

Abstract
Cullin-RING ligases (CRLs) are a family of E3 ubiquitin ligase complexes that rely on either RING-box 1 (RBX1) or sensitive to apoptosis gene (SAG), also known as RBX2, for activity. RBX1 and SAG are both overexpressed in human lung cancer; however, their contribution to patient survival and lung tumorigenesis is unknown. Here, we report that overexpression of SAG, but not RBX1, correlates with poor patient prognosis and more advanced disease. We found that SAG is overexpressed in murine KrasG12D-driven lung tumors and that Sag deletion suppressed lung tumorigenesis and extended murine life span. Using cultured lung cancer cells, we showed that SAG knockdown suppressed growth and survival, inactivated both NF-κB and mTOR pathways, and resulted in accumulation of tumor suppressor substrates, including p21, p27, NOXA, and BIM. Importantly, growth suppression by SAG knockdown was partially rescued by simultaneous knockdown of p21 or the mTOR inhibitor DEPTOR. Treatment with MLN4924, a small molecule inhibitor of CRL E3s, also inhibited the formation of KrasG12D-induced lung tumors through a similar mechanism involving inactivation of NF-κB and mTOR and accumulation of tumor suppressor substrates. Together, our results demonstrate that Sag is a Kras-cooperating oncogene that promotes lung tumorigenesis and suggest that targeting SAG-CRL E3 ligases may be an effective therapeutic approach for Kras-driven lung cancers.
Introduction
CRL (Cullin-RING ligase) complexes consist of a scaffold protein cullin (8 family members), an adaptor protein (few family members), a substrate-recognizing protein (many family members), and a RING component (2 family members) (1). Various combinations of these components constitute the largest family of E3 ubiquitin ligases, which are responsible for the ubiquitylation of about 20% of all ubiquitinylated proteins for targeted degradation by the 26S proteasome (1, 2). By promoting ubiquitylation and degradation of various substrates, CRLs regulate numerous biological processes, including cell-cycle progression, DNA damage response, signal transduction, and tumorigenesis (3, 4). The activity of CRL E3s requires (a) 1 of 2 RING components, RING-box 1 (RBX1), also known as regulator of cullins 1 (ROC1) or RBX2/ROC2/SAG (where SAG indicates sensitive to apoptosis gene), also known as RNF7 (RING finger protein 7) (5, 6) and (b) cullin neddylation (7, 8), a process catalyzed by NEDD8-activating enzyme (NAE) (E1), NEDD8-conjugating enzyme (E2), and NEDD8 ligase (E3) (9).
RBX1 was initially identified as an essential protein for the full activity of SKP1–Cul1–F-Box protein (SCF) E3 ligase, also known as CRL-1, the founding member of CRL (10–13). SAG, originally cloned in our laboratory, is a redox-inducible antioxidant protein (14) that was later characterized as the second member of the RBX/ROC RING component in SCF/CRL E3s (15). Although RBX1 and SAG are biochemically interchangeable in carrying out E3 ligase activity (15, 16), the in vivo physiological functions of both family members was found to be nonredundant. Our knockout studies revealed that disruption of either Rbx1 or Sag in mice caused embryonic lethality, although at different developmental stages (17, 18), suggesting that in vivo, the 2 family members potentially target different sets of nonoverlapping substrates.
As a redox-inducible antioxidant protein (14, 19), SAG protects cells from apoptosis induced by a variety of stimuli (for review see ref. 20). When forming a complex with other components of SCF/CRL E3 ubiquitin ligases, SAG has E3 ubiquitin ligase activity (15, 16, 21) and promotes the ubiquitylation and subsequent degradation of various cellular proteins, including p27 (22, 23), c-Jun (24), pro–caspase-3 (25), IκBα (16, 26), HIF-1α (27), NOXA (28), and NF1 (18) in a cell context–dependent manner. Whole animal studies revealed that SAG overexpression protects mouse brain tissues from ischemia/hypoxia-induced damage (29, 30). SAG transgenic expression in mouse skin inhibited tumor formation at the early stage by targeting c-Jun/AP1, but enhanced tumor growth at later stages in a DMBA-TPA carcinogenesis model by targeting IκBα to activate NF-κB (26) and promoted UVB-induced skin hyperplasia by targeting p27 (23). Finally, targeted disruption of Sag in the mouse caused embryonic lethality at E11.5–E12.5, which was associated with overall growth retardation, massive apoptosis, and reduced vasculogenesis (18). However, despite all this progress in our understanding of this RING protein, it has not been systematically shown whether Sag is an oncogenic-cooperating gene required for tumorigenesis, induced by a dominant oncogene. Furthermore, although SCF/CRL1 E3 ligase has been suggested as a valid cancer target (31, 32), no study has been conducted to elucidate its role in the development of lung cancer, in which SAG is often overexpressed and correlated with poor patient survival (28, 33).
Lung cancer is the leading cause of cancer-related death both in the USA and world wide, with non–small cell lung cancer (NSCLC) being the most common type and representing nearly 80% of all cases (34). Among the molecular changes found in NSCLC, mutational activation of Kras is one of the most common genetic alterations (35). We recently found that SAG is overexpressed in human NSCLC (28), and it was also previously reported in a study with limited samples that NSCLC patients with SAG overexpression have a poor prognosis (33). However, it is unknown whether SAG overexpression plays a causal role or is merely a consequence of lung tumorigenesis.
