressing WT FHC and in cells expressing mutant FHC 222 (Figure 3B), which suggests that CXCR4 regulation is an independent function of FHC, distinct from the protein’s primary iron-binding activity.
Figure 3. WT FHC and mutant FHC 222 similarly inhibit CXCR4 signaling in HOS cells.

(A) WT FHC, mutant FHC 222 FHC (Mut; non–iron-binding), or EV was transfected into HOS cells (left), and the specific inability of mutant FHC 222 to bind iron was confirmed by calcein assay (right), a measure of free intracellular iron levels. Calcein fluorescence was increased by treatment with DFO or by overexpressing WT FHC, but not mutant FHC 222. (B) Effects of these proteins on CXCL12 signaling were compared by treating cells with 20 nM CXCL12 for various times. WT FHC– and mutant FHC 222–expressing cells demonstrated similar reductions in CXCL12-induced Akt and CXCR4 activation (n = 4). *P < 0.05, #P < 0.01, §P < 0.001 versus empty vector. See complete unedited blots in the supplemental material.
CXCL12/CXCR4 regulates dendritic spine density in vitro and in vivo.
The CXCL12/CXCR4 axis protects neurons from various insults involved in HIV-induced neuronal damage and ensuing neurological disorders, such as exposure to the HIV-envelope protein gp120 or excessive glutamate stimulation (5, 48). A particularly important component of HAND is reduced complexity of the dendritic arbor and synaptic density, including loss of dendritic spines, which occurs early in the disease and is thought to contribute directly to cognitive loss (14, 38, 39). Additionally, CXCL12 has been shown to regulate p21-activated kinase (PAK) in T cells (49), which contributes to morphological changes in these cells and promotes dendritic spine formation in neurons. For these reasons, we investigated a direct role of CXCL12 in the regulation of dendritic spine density, both in vitro and in vivo. Rat cortical neurons in culture were first treated with CXCL12 (in the presence of glia), and a moderate but significant increase in spine density was observed 3 hours after a single dose (Figure 4A). Importantly, this effect of CXCL12 on spine density was confirmed at multiple time points in a pure neuronal culture lacking a glial feeder layer (Figure 4B), indicative of a direct effect of CXCL12 on neurons. Additionally, this effect of CXCL12 was blocked by the specific CXCR4 antagonist AMD3100 and by pertussis toxin, an inhibitor of Gαi signaling (Supplemental Figure 2), supporting the involvement of CXCR4 and its G protein–dependent signaling.
Figure 4. CXCL12 regulates dendritic spine density in vitro and in vivo.

(A) Rat cortical neurons were cultured for 21 days in the presence of a glial feeder layer. CXCL12 treatment (20 nM, 3 hours) increased dendritic spine density (n = 12). (B) Rat cortical neurons were cultured for 21 days as a pure neuronal population. CXCL12 treatment (20 nM) time-dependently increased dendritic spines (n = 7–29). (C) CXCL12 treatment (20 nM) also increased nuclear HDAC4 levels in pure neuronal cultures, as visualized by immunocytochemistry at 30 minutes and quantified over time (n = 12–34). Similar results were obtained by Western blot using nuclear extracts from neurons. (D) After 8 days in culture, transfection of a pure neuronal population with siRNA specifically decreased HDAC4, but not HDAC1, 36 hours after transfection. siRNA-mediated HDAC4 deficiency completely blocked the effects of CXCL12 on dendritic spines (n = 8–17). (E) The CXCR4 antagonist AMD3100 (1 μg i.c.v.) was injected into the left lateral ventricle of 3-week-old rats. 6 hours after injection, animals were sacrificed, and layer II/III pyramidal neurons were analyzed, revealing reduced total dendritic spine density (n = 15). (F) This effect of AMD3100 was similarly confirmed in a prolonged treatment model: implantation of 3-week-old rats with osmotic pumps delivering constant levels of AMD3100 (0.75 μg/h) for 4 days decreased total dendritic spine density (n = 15). *P < 0.05; **P < 0.01; ***P < 0.001. Scale bars: 5 μm (A); 10 μm (C, E, and F). See complete unedited blots in the supplemental material.
Previous studies have identified an important role of histone deacetylases (HDACs) in mediating the neuroprotective functions of CXCR4 activation (5). A particularly important role of HDAC4 in the regulation of dendritic spines by CXCL12/CXCR4 was suggested by prior findings that this HDAC promotes neuronal survival (50, 51) and synaptic plasticity (52) and that the transcription factor myocyte enhancer factor 2 (MEF2), the major nuclear target of HDAC4, is directly involved in structural synapse regulation (52, 53). Like the other class II HDACs, activity of HDAC4 is regulated by nuclear import/export; therefore, we hypothesized that increased nuclear HDAC4 levels mediate the effects of CXCL12 on spine density. Consistent with this model, neurons cultured as a pure population and treated with CXCL12 exhibited increased nuclear HDAC4 levels, as shown by immunocytochemistry and Western blot (Figure 4C). In order to assess the involvement of HDAC4 in CXCL12-induced spine alterations, HDAC4 levels were reduced by transfecting neurons with HDAC4-specific siRNA (Figure 4D). The resulting HDAC4 deficiency completely prevented the effects of CXCL12 on spine density, indicative of a crucial role of this HDAC in CXCL12-induced spine regulation. Additional studies are in progress to determine the mechanisms involved.
In order to confirm the spine-regulating effect of CXCL12 in vivo, in the context of opiate abuse and decreased CXCR4 signaling, we administered the CXCR4 antagonist AMD3100 to rats via i.c.v. injection. 6 hours after a single 1-μg injection of this drug, dendritic spine densities were analyzed among layer II/III pyramidal neurons of the prefrontal cortex, by reconstructing individual neurons in their entirety. This single dose of AMD3100 significantly decreased spine density (Figure 4E), which indicates that the basal level of CXCR4 signaling contributes to the regulation of spine density in vivo. In order to investigate the effects of more prolonged CXCR4 inhibition, as would occur with long-term opiate abuse, we implanted osmotic pumps to deliver constant levels of AMD3100 into the left lateral ventricle of the brain for 4 days, which similarly decreased dendritic spine density (Figure 4F). Together, these data identify the endogenous CXCL12/CXCR4 axis as a novel regulator of dendritic spines and suggest a specific neuropathological outcome of CXCR4 dysregulation highly relevant to opiate abuse and neuroAIDS.
To determine the functional consequences of spine density alterations, we used whole-cell patch-clamp electrophysiology to record from layer II/III pyramidal neurons in the prefrontal cortex. First, in current-clamp mode, we recorded action potentials (APs) in response to varying step currents in slices from animals that received i.c.v. injections of AMD3100 or vehicle, or from slices that had been exposed ex vivo to CXCL12 or vehicle. For both, there was no drug effect on spike number generation (AMD3100, P = 0.57; CXCL12, P = 0.71; data not shown). Furthermore, there was no drug effect on resting membrane potential, AP threshold, AP peak amplitude, AP half-width, 20%–80% rise time, and afterhyperpolarization (AHP) (Supplemental Table 4). Thus, drug exposure did not significantly alter cell health or basic functional properties. We next recorded spontaneous and miniature excitatory postsynaptic currents (sEPSCs and mEPSCs, respectively) while holding the cell at –70 mV. AMD3100 had no effect on the frequency (vehicle, 4.22 ± 0.70 Hz; AMD3100, 3.84 ± 0.65 Hz; P = 0.69; n = 6) or amplitude (vehicle, 18.87 ± 1.41 pA; AMD3100, 16.79 ± 0.80 pA; P = 0.10; n = 6) of sEPSCs (Figure 5A), which presumably result from APs of presynaptic neurons as well as spontaneous release of neurotransmitter at synapses. However, AMD3100 exposure significantly reduced the frequency of mEPSCs (vehicle, 3.47 ± 0.41 Hz; AMD3100, 2.27 ± 0.33 Hz; P < 0.04; n = 10) and induced a trend toward reduced mEPSC amplitude (vehicle, 18.3 ± 0.52 pA; AMD3100, 16.55 ± 0.55 pA; P = 0.068; n = 9) (Figure 5B). mEPSCs are the result of individual synaptic events, and thus their frequency may be an indication of the number of functional synapses. Therefore, these data are consistent with a reduced number of synapses, which would be expected with reduced dendritic spine density after AMD3100 treatment. Furthermore, the trend toward decreased mEPSC amplitude may indicate changes to postsynaptic receptors, in line with our previous studies (5), but this prediction requires further investigation. Similarly, we recorded sEPSCs and mEPSCs after exposing slices to CXCL12. We found no difference in the frequency of sEPSCs (vehicle, 2.07 ± 0.36 Hz; CXCL12, 1.85 ± 0.41 Hz; P = 0.64; n = 6 and 7, respectively) or mEPSCs (vehicle, 1.59 ± 0.24 Hz; CXCL12, 1.43 ± 0.25 Hz; P = 0.66; n = 8) (Figure 5, C and D). This lack of effect may be the result of an altered time course of CXCL12 on slices versus primary culture, but remains to be tested. Another factor is the potential effect of CXCL12 on GABAergic neurotransmission (54, 55), which may indirectly affect dendritic spines (56); GABA receptors are inhibited by picrotoxin present in the recording solution, but not in the culture studies.
Figure 5. Regulation of the CXCR4 axis alters the frequency and amplitude of EPSCs of layer II/III pyramidal neurons.

(A) AMD3100 injection i.c.v. had no effect on sEPSC frequency or amplitude (sample data not shown). (B) Sample traces of mEPSCs recorded at –70 mV in the presence of tetrodotoxin (1 μM) and picrotoxin (100 μM) in layer II/III pyramidal neurons from vehicle- and AMD3100-exposed animals. Quantification revealed AMD3100 exposure significantly decreased mEPSC frequency, but not amplitude. (C) Incubation of prefrontal cortical slices with CXCL12 (20 nM, 3 hours) induced no change in sEPSC frequency, but significantly increased sEPSC amplitude (sample data not shown). (D) Sample traces of mEPSCs recorded at –70 mV in the presence of tetrodotoxin (1 μM) and picrotoxin (100 μM) in layer II/III pyramidal neurons from vehicle- and CXCL12-exposed slices. Quantification revealed CXCL12 exposure had no significant effect on mEPSC frequency or amplitude. *P < 0.04; #P < 0.03.
In contrast, CXCL12 significantly increased sEPSC amplitude (vehicle, 23.34 ± 1.07 pA; CXCL12, 25.32 ± 0.43 pA; P = 0.027; n = 6 and 7, respectively), and there was a trend toward increased mEPSC amplitude (vehicle, 20.873 ± 1.21 pA; CXCL12, 25.25 ± 1.02 pA; P = 0.051; n = 8) (Figure 5, C and D). Together, these data suggest that the CXCL12/CXCR4 axis regulates functional connectivity of layer II/III pyramidal neurons and that stimulation of this receptor by the endogenous ligand may alter both spine density and synaptic strength.
Morphine decreases dendritic spine density through effects on neuronal FHC.
Opiates such as morphine have previously been shown to reduce dendritic spine density and synaptic plasticity (40), which may contribute to cognitive dysfunction among vulnerable populations, such as HIV-infected opiate abusers. Furthermore, our findings that constitutive CXCR4 signaling was important in regulating spine number and neuronal activity suggest a specific FHC-mediated pathway by which opiates alter neuronal function. We therefore explored the role of FHC in opiate-induced spine loss, first by confirming this effect both in vitro and in vivo. Consistent with our previous findings (27), morphine treatment significantly increased FHC levels in cultured rat cortical neurons (Figure 6A). Interestingly, this effect was associated with a significant decrease in the number of dendritic spines (Figure 6B), in line with reports showing collapse of spine and reduced mEPSCs in neurons exposed to morphine (40). We also observed a similar effect in vivo after prolonged morphine treatment: rats were implanted with slow-release morphine pellets that delivered morphine continuously for 4 days, which increased FHC levels in brain lysates and significantly decreased dendritic spine densities in the same animals (Figure 6, C and D). In order to evaluate a direct effect of FHC on spine density, we then transfected neurons to overexpress FHC. These neurons expressing high levels of FHC were shown to contain fewer dendritic spines (Figure 6E), indicative of a pathological role of FHC in neuronal function and survival. Additionally, we explored the role of FHC in morphine-induced spine loss by using FHC-specific shRNA. The ability of morphine to regulate spines was blocked in FHC-deficient neurons (Figure 6F), suggestive of an important role of FHC in neuronal dysfunction resulting from opiate use.
Figure 6. Morphine regulates dendritic spine density through effects on neuronal FHC.

(A and B) Rat cortical neurons cultured for 21 days were treated with morphine (1 μm, 24 hours), which both (A) increased FHC levels (n = 4) and (B) decreased dendritic spine density (n = 12). (C and D) Similarly, 3-week-old rats treated with sustained-release morphine pellets for 4 days exhibited (C) increased brain FHC levels (n = 4) and (D) decreased total dendritic spine density (n = 12). (E) FHC overexpression in the absence of morphine also decreased dendritic spine density, as measured in cultured rat cortical neurons transfected with a FHC-expressing plasmid or empty vector (n = 16). (F) The ability of morphine to regulate dendritic spine density was blocked in FHC-deficient neurons, as shown using FHC-specific or scrambled control shRNA. *P < 0.05; ***P < 0.001. Scale bars: 5 μm (B and E); 10 μm (D). See complete unedited blots in the supplemental material.
Discussion
These studies provide the first systematic characterization of FHC expression in human neurons and identify a novel mechanism contributing to neuronal dysfunction caused by opiate use and HIV infection. We observed increased levels of FHC within neurons of the prefrontal cortex associated with both opiate drug use and HIV/SIV infection in human and rhesus macaque tissue. FHC levels across treatment groups were shown to negatively correlate with pCXCR4 levels, which suggests that pathological induction of FHC by either opiate exposure or viral infection similarly inhibits CXCR4 signaling. We also identified a novel neuroprotective activity of CXCR4 in its ability to increase dendritic spine numbers, and we implicated FHC in morphine-induced spine loss, an important pathological component of HAND.