In this study, we used a Sag conditional KO mouse model in combination with the KrasG12D-lung cancer model (36) to determine the in vivo role of SAG in lung tumorigenesis, and found that Sag inactivation substantially suppressed KrasG12D-induced lung tumor formation by inactivation of NF-κB (via IκB), mTOR (via DEPTOR), and CDKs (via p21 and p27). Consistently, SAG knockdown suppressed, whereas SAG overexpression promoted (in a cell line-dependent manner), the growth and survival of lung cancer cells harboring the same Kras mutation at codon 12. Importantly, pharmaceutical inactivation of CRL E3 ligases by the small molecule MLN4924 also markedly inhibited lung tumor formation triggered by KrasG12D, again through inactivation of NF-κB, mTOR, and CDKs. Our study provides what we believe is the first in vivo demonstration that (a) Sag is a Kras-cooperative oncogene that is overexpressed in human lung cancer and (b) the CRL ligase inhibitor MLN4924, which inhibits both SAG- and RBX1-associated CRLs, has potential for future development as a class of anticancer drugs for the treatment of Kras-driven lung cancer patients where effective treatments are greatly needed.
Results
SAG overexpression in lung cancer tissues correlates with poor patient survival.
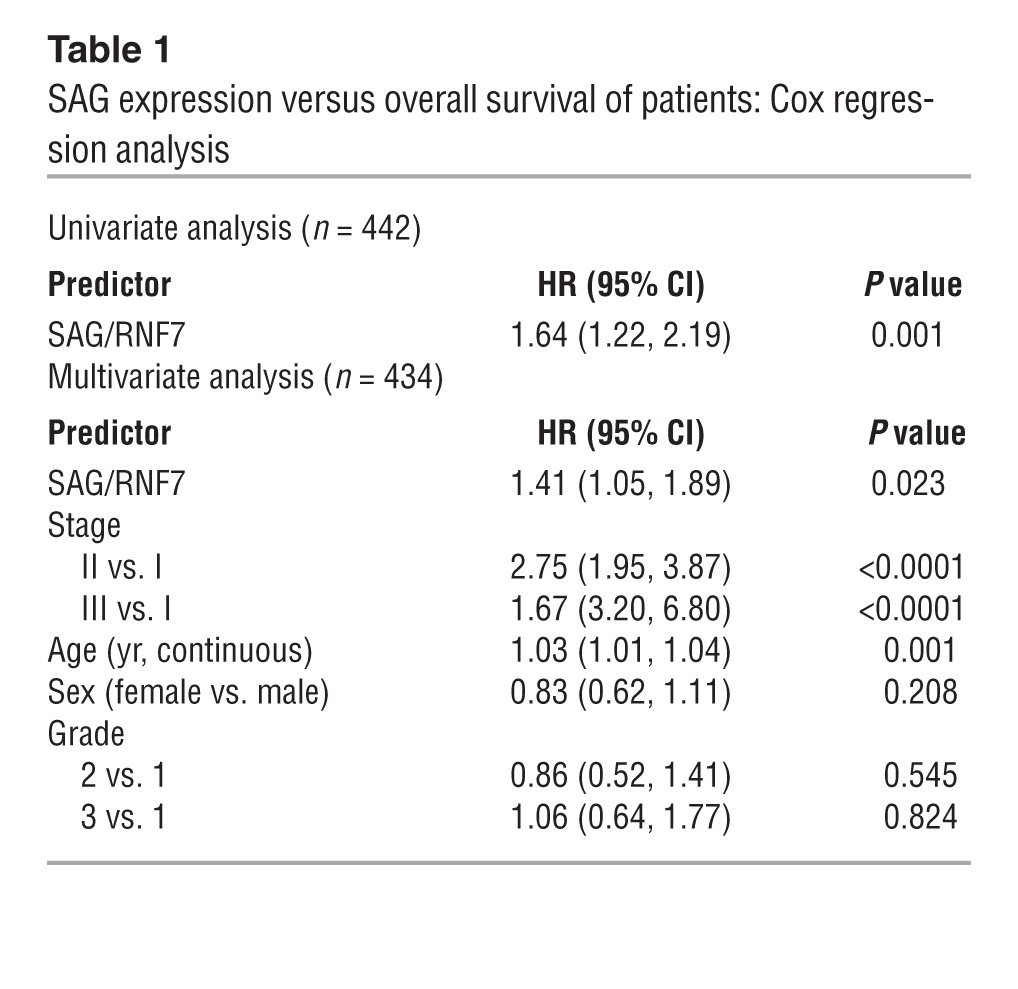
Our recent work showed that both RBX1/ROC1 and SAG/RBX2/ROC2 are overexpressed in human NSCLC, including adenocarcinomas and squamous cell carcinomas (28, 37). A previous study with limited sample size suggested that SAG overexpression in NSCLC correlated with worse prognosis (33). To confirm and extend this work, we performed a large-scale analysis of SAG mRNA expression and prognosis using data from 442 lung adenocarcinoma patients (38). As shown in Figure 1 and Table 1, patients with high SAG mRNA levels had a statistically significant lower survival probability, whereas those with low SAG levels had better survival (P = 0.001) (Figure 1A). Consistent with this, high SAG expression was significantly correlated with larger tumor size (Figure 1B) and with poor tumor differentiation (Figure 1C). Furthermore, SAG remained a significant independent prognostic factor after adjusting for stage, age, sex, and grade (HR = 1.41; 95% CI: 1.05-1.89; P = 0.023) (Table 1). In contrast, RBX1 expression was not associated with patient survival or disease progression (Figure 1D and Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI70297DS1). Taken together, these results show that overexpression of SAG, but not RBX1, is directly associated with and may potentially be causally related to lung cancer development and that SAG overexpression may serve as a biomarker for prognosis of lung cancer patients. We, therefore, focused our remaining studies on SAG.