Our previous studies demonstrated the regulation of FHC by morphine and the specific involvement of MORs in this effect (27, 57); in vivo or in vitro deletion of FHC in neurons abolished the inhibitory action of morphine exposure on CXCL12/CXCR4 signaling, emphasizing the importance of the FHC/CXCR4 interaction promoted by morphine. Here, we extend these findings to show a similar effect caused by HIV infection in the absence of opiate exposure. The combined effect of morphine and HIV/SIV infection on FHC levels was similar to that caused by each condition alone, suggestive of no additive effect; the mechanisms involved in these pathways may converge or initiate feedback, thereby preventing further increases in FHC. It is also possible that HIV and opiates in combination prolongs the duration of FHC changes induced by opiates alone (27). Indeed, ongoing studies in our laboratory suggest that IL-1β and TNF-α are responsible for neuronal FHC induction by HIV and that both transcriptional and posttranscriptional events are involved in this process. These experiments showed greater levels of FHC in cortical neurons treated with IL-1β (or TNF-α) and a similar — but glia-dependent — increase in neuronal cultures exposed to HIVgp120, which was inhibited by IL-1β–neutralizing antibodies or receptor antagonists (L. Festa, unpublished observations). In any case, the decrease in neuronal pCXCR4 levels associated with HIV and SIV infection occurred despite increases in total CXCR4 levels (Figures 1 and 2). These observations suggest a greater degree of receptor inhibition as well as a potentially additive effect of HIV infection and opiate use on CXCR4 signaling, ultimately rendering neurons more vulnerable to insults. Our data also showed important correlations between neuronal FHC levels and cognitive function: among HIV patients, neuronal FHC levels correlated significantly with MSK scores, a clinical measure of cognitive impairment (58, 59). Additionally, the correlations between disease severity and FHC/pCXCR4 alterations observed among SIV-infected macaques suggested that FHC alterations, which could potentially be detected in CSF or peripheral blood samples, may be a useful biomarker to assess HIV neuropathology. However, this requires further investigation both in humans and in nonhuman models.
Another important finding of these studies was the iron independence of FHC’s CXCR4-regulatory activity (Figure 3). The predominant function of this protein is to bind, oxidize, and sequester free reactive iron, and alterations of this critical activity could have severe consequences on cellular iron availability and oxidative stress. A mechanistic distinction between CXCR4 regulation and iron-binding activity of FHC implies the ability to independently target these functions: enhancing CXCR4 signaling by preventing its association with FHC should be possible without necessarily disrupting FHC’s critical iron-binding functions. An intriguing target for specifically blocking this association may be phosphorylation of FHC at Ser178, which is essential for its association with CXCR4 (29) and may not affect its iron-binding activity.
Our finding that CXCL12/CXCR4 signaling positively regulated the density of dendritic spines (Figure 4) represents a novel mechanism by which this chemokine/receptor pair promotes the survival and function of mature neurons. The evidence accumulated thus far suggests that this effect depends on G protein–mediated CXCR4 signaling (Supplemental Figure 2), HDAC4 expression (Figure 4, C and D), and interaction of HDAC4 with the transcription factor MEF2 (data not shown), but further investigation is necessary to identify the specific pathways involved. Due to their critical roles in postsynaptic calcium buffering and synaptic plasticity, spines are essential and dynamic components of neuronal physiology. Previous studies have demonstrated neuroprotective effects of CXCL12/CXCR4 in regulating the antiapoptotic kinase Akt (60) and various cell cycle proteins, whose aberrant expression in postmitotic neurons leads to cell death (2–4). This signaling pair also decreases excitotoxicity by regulating the subunit composition of extrasynaptic NMDA receptors (5) and promotes tissue repair by regulating differentiation and migration of progenitor cells in models of multiple sclerosis (7, 61) and ischemia (8, 62). Additionally, CXCL12 has been shown to block a reduction in hippocampal dendritic spines caused by the neurotoxin amyloid β (63), although this study did not report any changes in spine density in the absence of amyloid β. Here, we showed that loss of basal CXCR4 signaling in vivo resulted in reduced cortical spine density. Concomitant electrophysiological alterations confirmed the functional relevance of these structural changes. In line with these data, stimulation of CXCR4 by exogenous CXCL12 increased spine density in cultured neurons. A reduction in this basal CXCR4 signaling likely underlies FHC-mediated spine loss, as we observed after FHC overexpression or induction by morphine (Figure 6). The loss of CXCR4-mediated signaling may be particularly detrimental in HIV patients, due to the reported role of specific MMPs in HIV-associated neurodegeneration (64). Proteolytic processing of CXCL12 by MMP2 generates a cleaved form of the chemokine (CXCL12 5-67), which is not only deficient in CXCR4 binding, but also triggers neurotoxic pathways via the CXCR3 receptor (65). Opiates could further exacerbate this process by influencing MMP release from microglia, although this remains to be tested. On the other hand, our present data showed that CXCL12-induced changes in spine density mainly depend on engagement of neuronal CXCR4, highlighting the role of the FHC/CXCR4 interaction.
The extent to which FHC induction alters the HIV coreceptor function of CXCR4 was not investigated here. However, previous studies show that prolonged exposure to morphine or DAMGO increases CXCR4 and CCR5 expression in T cells and monocytes, respectively, resulting in a greater number of HIV-infected cells (66). In line with this, studies in SIV-infected macaques indicate that prolonged morphine exposure augments viral replication and SIV-infected cell number (67). While it is generally accepted that opiates accelerate HIV neuropathogenesis, the mechanisms are still not completely understood.
In conclusion, our implication of FHC in morphine-induced spine loss both identifies a pathway involved in this known effect (40, 68, 69) and elucidates an important contribution of constitutive CXCL12/CXCR4 signaling to basic neuronal function. Together, our findings support a model in which elevated neuronal FHC resulting from opiate use or HIV infection contributes to neuronal dysfunction by altering the neuroprotective actions of the CXCL12/CXCR4 axis. Future studies aiming to characterize the molecular mechanisms involved in the FHC/CXCR4 interaction and its regulation by opiates may lead to important therapeutic discoveries in the field of HAND and other neuropathologies.
Methods
Human population.
Postmortem human brain tissue was acquired from the National NeuroAIDS Tissue Consortium (NNTC). The human population studied (N = 51) was divided among individuals with a history of drug abuse (including opiate abuse) and/or documented HIV, with a degree of HIV-associated neurological impairment (59) ranging from normal (MSK 0) to profoundly impaired (MSK 4) (Supplemental Tables 1 and 2). The tissue acquisition protocol was as previously described (70). Briefly, within the frontal cortex, Broadman area 8 and underlying white matter was removed at autopsy. Patient information was collected regarding HIV status, neurocognitive impairment, and relevant clinical measures pertinent to disease progression and comorbidities. The following demographic and clinical information was obtained: age at death, race, sex, HIV status, CD4+ cell count, CD8+ cell count, PB viral load, CSF viral load, neurocognitive impairment, and drug abuse history (opiates, cocaine, stimulants, cannabis, methamphetamine, sedatives, hallucinogens, and alcohol). Subjects were studied longitudinally with neurologic, psychiatric, and neurocognitive evaluations at 6-month intervals. Psychiatric disorders were determined in the context of underlying substance abuse using the Psychiatric Research Interview for Substance and Mental Disorders (PRISM) scale (71). The majority of patients were undergoing combination antiretroviral therapy (cART) regimens; only a small number were on a structured treatment interruption (70). Additional control samples (matching the diseased population, at least for age and PMI) were obtained from the DUCOM tissue procurement facility. Variability of FHC and CXCR4 staining within the normal subjects was comparable.
Nonhuman primate population.
20 Indian rhesus macaques, housed and treated at BIOQUAL Inc., were studied in conjunction with the human cohort to determine the effects of a fixed dosage regimen of i.v. morphine with and without concurrent SIV infection (Supplemental Table 3). After quarantine, all animals received a single 0.5-ml tetanus vaccine and quarterly TB tests. TB tests were given 3 times, biweekly, accompanied with doses of praziquantel and ivermectin for deworming. Animals were given ad libitum monkey chow (Harlan Tekland chow no. 8714) supplemented with fruit, vegetables, and feeding devices consisting of peanut butter and cereals. All treatment group animals received 5 Prima Treats to supplement dietary intake. Treatment groups were as follows: control (n = 11), SIV only (n = 4), morphine only (n = 3), both SIV inoculated and morphine administered (n = 2). The neurovirulent SIV viral strain SIVMac251 was diluted in 0.9% NaCl and administered i.v. through the saphenous vein, at a final dose of 5 mg/kg. Morphine treatment began with a dose of 3 mg/kg body weight, administered 3 times daily for 2 weeks. This dose was increased to 5 mg/kg, administered 3 times daily, for the remaining time (total treatment time, 90–110 days). Animals were then sacrificed, and the frontal cortex was removed, fixed in formalin, and paraffin embedded for immunohistochemistry.
Immunohistochemistry.
Postmortem frontal cortex tissue of both humans and rhesus macaques was formalin fixed and paraffin embedded for immunohistochemistry analyses. The dual staining immunohistochemistry protocol was previously described in greater detail (72). Tissue was sequentially dual-stained with the neuronal marker MAP2 (Chemicon, 1:250) and the protein of interest: FHC (Abcam, 1:50), pCXCR4 (Abcam, 1:50), or CXCR4 (1:100). After rehydration, endogenous peroxidase activity was quenched with hydrogen peroxidase and methanol for 30 minutes at room temperature, followed by antigen retrieval in a citrate buffer at 95°C for 1 hour (DAKO). Tissue was blocked with avidin-biotin (Vector Labs) and blocking buffer before incubation with primary antibody overnight at 4°C in a humidity chamber. Secondary antibody incubation (Jackson ImmunoResearch, 1:250) was performed for 2 hours at room temperature, then amplified with the avidin-biotin complex kit (Vector Labs). Visualization of MAP2 was performed with hydrogen peroxidase–conjugated NovaRed (Vector Labs), while the additional protein of interest (FHC, pCXCR4, or CXCR4) was visualized with alkaline phosphatase–conjugated VectorBlue (Vector Labs).
Multispectral imaging.
Imaging and analysis of dual-stained human and rhesus macaque cortex was previously described in detail (72). Briefly, brightfield microscopy coupled with multispectral image analysis was performed to identify individual MAP2+ neurons within the cortex and measure the absolute OD of the given antigen of interest (FHC, CXCR4, or pCXCR4) within each individual MAP2+ neuron. The absolute OD is a semiquantitative measure of protein expression on a cell-by-cell basis.
Neuronal cultures.
Rat cortical neurons were cultured in the presence of a glial feeder layer, using the previously described bilaminar cell culture model (73). Due to the presence of glia, this culture system largely preserves the in vivo mechanisms of neuronal growth and differentiation, but also allows for direct studies on neurons, which were separated from glia at the time of analysis. Selected experiments were performed using glia-free neuronal cultures as indicated, using previously described protocols (27). Neurons were transfected at the time of plating by Nucleofection (Amaxa), or after 8 days in culture by lipofectamine (Invitrogen), as described previously (4, 27). Unless otherwise indicated, all experiments were conducted at 21 DIV. HDAC4 siRNA and scrambled control siRNA were obtained from Ambion (catalog nos. s170677 and 4390843, respectively), and FHC shRNA and scrambled control shRNA were from GenScript (custom order; shRNA vector also expressed GFP).
HOS cells.
CXCR4-expressing HOS cells (HOS.CXCR4) were obtained from N. Landau through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (Rockville, Maryland, USA). Cells were grown in DMEM containing 10% FBS, 50 μg/ml gentamicin, and 1 μg/ml puromycin. Experiments were conducted at approximately 75% confluence, and cells were serum-starved for 24 hours before treatment. Transfection was performed by Nucleofection, and transfected cells were selected with 400 μg/ml G-418 to generate stable cell lines.
Western blot.
Total and nuclear extracts were obtained by standard protocols, as we previously reported (27, 74), with minor modifications. Briefly, for total extracts, cells or tissues were lysed in lysis buffer containing 150 mM NaCl, 50 mM Tris, 0.5% Na deoxycholate, 0.1% SDS, 10 mM Na4P2O7, 5 mM EDTA, 1% Triton-X, 1 mM DTT, and protease/phosphatase inhibitors. Equal amounts of protein (40–50 μg) were loaded into each lane for SDS-PAGE and transferred to PVDF membranes for immunoblotting. For nuclear extracts, a hypotonic buffer solution (20 mM Tris-HCl, pH 7.4; 10 mM NaCl; and 3 mM MgCl2) was used to isolate nuclei; 0.05% NP-40 was added toward the end of the incubation period. Nuclei were then lysed using a triple detergent buffer (100 mM Tris, pH 7.4; 2 mM Na3VO4; 100 mM NaCl; 1% Triton X-100; 1 mM EDTA; 10% glycerol; 1 mM EGTA; 0.1% SDS; 1 mM NaF; 0.5% deoxycholate; and 20 nM Na4P2O7). Protease and phosphatase inhibitors were added to both buffers. Histone H3 was used as a nuclear marker; 20 μg protein was loaded into each lane. Antibodies used were as follows: anti-FHC (H-53, Santa Cruz Biotechnology, 1:1,000), anti-pAkt (Ser473, Cell Signaling Technology, 1:2,000), anti-pCXCR4 (Ser339, Abcam, 1:1,000), anti-HDAC4 (A-4004, Epigentek, 1:1,000), anti-HDAC1 (10E2, Santa Cruz Biotechnology, 1:1,000), anti–β-actin (Sigma-Aldrich, 1:5,000), and anti–histone H3 (9715, Cell Signaling, 1:1,000).
Calcein assay.
HOS cells transfected with EV, WT FHC, or mutant FHC 222 mutant were plated in 96-well plates at 20,000 cells per well. After 24 hours of serum starvation, vehicle or the iron chelator DFO (100 μM) were added to the untreated EV control wells and to the DFO-treated positive control wells. 6 hours later, all cells were washed with PBS and loaded with the iron-binding dye calcein (200 μg/ml) for 20 minutes. Cells were then washed again in PBS, and calcein fluorescence was determined (excitation 485 nm, emission 535 nm) as a measure of the labile iron pool (47).