Figure 1. Overexpression of SAG, but not RBX1, correlates with poor survival of lung cancer patients, increased tumor size, and poor differentiation.

(A) SAG mRNA expression versus the survival of lung cancer patients: Affymetrix U133A microarray data including 442 lung adenocarcinomas was downloaded from Shedden et al. (38). Robust multi-array average (RMA) normalized (62) and log2 transformed data were used in this study. Survival functions were estimated by the Kaplan-Meier method for patients with low and high expressions SAG (A) and RBX1 (D). Continuous expression levels were dichotomized using median splits (n = 220 and n = 222 in low and high groups in SAG [A]; n = 222 and n = 220 in Low and High groups in RBX1 [D], respectively). Patients with higher SAG levels had a worse overall survival (HR = 1.64; 95% CI: 1.22–2.19; P = 0.001, log-rank test). (B) SAG mRNA was higher in T3–T4 tumors as compared with T1 tumors (P < 0.001, t test). (C) SAG mRNA was higher in poorly differentiated tumors as compared with well-differentiated tumors (P < 0.001, t test). Definition of boxplot: the bounds of the box represent the middle 50% of all value, the line within the box is the median value, the upper line of the box is the value at 25%, the lower line of the box is the value at 75%. The upper and lower whiskers represent the maximum and minimum values. Few outlying values are located outside of whiskers. (D) RBX1 mRNA expression was not related to the survival of lung cancer patients (P = 0.53, log-rank test).
Table 1.
SAG expression versus overall survival of patients: Cox regression analysis
Sag deletion inhibits KrasG12D-induced lung tumorigenesis and extends life-span in the mouse.
We next determined whether SAG overexpression seen in human lung cancer patients is causally related to, or merely the consequence of lung tumorigenesis, using a well-established LSL-KrasG12D (Lox-STOP-Lox KrasG12D) conditional mouse lung tumor model, in which KrasG12D is activated by Cre-recombinase via the removal of the STOP element to induce epithelial hyperplasia, adenomas, and adenocarcinomas in the lung (36). We first confirmed that SAG is indeed overexpressed in lung tumors induced by KrasG12D in 3 individual mice (Supplemental Figure 1). We then generated a Sag conditional KO mouse model (Sagfl/+, Supplemental Figure 2) and crossed it with Saggt/+ gene-trap mice (18) to generate Saggt/fl mice (1 gene-trap Sag-null allele, gt and 1 Sag-floxed allele, fl). Two-step crossing with LSL-KrasG12D mice generated compound mice with the following 4 genotypes: (a) KrasG12D;Sag+/+ (positive control), (b) LSL-KrasG12D;Saggt/+, (c) LSL-KrasG12D;Saggt/fl, and (d) Saggt/fl (negative and toxicity control). The mice were intratracheally administrated with Ad-Cre to activate KrasG12D and simultaneously inactivate Sag in group c mice. Mice were euthanized 12 weeks after infection. As expected, both LSL-KrasG12D;Sag+/+ and LSL-KrasG12D;Saggt/+ mice developed substantial numbers of adenomas with few adenocarcinoma (classified as tumors), consistent with our previous observation that heterozygous Sag deletion had no phenotype compared with WT mice (18). Remarkably, Sag inactivation in group c mice significantly reduced lung tumor burden induced by KrasG12D, although the number of hyperplastic loci was not affected by Sag inactivation (Figure 2, A and B, and Supplemental Figure 3, A and B). It is worth noting that Sag inactivation in adult mice (group d) had no toxic effects on normal lung tissues (Figure 2A). The results clearly suggest that Sag inactivation may not affect tumor initiation but remarkably inhibits the progression from hyperplasia to adenomas. Thus, Sag is required for the progression of lung tumor development triggered by KrasG12D activation.
Figure 2. Sag inactivation inhibits KrasG12D-induced lung tumorigenesis.

(A) Morphological observation of 1 lobe of lung with indicated genotypes 12 weeks after Ad-Cre administration to activate KrasG12D and/or inactivate Sag. Scale bars: 100 μm. (B) Quantification of mouse lung tumors (mostly adenomas) developed in indicated numbers of mice with indicated genotypes at 12 weeks after Ad-Cre administration. **P < 0.01. (C) Kaplan-Meier survival curve of compound mice with genotype of KrasG12D;Saggt/+ (n = 20) versus KrasG12D;Saggt/fl (n = 24) for up to 60 weeks after administration of Ad-Cre.