Dendritic spine analysis.
For in vitro experiments, neurons were cultured in the presence of glia for 21 days, then fixed and stained for MAP2 (Millipore, 1:1,000; detected with goat anti-rabbit Alexa Fluor 568, Invitrogen, 1:250) and counterstained with phalloidin (Invitrogen, 25 μg/ml). For FHC shRNA experiments, neurons were identified and analyzed using incorporated GFP expression. Images were acquired with a Zeiss LSM5 laser scanning confocal microscope using a ×63 objective. Dendritic spines were defined as phalloidin- or GFP-positive protrusions clearly connected to the dendrite by a thin shaft. For each neuron, 3–4 segments totaling at least 200 μM in length were added to measure spine density, and each neuron was represented as a single data point. Each experiment included 3–4 coverslips from each of 2 or more separate preparations. For in vivo experiments, dendritic spines were visualized using a Golgi stain kit according to the manufacturer’s instructions (FD NeuroTechnologies). Brains were rapidly removed, rinsed in H2O, and incubated in the Golgi stain for 2 weeks. Tissue was then cryoprotected in sucrose at 4°C for 7 days and frozen in isopentane. Brains were sectioned by cryostat at a thickness of 180 μm and mounted on gelatin-coated microscope slides. Sections were allowed to dry overnight, and a signal intensification step was performed before dehydrating, clearing in xylene, and coverslipping with Permount (Fisher Scientific). Spine analysis was performed using Neurolucida software equipped with a ×100 objective (Supplemental Figure 3A). Pyramidal neurons in layer II/III prefrontal cortex were identified and reconstructed in their entirety, and total spine density for each neuron was reported as a single data point. 3 neurons per brain were analyzed, and 4–5 brains per experimental group were used.
Immunocytochemistry.
Neurons were fixed in 4% paraformaldehyde, stained with MAP2 (Millipore, 1:1,000) and HDAC4 (Epigentek, 1:150), and detected with cy2- and cy3-conjugated secondary antibodies. Cell nuclei were visualized with Hoechst (Invitrogen, 1:2,000), and images were acquired with a Zeiss LSM5 laser scanning confocal microscope (Imager.Z1m), mounting ×10, ×20, ×40, and ×63 objectives.
Morphine pellet implantation.
Slow-release morphine or placebo pellets were implanted s.c. in 3-week-old rats, following an escalating dose protocol. Animals were anesthetized with a mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg), and a small incision was made through the skin to produce a small pocket under the skin. A single pellet of morphine (25 mg) or vehicle was placed inside, and the incision was closed using 7-mm wound clips. 48 hours after the initial surgery, 2 additional pellets of morphine or vehicle were implanted in a similar manner. 48 hours after the second surgery, animals were sacrificed, and brain tissue was rapidly removed and processed.
i.c.v. injections.
3-week-old rats received injections of AMD3100 (1 μg in 5 μl) or PBS into the left lateral ventricle. Animals were anesthetized with isofluorane, and stereotaxic coordinates were zeroed to the interaural line. The following coordinates were used to locate the lateral ventricle: 5.5 mm anterior, 1.4 mm lateral, 6.4 mm dorsal. 5 μl drug or PBS was mechanically injected over 10 minutes through a 33-gauge Hamilton needle. 6 hours after each injection, animals were sacrificed, and brains were rapidly removed and processed. Correct targeting of the lateral ventricle was verified by a similar injection of 2% Evans blue dye (Supplemental Figure 3B).
Osmotic pump implantation.
Micro-osmotic pumps (Alzet), together with brain infusion kits (Alzet, Brain Infusion Kit 2), were implanted in 3-week-old rats to deliver AMD3100 (0.75 μg/h) or PBS to the left lateral ventricle of the brain for a prolonged time period (4 days). The surgical procedure was similar to that of the i.c.v. injections above, except a cannula needle and osmotic pump replaced the Hamilton needle and syringe. The pump was placed in a subcutaneous pocket in the midscapular area, and a cannula was attached to the skull using a cyanoacrylate adhesive (Alzet). After 96 hours of continuous drug delivery (0.5 μl/h), animals were sacrificed, and brains were rapidly removed and processed.
Electrophysiology.
Experiments were conducted as previously described (75). For AMD3100 experiments, 6 hours after i.c.v. infusion of AMD3100 (1 μg in 5 μl) or PBS, animals were anesthetized with euthasol (0.2 ml/kg i.p.) and perfused with ice-cold, oxygenated artificial CSF (ACSF; 124 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 2 mM CaCl2, 1 mM MgSO4, 26 mM NaHCO3, and 10 mM dextrose, pH 7.4). Brains were removed, and horizontal sections through the prefrontal cortex were cut (300 μm) into an ice-cold bath of oxygenated ACSF using a vibrotome tissue slicer (Leica Microsystems). Slices were transferred to a holding chamber, submerged in oxygenated ACSF at 35°C for 1 hour, and then kept at room temperature until use for recordings. For CXCL12 experiments, prefrontal slices were generated as above, except that slices were transferred into a holding chamber containing vehicle or 20 nM CXCL12 and incubated for 3 hours before recordings began. Slices were placed into a recording chamber mounted on an Olympus upright microscope (BX51), where they were bathed in oxygenated ACSF at a flow rate of 2–3 ml/min. Neurons were visualized with infrared differential interference video microscopy. Somatic whole-cell voltage-clamp recordings were obtained from visually identified layer II/III pyramidal cells using patch electrodes with an open tip resistance of 5–7 M⁄ and filled with a potassium gluconate internal solution (120 mM potassium gluconate, 20 mM KCl, 4 mM ATP-Na, 0.3 mM Na2GTP, 5 mM Na-phosphocreatine, 0.1 mM EGTA, 10 mM HEPES, pH 7.3, 305 mosmol/l). Whole-cell current-clamp was used to record APs in response to varying step currents from –300 pA to +400 pA with a 50-pA increment. The recording was then switched into voltage-clamp mode with membrane potentials held at –70 mV in the presence of picrotoxin (100 μM) to record sEPSCs, or in the presence of both picrotoxin and tetrodotoxin (1.0 μM) to record mEPSCs.
All experiments were conducted with an Axon MultiClamp 700B amplifier (Molecular Devices), and data were acquired using pCLAMP 9.2 software and analyzed using Clampfit 10.2 (Molecular Devices). A typical sEPSC/mEPSC was selected to create a sample template for the event detections within a 5-minute data period. The frequency (number of events) and amplitude of the individual events were examined, with the threshold set at the medium level (i.e., 5) in Clampfit. Detected events were then visually inspected to ensure specificity. APs recorded in current-clamp mode were used to measure resting membrane potential, input resistance, AP threshold, AP half-width, peak AP amplitude, and AHP size. Additionally, the number of APs produced in response to varying step currents was counted, and repeated-measures ANOVA was used to determine statistical significance. All other data were analyzed with 2-tailed Student’s t test with 95% confidence and presented as mean ± SEM.
Materials.
Cell culture media were purchased from Invitrogen, and Holtzman rats for both cell culture and in vivo experiments were obtained from Harlan. Morphine sulfate, DFO, AMD3100, and pertussis toxin were purchased from Sigma-Aldrich, and recombinant CXCL12 was from Peprotech. Morphine pellets were obtained from NIDA, and mutant/WT FHC plasmids were a gift from P. Arosio (University of Brescia, Brescia, Italy).
Statistics.
Data are represented as mean ± SEM. Both human and nonhuman primate databases were generated using the SPSS statistical package (IBM). Mean differences in OD among human disease groups were calculated by 1-way ANOVA followed by Bonferroni’s multiple-comparison test. Rhesus macaque treatment groups were analyzed by unpaired 2-tailed t test. Graphpad Prism was used for these analyses; a P value less than 0.05 was considered significant. The correlation between FHC and pCXCR4 ODs (Figure 1D) and between average FHC OD and age at death (Figure 1E) were calculated as Pearson r using Graphpad Prism and SPSS, respectively. Western blot densitometry and dendritic spine densities were compared by 1-way ANOVA followed by Newman-Keuls post-hoc test, unless otherwise indicated.
Study approval.
Rat studies were reviewed and approved by the Drexel University IACUC. Human brain tissue studies were reviewed and approved by the Drexel University IRB. All subjects were required to sign informed consent forms. Macaque studies were reviewed and approved by the Bioqual Inc. Animal Care and Use Committee (Rockville, Maryland, USA).
Supplementary Material
Acknowledgments
The authors thank Paolo Arosio and Maura Poli (University of Brescia) for the mutant and WT FHC plasmids; Padmavathi Ponnuru and Saori Shimizu (Drexel University) for help setting up the calcein assay and MAP2/CXCR4 immunostaining, respectively; and Alessandro Fatatis (Drexel University) and David Sulzer (Columbia University) for discussion and critical reading of the manuscript. This work was supported by NIH grants DA15014 and DA32444 (to O. Meucci); DA023860 (to J. Rappaport and T. Fischer-Smith); and MH 085666 (to W-.J. Gao). J. Pitcher and A. Abt were fellows of the “Interdisciplinary and Translational Research Training in neuroAIDS” (T32-MH078795). M. Snyder was the recipient of a NARSAD Young Investigator Award from the Brain and Behavior Research Foundation.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(2):656–669. doi:10.1172/JCI70090.
Jaclyn Myers’s present address is: The Wistar Institute Vaccine Center, Philadelphia, Pennsylvania, USA.
References
- 1.Tiveron MC, Cremer H. CXCL12/CXCR4 signalling in neuronal cell migration. Curr Opin Neurobiol. 2008;18(3):237–244. doi: 10.1016/j.conb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Shimizu S, Khan MZ, Hippensteel RL, Parkar A, Raghupathi R, Meucci O. Role of the transcription factor E2F1 in CXCR4-mediated neurotoxicity and HIV neuropathology. Neurobiol Dis. 2007;25(1):17–26. doi: 10.1016/j.nbd.2006.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan MZ, Brandimarti R, Musser BJ, Resue DM, Fatatis A, Meucci O. The chemokine receptor CXCR4 regulates cell-cycle proteins in neurons. J Neurovirol. 2003;9(3):300–314. doi: 10.1080/13550280390201010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan MZ, Brandimarti R, Shimizu S, Nicolai J, Crowe E, Meucci O. The chemokine CXCL12 promotes survival of postmitotic neurons by regulating Rb protein. Cell Death Differ. 2008;15(10):1663–1672. doi: 10.1038/cdd.2008.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicolai J, Burbassi S, Rubin J, Meucci O. CXCL12 inhibits expression of the NMDA receptor’s NR2B subunit through a histone deacetylase-dependent pathway contributing to neuronal survival. Cell Death Dis. 2010;1:e33. doi: 10.1038/cddis.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Opatz J, et al. SDF-1 stimulates neurite growth on inhibitory CNS myelin. Mol Cell Neurosci. 2009;40(2):293–300. doi: 10.1016/j.mcn.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Patel JR, McCandless EE, Dorsey D, Klein RS. CXCR4 promotes differentiation of oligodendrocyte progenitors and remyelination. Proc Natl Acad Sci U S A. 2010;107(24):11062–11067. doi: 10.1073/pnas.1006301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robin AM, et al. Stromal cell-derived factor 1alpha mediates neural progenitor cell motility after focal cerebral ischemia. J Cereb Blood Flow Metab. 2006;26(1):125–134. doi: 10.1038/sj.jcbfm.9600172. [DOI] [PubMed] [Google Scholar]
- 9.Reaux-Le Goazigo A, Van Steenwinckel J, Rostene W, Melik Parsadaniantz S. Current status of chemokines in the adult CNS. Prog Neurobiol. 2013;104:67–92. doi: 10.1016/j.pneurobio.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Li M, Ransohoff RM. Multiple roles of chemokine CXCL12 in the central nervous system: a migration from immunology to neurobiology. Prog Neurobiol. 2008;84(2):116–131. doi: 10.1016/j.pneurobio.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tyor WR, et al. Cytokine expression in the brain during the acquired immunodeficiency syndrome. Ann Neurol. 1992;31(4):349–360. doi: 10.1002/ana.410310402. [DOI] [PubMed] [Google Scholar]
- 12.Ferrarese C, et al. Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology. 2001;57(4):671–675. doi: 10.1212/WNL.57.4.671. [DOI] [PubMed] [Google Scholar]
- 13.An SF, Giometto B, Scaravilli T, Tavolato B, Gray F, Scaravilli F. Programmed cell death in brains of HIV-1-positive AIDS and pre-AIDS patients. Acta Neuropathol. 1996;91(2):169–173. doi: 10.1007/s004010050409. [DOI] [PubMed] [Google Scholar]
- 14.Everall IP, et al. Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. HNRC Group. HIV Neurobehavioral Research Center. Brain Pathol. 1999;9(2):209–217. doi: 10.1111/j.1750-3639.1999.tb00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8(1):33–44. doi: 10.1038/nrn2040. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5(1):69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- 17.Kraft-Terry SD, Buch SJ, Fox HS, Gendelman HE. A coat of many colors: neuroimmune crosstalk in human immunodeficiency virus infection. Neuron. 2009;64(1):133–145. doi: 10.1016/j.neuron.2009.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hauser KF, et al. Impact of opiate-HIV-1 interactions on neurotoxic signaling. J Neuroimmune Pharmacol. 2006;1(1):98–105. doi: 10.1007/s11481-005-9000-4. [DOI] [PubMed] [Google Scholar]
- 19.Hauser KF, Fitting S, Dever SM, Podhaizer EM, Knapp PE. Opiate Drug Use and the Pathophysiology of NeuroAIDS. Curr HIV Res. 2012;10(5):435–452. doi: 10.2174/157016212802138779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nath A. Human immunodeficiency virus-associated neurocognitive disorder: pathophysiology in relation to drug addiction. Ann N Y Acad Sci. 2010;1187:122–128. doi: 10.1111/j.1749-6632.2009.05277.x. [DOI] [PubMed] [Google Scholar]