We noticed that adenomas did develop in some LSL-KrasG12D;Saggt/fl mice (Supplemental Figure 3C). We reasoned that this could be due to incomplete deletion of the floxed Sag allele, a phenomenon often seen in compound KrasG12D mouse models (39, 40). To test this, we microdissected 2 tumors via laser capture and performed PCR-based genotyping. As shown in Supplemental Figure 3D, in both tumor cases, we detected floxed Sag allele, indicating incomplete excision. Activation of KrasG12D was also confirmed in these tumors (Supplemental Figure 3E). We further dissected 10 individual larger tumors that were developed 16 weeks after Ad-Cre administration in 10 individual LSL-KrasG12D;Saggt/fl mice and found the floxed Sag allele remained in every single case (Supplemental Figure 3F). Thus, development of the lung tumors in some LSL-KrasG12D;Saggt/fl mice is due to incomplete Sag deletion.
We next determined the effect of Sag inactivation on overall mouse survival upon KrasG12D activation by comparison of the survival probability between LSL-KrasG12D;Saggt/+ and LSL-KrasG12D;Saggt/fl mice. Consistent with published reports (40, 41), while LSL-KrasG12D;Saggt/+ mice had a median time to death of 27.6 weeks, with all mice dying by 33 weeks after KrasG12D activation, the LSL-KrasG12D;Saggt/fl mice had a median time to death of 37.9 weeks, with 4 mice remaining alive after 60 weeks. This difference is statistically significant, with a log-rank P value of less than 0.0001 (Figure 2C). Taken together, our results clearly demonstrate the requirement of Sag for lung tumor development in this KrasG12D model, suggesting that targeting SAG E3 ligase could be an effective approach for the treatment of lung cancer associated with Kras mutation.
Sag deletion suppresses proliferation and causes accumulation of tumor suppressor substrates.
We then determined the rate of proliferation and apoptosis in hyperplasia or adenomas derived from KrasG12D;Saggt/fl mice as compared with those from KrasG12D;Saggt/+ and found a reduced proliferation, as reflected by Ki67 staining (Figure 3), but not apoptosis, as indicated by lack of cleaved caspase-3 staining (not shown). Immunohistochemistry staining also showed in KrasG12D;Saggt/fl hyperplasia/adenomas the increased levels of several tumor suppressor proteins known to be SAG substrates, including IκB (with corresponding inhibition of p65 nuclear translocation), p21, and p27. Reduction of mTORC1 pathway, as reflected by a reduced pS6K staining, was also observed (Figure 3). Thus, inhibition of tumor progression by Sag deletion is attributable to reduced proliferation as a result of accumulation of tumor suppressor substrates.
Figure 3. Sag deletion suppresses proliferation by inducing accumulation of tumor suppressor substrates.

(A) Immunohistochemical staining of lung tumor tissues. Lung tissues from KrasG12D;Saggt/+ (Saggt/+) or KrasG12D;Saggt/fl (Saggt/fl) mice were fixed, sectioned, and stained with indicated Abs. Representative areas of lung tumors are shown. Scale bars: 100 μm. (B) Staining quantification: positively stained cells were counted out of a total of 500 cells on average from 3 independent tumors derived from 3 mice per group. *P < 0.05; **P < 0.01.
SAG knockdown inhibits the growth and survival of human lung cancer cells via inactivation of NF-κB and mTOR and accumulation of p21, p27, NOXA, and BIM.
We further used in vitro cell culture models to elucidate the role of SAG in the growth and survival of lung cancer cells. We first used the human lung cancer line A549, which harbors a Kras mutation at the 12th codon position (G12S), identical to that in our mice model (41, 42). Indeed, SAG knockdown via lentivirus-based siRNA caused significant reduction in (a) monolayer growth, (b) clonogenic survival, and (c) anchorage-independent soft-agar growth (Figure 4, A–C). Similar growth suppression was also observed when an independent SAG-targeting siRNA oligonucleotide was used to knock down SAG in A549 cells (Supplemental Figure 4, A–C). We further knocked down SAG in 2 additional lung cancer lines, A427 and H358, which harbor the same Kras mutation at codon 12 (G12D for A427 and G12C for H358; ATCC), and found a similar level of growth suppression (Supplemental Figure 5, A and B, and Supplemental Figure 6A). Note that H358 cells are unable to form colonies in both monolayer culture and in soft agar, whereas A427 cells are unable to form colonies in soft agar in our experimental conditions. Taken together, our results clearly showed that SAG is required for the growth and survival of lung cancer cells as well as for the maintenance of the tumor cell phenotype in A549 cells.
Figure 4. SAG siRNA knockdown inhibits growth and survival of human lung cancer cells via inactivation of NF-κB and mTOR.

(A–C). Growth of A549 human lung tumor cells upon SAG siRNA knockdown. Human lung cancer A549 cells were subjected to lentivirus-based siRNA silencing of SAG (Lt-SAG) along with scramble siRNA control (Lt-Con). Cells were tested by ATP-lite based proliferation assay (n = 3) (A); clonogenic survival assay (n = 3) (B); and soft agar assay (n = 3) (C). (D) Accumulation of tumor-suppressive proteins upon SAG knockdown: A549 cells were infected with Lt-SAG or Lt-Con, followed by immunoblotting using indicated Abs. (E) SAG knockdown blocks p65 nuclear translocation. Cells upon SAG knockdown were treated with or without TNF-α (100 ng/ml) for 1 hour, followed by nuclear fractionation and immunoblotting. Caspase-3 and PARP were used to demonstrate the purity of cytoplasm and nuclear fractions, respectively. (F). p65 DNA binding: nuclear fraction from indicated treatment is subjected to DNA-binding assay using TransAM NF-κB assay kit. (G). Luciferase reporter–based NF-κB activity assay. Lentiviral infected cells were transiently transfected with pNifty plasmid, along with renilla for transfection efficiency control. Cells were untreated or treated with TNF-α (10 ng/ml) and assayed for luciferase activity (n = 3). *P < 0.05; **P < 0.01.
To explore the potential mechanisms by which SAG knockdown suppresses the growth of lung cancer cells, we first focused on Kras itself, since Kras activity is subjected to ubiquitylation regulation (43), whereas Ras proteins are subjected to degradation by SCFβ-TrCP (44). Compared with control human bronchial epithelial BEAS-2B cells, in which no active Kras activity was detected, Kras activity was very high in all 3 lung cancer cell lines, A549, H358, and A427 (Supplemental Figure 4D). However, SAG knockdown affected neither the activity nor the protein levels of Kras (Supplemental Figure 4D), indicating that growth suppression by SAG knockdown is not mediated via modulation of Kras itself.
We next determined the potential accumulation of known substrates of SAG E3 ligase (20). SAG knockdown caused accumulation of phosphor-IκBα, an indicator of the lack of IκBα degradation (Figure 4D and Supplemental Figure 4E). Follow-up studies revealed that upon SAG knockdown, TNF-α–induced p65 nuclear localization, p65 DNA binding, and NF-κB transactivation were all significantly reduced (Figure 4, E–G), indicating blockage of NF-κB activation. We also found that SAG knockdown caused accumulation of DEPTOR, a naturally occurring inhibitor of mTOR (45) and a physiological substrate of SCFβTrCP (46), with the associated inactivation of mTOR signals, as reflected by reduced phosphorylation of S6K1 and 4E-BP1 (Figure 4D). Finally, SAG knockdown caused the accumulation of several tumor suppressor substrates, including p21, p27, NOXA, and BIM, but not oncogenic substrates, such as c-Myc, cyclin D1, and NOTCH-1 (Figure 4D and Supplemental Figure 4E). Similar results were seen when SAG was knocked down in A427 and H358 cells, with accumulation of tumor suppressor substrates (Supplemental Figure 5C and Supplemental Figure 6B), inhibition of p65 nuclear localization (Supplemental Figure 5D and Supplemental Figure 6C), and inactivation of NF-κB transcriptional activity (Supplemental Figure 5E and Supplemental Figure 6D). Taken together, our results indicated that the suppression in growth and survival of lung cancer cells induced by SAG knockdown is associated with inactivation of NF-κB and mTOR signaling pathways as well as selective accumulation of tumor suppressor substrates.
Growth suppression by SAG knockdown is abrogated by silencing of p21 or DEPTOR.
Two recent studies clearly demonstrate a requirement of the NF-κB pathway in the development of KrasG12D-driven lung cancer, with one study using the same LSL-KrasG12D mouse model (47, 48). We therefore focused our cell-based rescue experiment on p21 and DEPTOR and determined whether accumulated p21 or DEPTOR is causally related to growth suppression induced by SAG knockdown. As shown in Figure 5, simultaneous knockdown of p21 or DEPTOR with SAG in A549 cells (Figure 5A) partially reversed the growth suppression in monolayer proliferation (Figure 5B), clonal survival (Figure 5C), and soft agar-independent growth (Figure 5D). Complete rescue by DEPTOR knockdown is likely attributable to a growth-stimulating effect upon DEPTOR knockdown alone. Similar partial rescue effects were also observed in H358 cells (Supplemental Figure 7). Thus, accumulation of p21 or DEPTOR upon SAG knockdown plays at least in part a causal role in growth suppression.
Figure 5. Knockdown of p21 or DEPTOR partially rescues growth suppression triggered by SAG knockdown.

A549 cells were first infected with Lt-SAG to knockdown SAG along with the control (Lt-Con), followed by transfection with siRNA oligonucleotides, targeting p21 or DEPTOR, respectively. A portion of cells (combined from multiple infections/transfections) were harvested for immunoblotting (A); the other portions for monolayer growth for 5 days, followed by ATP-lite proliferation assay (B, n = 3); clonogenic assay for survival (C, n = 3); or soft agar assay for anchorage-independent growth (D, n = 3). *P < 0.05; **P < 0.01.
SAG ectopic expression reduces the levels of tumor suppressor substrates and promotes cell growth in a cell line–dependent manner.
Given that SAG is overexpressed in both human and mouse lung cancer tissues, we next investigated potential growth stimulating effect of SAG overexpression. We ectopically expressed SAG at a level comparable to that of endogenous SAG in all 3 lung cancer cell lines and found that SAG overexpression had a cell line–dependent effect on the substrate levels and cell growth. In A427 cells, SAG ectopic expression reduced the levels of tumor suppressor substrates, including pIκB, Deptor, p21 and p27 (Figure 6A), and consistently promoted cell growth in monolayer culture and increased clonogenic survival (Figure 6, B and C). In A549 and H358 cells, however, SAG ectopic expression had no effect on its substrate levels or cell growth (Supplemental Figure 8). Thus, SAG overexpression promotes the degradation of tumor suppressor substrates and cell growth in a cell line–dependent manner.