- 21.Bell JE, Arango JC, Anthony IC. Neurobiology of multiple insults: HIV-1-associated brain disorders in those who use illicit drugs. J Neuroimmune Pharmacol. 2006;1(2):182–191. doi: 10.1007/s11481-006-9018-2. [DOI] [PubMed] [Google Scholar]
- 22.Arango JC, Simmonds P, Brettle RP, Bell JE. Does drug abuse influence the microglial response in AIDS and HIV encephalitis? AIDS. 2004;18(suppl 1):S69–S74. doi: 10.1097/00002030-200406003-00013. [DOI] [PubMed] [Google Scholar]
- 23.Bell JE, Brettle RP, Chiswick A, Simmonds P. HIV encephalitis, proviral load and dementia in drug users and homosexuals with AIDS. Effect of neocortical involvement. Brain. 1998;121(pt 11):2043–2052. doi: 10.1093/brain/121.11.2043. [DOI] [PubMed] [Google Scholar]
- 24.Finley MJ, et al. Bi-directional heterologous desensitization between the major HIV-1 co-receptor CXCR4 and the kappa-opioid receptor. J Neuroimmunol. 2008;197(2):114–123. doi: 10.1016/j.jneuroim.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel JP, Sengupta R, Bardi G, Khan MZ, Mullen-Przeworski A, Meucci O. Modulation of neuronal CXCR4 by the micro-opioid agonist DAMGO. J Neurovirol. 2006;12(6):492–500. doi: 10.1080/13550280601064798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pello OM, et al. Ligand stabilization of CXCR4/delta-opioid receptor heterodimers reveals a mechanism for immune response regulation. Eur J Immunol. 2008;38(2):537–549. doi: 10.1002/eji.200737630. [DOI] [PubMed] [Google Scholar]
- 27.Sengupta R, et al. Morphine increases brain levels of ferritin heavy chain leading to inhibition of CXCR4-mediated survival signaling in neurons. J Neurosci. 2009;29(8):2534–2544. doi: 10.1523/JNEUROSCI.5865-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burbassi S, Sengupta R, Meucci O. Alterations of CXCR4 function in mu-opioid receptor-deficient glia. Eur J Neurosci. 2010;32(8):1278–1288. doi: 10.1111/j.1460-9568.2010.07402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li R, Luo C, Mines M, Zhang J, Fan GH. Chemokine CXCL12 induces binding of ferritin heavy chain to the chemokine receptor CXCR4, alters CXCR4 signaling, and induces phosphorylation and nuclear translocation of ferritin heavy chain. J Biol Chem. 2006;281(49):37616–37627. doi: 10.1074/jbc.M607266200. [DOI] [PubMed] [Google Scholar]
- 30.Orino K, Lehman L, Tsuji Y, Ayaki H, Torti SV, Torti FM. Ferritin and the response to oxidative stress. Biochem J. 2001;357(pt 1):241–247. doi: 10.1042/0264-6021:3570241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheepsunthorn P, Palmer C, Connor JR. Cellular distribution of ferritin subunits in postnatal rat brain. J Comp Neurol. 1998;400(1):73–86. doi: 10.1002/(SICI)1096-9861(19981012)400:1<73::AID-CNE5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 32.Connor JR, Menzies SL, St Martin SM, Mufson EJ. Cellular distribution of transferrin, ferritin, and iron in normal and aged human brains. J Neurosci Res. 1990;27(4):595–611. doi: 10.1002/jnr.490270421. [DOI] [PubMed] [Google Scholar]
- 33.Han J, Day JR, Connor JR, Beard JL. H and L ferritin subunit mRNA expression differs in brains of control and iron-deficient rats. J Nutr. 2002;132(9):2769–2774. doi: 10.1093/jn/132.9.2769. [DOI] [PubMed] [Google Scholar]
- 34.McNeill A, Chinnery PF. Neuroferritinopathy: update on clinical features and pathogenesis. Curr Drug Targets. 2012;13(9):1200–1203. doi: 10.2174/138945012802002375. [DOI] [PubMed] [Google Scholar]
- 35.Vidal R, et al. Intracellular ferritin accumulation in neural and extraneural tissue characterizes a neurodegenerative disease associated with a mutation in the ferritin light polypeptide gene. J Neuropathol Exp Neurol. 2004;63(4):363–380. doi: 10.1093/jnen/63.4.363. [DOI] [PubMed] [Google Scholar]
- 36.Torti SV, et al. The molecular cloning and characterization of murine ferritin heavy chain, a tumor necrosis factor-inducible gene. J Biol Chem. 1988;263(25):12638–12644. [PubMed] [Google Scholar]
- 37.Wei Y, Miller SC, Tsuji Y, Torti SV, Torti FM. Interleukin 1 induces ferritin heavy chain in human muscle cells. Biochem Biophys Res Commun. 1990;169(1):289–296. doi: 10.1016/0006-291X(90)91466-6. [DOI] [PubMed] [Google Scholar]
- 38.Masliah E, et al. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann Neurol. 1997;42(6):963–972. doi: 10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- 39.Sa MJ, Madeira MD, Ruela C, Volk B, Mota-Miranda A, Paula-Barbosa MM. Dendritic changes in the hippocampal formation of AIDS patients: a quantitative Golgi study. Acta Neuropathol. 2004;107(2):97–110. doi: 10.1007/s00401-003-0781-3. [DOI] [PubMed] [Google Scholar]
- 40.Liao D, Lin H, Law PY, Loh HH. Mu-opioid receptors modulate the stability of dendritic spines. Proc Natl Acad Sci U S A. 2005;102(5):1725–1730. doi: 10.1073/pnas.0406797102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pitcher J, Shimizu S, Burbassi S, Meucci O. Disruption of neuronal CXCR4 function by opioids: preliminary evidence of ferritin heavy chain as a potential etiological agent in neuroAIDS. J Neuroimmunol. 2010;224(1–2):66–71. doi: 10.1016/j.jneuroim.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Busillo JM, Armando S, Sengupta R, Meucci O, Bouvier M, Benovic JL. Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J Biol Chem. 2010;285(10):7805–7817. doi: 10.1074/jbc.M109.091173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woerner BM, Warrington NM, Kung AL, Perry A, Rubin JB. Widespread CXCR4 activation in astrocytomas revealed by phospho-CXCR4-specific antibodies. Cancer Res. 2005;65(24):11392–11399. doi: 10.1158/0008-5472.CAN-05-0847. [DOI] [PubMed] [Google Scholar]
- 44.Mondal D, Williams CA, Ali M, Eilers M, Agrawal KC. The HIV-1 Tat protein selectively enhances CXCR4 and inhibits CCR5 expression in megakaryocytic K562 cells. Exp Biol Med (Maywood). 2005;230(9):631–644. doi: 10.1177/153537020523000905. [DOI] [PubMed] [Google Scholar]
- 45.Chen S, et al. Transforming growth factor-beta1 increases CXCR4 expression, stromal-derived factor-1alpha-stimulated signalling and human immunodeficiency virus-1 entry in human monocyte-derived macrophages. Immunology. 2005;114(4):565–574. doi: 10.1111/j.1365-2567.2004.02110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Centers for Disease Control and Prevention. HIV Surveillance Report, 2010. Vol. 22. http://www.cdc.gov/hiv/library/reports/surveillance/index.html . Published March 2012. Accessed November 18, 2013.