Figure 6. SAG overexpression reduces tumor suppressor substrates and promotes tumor cell growth.

A427 cells were transfected with FLAG-tagged SAG construct, along with pcDNA3 vector control. Stable clones after G418 selection were pooled and subjected to immunoblotting with indicated Abs (A), and assays for monolayer growth by ATP-lite (n = 4) (B), clonogenic survival (n = 5) (C, left panel: representative images; right panel: quantitative measure), and MLN4924 sensitivity by IC50 determination (n = 3) (D). Original magnification, ×1 (C). **P < 0.01. End. SAG, endogenous SAG.
MLN4924, a small molecule inhibitor of CRL ligase, inhibits lung tumor formation in vivo and lung cancer cell growth in vitro by blockage of NF-κB and mTOR and accumulation of p21 and p27.
SAG is the RING component of SCF-CRL E3 ligases, required for its enzymatic activity. Activity of CRLs also requires cullin neddylation (3, 49). MLN4924 is a recently discovered small molecule inhibitor of NAE, whose inhibition blocks cullin neddylation, thus having a broader effect by inactivating both SAG-associated and RBX1-associated CRL E3s (2, 50). We used MLN4924 as a pharmaceutical approach of SAG (as well as RBX1) targeting and examined its anti–lung cancer activity. To this end, LSL-KrasG12D mice were treated with MLN4924 for 4 weeks using a nontoxic dose regimen (60 mg/kg, once a day, 5 days per week) (2, 51) beginning at 13 weeks after Ad-Cre administration and when tumors had already formed (Figure 2A and Supplemental Figure 3A). Lung tissues were harvested after 16 weeks, sectioned, and examined for tumors. We found that MLN4924 treatment significantly reduced the tumor burden, with a remarkable reduction in both number of hyperplastic areas and adenomas as well as the size of the tumors (Figure 7, A and B, and Supplemental Figure 9, A and B), indicating potential anti–lung cancer activity of MLN4924. Using immunohistochemistry analysis, we found that, compared with vehicle controls, lung tumor tissues from MLN4924-treated mice had (a) decreased proliferation, reflected by reduced Ki67 staining (Figure 7C), but not apoptosis (not shown); (b) increased staining of pIκBα; (c) reduced number of cells with p65 nuclear staining; (d) increased staining of p21 and p27; and (e) reduced staining of pS6K1 (Figure 7, C and D). We further found that only 1 week of MLN4924 treatment is sufficient to reduce proliferation, as evidenced by decreased BrdU incorporation and Ki67 staining (Supplemental Figure 9C), but had no effect on apoptosis (not shown). Taken together, these results further support the notion that inactivation of both SAG- and RBX1-associated CRL E3s inhibits the growth of Kras-induced lung tumors by inhibiting, at least in part, NF-κB and mTOR signaling pathways.
Figure 7. Inactivation of CRL E3s by MLN4924 inhibits KrasG12D-induced lung tumorigenesis by blocking NF-κB and mTOR pathways.

(A and B) Morphological appearance of 1 lobe of lung tissue from KrasG12D;Sag+/+ mice 16 weeks after Ad-Cre administration, with last 4 week of MLN4924 treatment (s.c. 60 mg/kg, 5 days per week for 4 weeks) or vehicle control, as indicated. Scale bars: 100 μM (A). Quantified data are shown. *P < 0.05; **P < 0.01 (B). (C) Immunohistochemical staining of lung tumor tissues. Lung tissues from MLN4924- and vehicle-treated mice were stained with indicated Abs. Representative areas of lung tumors are shown. Scale bars: 100 μm. (D) Staining quantification: positively stained cells were counted out of a total of 500 cells in average from 3 independent tumors derived from 3 mice per group. *P < 0.05; **P < 0.01.
We next determined the effect of MLN4924 on the growth of 3 human lung cancer cell lines and found that MLN4924 effectively inhibited proliferation of tumor cells with an IC50 value ranging from 0.2 μM (A427) to 0.7 μM (A549) (Figure 8A and Supplemental Figure 10A), as well as clonogenic survival with A427 being more sensitive than A549 cells (IC50, ∼10 vs. ∼50 nM) (Figure 8B and Supplemental Figure 10B). MLN4924 also effectively inhibited the growth of A549 cells in soft agar with an IC50 value of approximately 100 nM (Figure 8C). Furthermore, we found that SAG overexpression increased the sensitivity of A427 cells to MLN4924, with IC50 value reduced by 50% (Figure 6D), but had no effect on A549 and H358 cells (data not shown). Mechanistically, and analogous to Sag knockdown, MLN4924 treatment, which inhibits CUL1 neddylation, caused significant accumulation of pIκBα in all 3 lines (Figure 8D and Supplemental Figure 10C) and completely blocked NF-κB nuclear translocation in A549 cells (Figure 8E), indicating NF-κB inactivation. The effect of MLN4924 treatment on DEPTOR was both cell line (Figure 8D and Supplemental Figure 10C) and treatment time dependent (Figure 8F and Supplemental Figure 10D). Specifically, significant DEPTOR accumulation was seen in H358 cells within 24 hours of MLN4924 treatment, whereas in A529 and A427 cells, accumulation was not observed until 36 to 60 hours. Accumulation of DEPTOR in A549 cells upon prolonged cell culture (DMSO treatment for 48–60 hours) was likely due to serum consumption and increased cell density (45, 52). Nevertheless, in every single cell line, DEPTOR accumulation was accompanied by the inactivation of mTORC1, as reflected by reduced phosphorylation of S6K1 or 4E-BP1 (Figure 8, D and F, Supplemental Figure 10, C and D). Furthermore, in all 3 lines tested, MLN4924 treatment caused remarkable accumulation of tumor suppressors p21 and p27, with accumulation of NOXA and BIM varying among the lines (Figure 8D and Supplemental Figure 10C). Accumulation of oncogenic substrates by MLN4924 varied among 3 lines as well, with no accumulation of c-Jun and cyclin D in A549, but they did accumulate in 2 other lines (Figure 8D and Supplemental Figure 10C). Taken together, our results demonstrate that inactivation of CRL E3 ligases suppresses lung cancer growth in both in vivo animal and in vitro cell culture models with mechanism(s) involving blockage of NF-κB/mTOR signals and accumulation of p21 and p27.
Figure 8. MLN4924 suppresses growth of lung cancer cells by targeting NF-κB and mTOR pathways and by accumulating tumor suppressor substrates.

(A–C) A549 cells were treated with MLN4924 at the indicated concentrations. Growth suppression effect was tested by monolayer growth (n = 3) (A), clonogenic survival (n = 4) (B), and soft agar (C, n = 2). **P < 0.01. (D). Accumulation of tumor-suppressive proteins: A549 cells were treated with indicated concentrations of MLN4924 for 24 hours, followed by immunoblotting using indicated Abs. (E). MLN4924 treatment blocks p65 nuclear translocation. Cells after 24-hour treatment of MLN4924 (1 μM) were exposed to TNF-α (100 ng/ml) or control vehicle for 1 hour, followed by nuclear fractionation and immunoblotting. Caspase-3 and PARP were used to demonstrate the purity of cytoplasm and nuclear fractions. (F) Time-dependent induction of DEPTOR and inactivation of mTORC1: cells were treated with 1 μM MLN4924 as well as DMSO for indicated periods and subjected to immunoblotting.
Discussion
Lung cancer, primarily NSCLC, is the leading cause of cancer-related deaths (34), with Kras mutations being one of the most common genetic alterations (35). Our recent studies revealed that both RBX1 and SAG are overexpressed in non–small cell lung carcinomas (28, 37) and SAG overexpression is likely mediated through transcriptional activation by AP1 (24) or HIF1 (27). In this study, we correlated their overexpression with patient survival using more than 400 lung cancer cases and found that overexpression of SAG, but not RBX1, is associated with higher stages of disease, poor tumor differentiation, and poor patient prognosis. It is noteworthy that due to very limit number of cases in which the status of Kras mutation is known among these more than 400 lung cancer tissues, we were not able to find an association between SAG overexpression and Kras mutation status. Nevertheless, our study provides supporting evidence that SAG overexpression has biological consequences and could serve as a prognostic biomarker as well as the biomarker of patient selection for MLN4924 treatment.
Overexpression of a specific gene in human cancers does not predict whether the overexpression is causally related to, or just the consequence of, tumorigenesis. In the case of SAG, it has never been mechanistically pursued whether SAG is required for lung cancer initiation and progression or for the maintenance of lung cancer cell phenotypes or simply as the consequence of tumorigenesis. Here, we addressed this important issue using a KrasG12D-driven mouse lung tumorigenesis model for 2 reasons: first, it recapitulates nicely the entire process of human lung tumorigenesis with sequential formation of lesions such as hyperplasia, adenomas, and eventually adenocarcinoma in a manner dependent on the length of KrasG12D activation (36, 40); second, SAG is overexpressed in this tumor model. By the use of a compound mouse model harboring LSL-KrasG12D and Sag conditional knockout alleles (LSL-KrasG12D;Saggt/fl), where KrasG12D activation and Sag deletion occur concomitantly upon intratracheal instillation of the Ad-Cre virus, we showed that Sag inactivation inhibited KrasG12D-induced lung tumorigenesis, as evidenced by reducing tumor burden both in tumor size and number and significantly extending the life span of the mice (Figure 2). The suppressive effect of Sag deletion is likely attributable to inactivation of the NF-κB and mTOR pathways and accumulation of p21 and p27 tumor suppressors (Figure 3). Furthermore, the suppression occurred mainly during hyperplasia-to-adenoma progression under our experimental conditions for a period of 12 weeks after KrasG12D activation, which is consistent with the observation that SAG overexpression is associated with later, but not early, stages of human lung cancer (Figure 1). It is worth noting that the in vivo anti-Kras activity of Sag inactivation/deletion was observed in the presence of WT Rbx1/Roc1, the only other member of the RBX/ROC family, indicating that RBX1/ROC1 cannot compensate for the loss of SAG/RBX2/ROC2. These results are consistent with our recent observation using genetic knockout approaches that there is no in vivo redundancy during embryogenesis between these 2 family members of the RING component of CRL E3 ubiquitin ligases (5, 17, 18). Thus, SAG and RBX1 play the nonredundant roles at least during mouse embryogenesis and in KrasG12D-induced lung tumorigenesis.