- 47.Cozzi A, Corsi B, Levi S, Santambrogio P, Albertini A, Arosio P. Overexpression of wild type and mutated human ferritin H-chain in HeLa cells: in vivo role of ferritin ferroxidase activity. J Biol Chem. 2000;275(33):25122–25129. doi: 10.1074/jbc.M003797200. [DOI] [PubMed] [Google Scholar]
- 48.Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci U S A. 1998;95(24):14500–14505. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Volinsky N, Gantman A, Yablonski D. A Pak- and Pix-dependent branch of the SDF-1alpha signalling pathway mediates T cell chemotaxis across restrictive barriers. Biochem J. 2006;397(1):213–222. doi: 10.1042/BJ20051655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D’Mello SR. HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev Neurobiol. 2008;68(8):1076–1092. doi: 10.1002/dneu.20637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bolger TA, Yao TP. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J Neurosci. 2005;25(41):9544–9553. doi: 10.1523/JNEUROSCI.1826-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim MS, et al. An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J Neurosci. 2012;32(32):10879–10886. doi: 10.1523/JNEUROSCI.2089-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cole CJ, et al. MEF2 negatively regulates learning-induced structural plasticity and memory formation. Nat Neurosci. 2012;15(9):1255–1264. doi: 10.1038/nn.3189. [DOI] [PubMed] [Google Scholar]
- 54.Bhattacharyya BJ, et al. The chemokine stromal cell-derived factor-1 regulates GABAergic inputs to neural progenitors in the postnatal dentate gyrus. J Neurosci. 2008;28(26):6720–6730. doi: 10.1523/JNEUROSCI.1677-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guyon A, Nahon JL. Multiple actions of the chemokine stromal cell-derived factor-1alpha on neuronal activity. J Mol Endocrinol. 2007;38(3):365–376. doi: 10.1677/JME-06-0013. [DOI] [PubMed] [Google Scholar]
- 56.Chiu CQ, Lur G, Morse TM, Carnevale NT, Ellis-Davies GC, Higley MJ. Compartmentalization of GABAergic inhibition by dendritic spines. Science. 2013;340(6133):759–762. doi: 10.1126/science.1234274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abt AC, Meucci O. Regulation of neuronal ferritin heavy chain, a new player in opiate-induced chemokine dysfunction. J Neuroimmune Pharmacol. 2011;6(4):466–476. doi: 10.1007/s11481-011-9278-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gandhi NS, et al. Comparison of scales to evaluate the progression of HIV-associated neurocognitive disorder. HIV Ther. 2010;4(3):371–379. doi: 10.2217/hiv.10.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Price RW, Brew BJ. The AIDS dementia complex. J Infect Dis. 1988;158(5):1079–1083. doi: 10.1093/infdis/158.5.1079. [DOI] [PubMed] [Google Scholar]
- 60.Khan MZ, et al. Apoptotic and antiapoptotic effects of CXCR4: is it a matter of intrinsic efficacy? Implications for HIV neuropathogenesis. AIDS Res Hum Retroviruses. 2004;20(10):1063–1071. doi: 10.1089/aid.2004.20.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carbajal KS, Miranda JL, Tsukamoto MR, Lane TE. CXCR4 signaling regulates remyelination by endogenous oligodendrocyte progenitor cells in a viral model of demyelination. Glia. 2011;59(12):1813–1821. doi: 10.1002/glia.21225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen J, et al. Transfusion of CXCR4-primed endothelial progenitor cells reduces cerebral ischemic damage and promotes repair in db/db diabetic mice. PLoS One. 2012;7(11):e50105. doi: 10.1371/journal.pone.0050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Raman D, Milatovic SZ, Milatovic D, Splittgerber R, Fan GH, Richmond A. Chemokines, macrophage inflammatory protein-2 and stromal cell-derived factor-1alpha, suppress amyloid beta-induced neurotoxicity. Toxicol Appl Pharmacol. 2011;256(3):300–313. doi: 10.1016/j.taap.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang K, et al. HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat Neurosci. 2003;6(10):1064–1071. doi: 10.1038/nn1127. [DOI] [PubMed] [Google Scholar]
- 65.Vergote D, et al. Proteolytic processing of SDF-1alpha reveals a change in receptor specificity mediating HIV-associated neurodegeneration. Proc Natl Acad Sci U S A. 2006;103(50):19182–19187. doi: 10.1073/pnas.0604678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steele AD, Henderson EE, Rogers TJ. Mu-opioid modulation of HIV-1 coreceptor expression and HIV-1 replication. Virology. 2003;309(1):99–107. doi: 10.1016/S0042-6822(03)00015-1. [DOI] [PubMed] [Google Scholar]
- 67.Kumar R, et al. Chronic morphine exposure causes pronounced virus replication in cerebral compartment and accelerated onset of AIDS in SIV/SHIV-infected Indian rhesus macaques. Virology. 2006;354(1):192–206. doi: 10.1016/j.virol.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 68.Fitting S, et al. Interactive comorbidity between opioid drug abuse and HIV-1 Tat: chronic exposure augments spine loss and sublethal dendritic pathology in striatal neurons. Am J Pathol. 2010;177(3):1397–1410. doi: 10.2353/ajpath.2010.090945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miller EC, et al. Differential modulation of drug-induced structural and functional plasticity of dendritic spines. Mol Pharmacol. 2012;82(2):333–343. doi: 10.1124/mol.112.078162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morgello S, et al. The National NeuroAIDS Tissue Consortium: a new paradigm in brain banking with an emphasis on infectious disease. Neuropathol Appl Neurobiol. 2001;27(4):326–335. doi: 10.1046/j.0305-1846.2001.00334.x. [DOI] [PubMed] [Google Scholar]
- 71.Hasin DS, Trautman KD, Miele GM, Samet S, Smith M, Endicott J. Psychiatric Research Interview for Substance and Mental Disorders (PRISM): reliability for substance abusers. Am J Psychiatry. 1996;153(9):1195–1201. doi: 10.1176/ajp.153.9.1195. [DOI] [PubMed] [Google Scholar]
- 72.Pitcher J, Wurth R, Shimizu S, Meucci O. Chemokines: Methods and Protocols. 2013. Multispectral imaging automated laser capture microdissection of human cortical neurons: a quantitative study of CXCR4 expression. In: Cardona AE, Ubogu EE, eds. pp. 31–48. New York, New York, USA: Humana Press; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shimizu S, Abt A, Meucci O. Bilaminar co-culture of primary rat cortical neurons and glia. J Vis Exp. 2011;(57):3257. doi: 10.3791/3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cook A, Hippensteel R, Shimizu S, Nicolai J, Fatatis A, Meucci O. Interactions between chemokines: regulation of fractalkine/CX3CL1 homeostasis by SDF/CXCL12 in cortical neurons. J Biol Chem. 2010;285(14):10563–10571. doi: 10.1074/jbc.M109.035477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang HX, Gao WJ. Development of calcium-permeable AMPA receptors and their correlation with NMDA receptors in fast-spiking interneurons of rat prefrontal cortex. J Physiol. 2010;588(pt 15):2823–2838. doi: 10.1113/jphysiol.2010.187591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.