To gain mechanistic insight into SAG action, we used the loss-of-function approach in 3 lung cancer cell lines harboring the same codon 12 Kras mutation and found that siRNA-based Sag knockdown caused in general accumulation of (a) pIκBα to inactivate NF-κB, which is required for Kras-mediated lung tumorigenesis (47, 48), (b) DEPTOR, a naturally occurring inhibitor of mTOR to block the mTOR signaling pathway, and (c) tumor suppressors p21 and p27 to block cell-cycle progression. Interestingly, although oncogenes such as cyclin D1, c-Myc, and Notch-1 are known substrates of SCF/CRL-1 E3 ligase (4, 5, 53), none of these oncogenic proteins were found to accumulate upon Sag knockdown (Figure 4D, Supplemental Figure 4E, and Supplemental Figure 5, B and C). Thus, it appears that Sag inactivation selectively accumulates tumor-suppressive substrates to block KrasG12D-induced lung tumorigenesis. This notion was further supported by our rescue experiment in which knockdown of either p21 or DEPTOR abrogated the growth suppression triggered by SAG knockdown in both A549 and H358 lung cancer cells (Figure 5 and Supplemental Figure 7). Consistently, our gain-of-function experiment showed that SAG overexpression caused reduction of tumor suppressor substrates, such as pIκBα, DEPTOR, p21, and p27, and consequent growth suppression in A427 cells, but not in A549 or H358 cells (Figure 6 and Supplemental Figure 8). Lack of SAG effect in the latter 2 lines is unlikely due to the level of SAG overexpression, which was at the highest level in H358 cells. Rather it might be explained by the likelihood that SAG is not limited in these lines or unavailability of associated kinase/F-box proteins, since SAG-mediated degradation of a given substrate depends upon the accessibility of a kinase to phosphorylate the substrate and availability of an F-box protein to bind to phosphorylated substrate.
To explore the therapeutic application of our finding in a Sag conditional knockout mouse model, we used the same KrasG12D lung cancer model and tested anti–lung cancer efficacy of MLN4924, an investigational small molecule inhibitor of both SAG-associated and RBX1-associated CRL E3 ligases via cullin deneddylation (2), which is currently in several phase 1 clinical trials directed against several human malignancies (54–56). Our results clearly demonstrate a therapeutic value of the compound administrated when tumors had been formed following Kras activation for 12 weeks (Figure 7 and Supplemental Figure 9). It is anticipated that compared with permanent Sag deletion (even though some incomplete Sag deletion can be seen), the anti–lung cancer effect of MLN4924 is less, since the compound was only given for the last 4 weeks and the drug effect is temporary. Mechanistically, and similar to SAG KO or knockdown, MLN4924 treatment also blocks NF-κB and mTOR pathways and activates tumor suppressive p21, p27, NOXA, and BIM (Figures 7 and 8, and Supplemental Figures 9 and 10). Apparently, the effect of SAG knockdown or MLN4924 treatment on known substrates of SAG-SCF E3 ligase occurs in a cell context, temporal, and spatially dependent manner (5, 31). Selective accumulation of tumor suppressor proteins that target multiple oncogenic pathways in lung cancer cells makes MLN4924 an appealing anti–lung cancer agent. It should be noted that MLN4924 is an NAE inhibitor and would probably inhibit other cellular neddylation reactions (49, 57), although cullins are the only known physiological substrates (49, 58). Nevertheless, given the fact that MLN4924 phenocopies Sag deletion biologically and mechanistically, the major effect of MLN4924 against lung cancer is through the inhibition of CRL E3s. Furthermore, MLN4924 is the first-in-class and only CRL E3 ligase inhibitor currently in phase I clinical trials for anticancer therapy (54, 56). Our study using both genetic and pharmaceutical approaches provides sound rationale for future development of MLN4924 as a class of drug against deadly lung cancer, particularly those resulting from Kras mutation and SAG overexpression.
In summary, our study supports the following model. During lung tumorigenesis, SAG is induced in response to various stresses, such as ROS, hypoxia, and likely KrasG12D activation through transcription activation by AP1 and HIF1 (20). Increased SAG then recruits other components of SCF/CRL E3s (e.g., SCFβTrCP) to promote the degradation of tumor suppressor proteins, such as IκBα, DEPTOR, p21, p27, NOXA, and BIM, leading to activation of the NF-κB and mTOR pathways, cell-cycle progression, and apoptosis inhibition, respectively, which then cooperates with KrasG12D to induce lung tumorigenesis. Sag inactivation by genetic deletion or SAG-CRL inhibition by small molecule MLN4924 abrogates these oncogenic processes by inducing the accumulation of tumor suppressive proteins to antagonize Kras effects, eventually leading to the suppression of lung tumor development (Figure 9). Our study, therefore, provides (a) convincing evidence of SAG-SCF/CRL-1 E3 as a valid anti–lung cancer target and (b) sound rationale for future clinical trials of MLN4924 as a class of anti–lung cancer agent that abrogates multiple oncogenic pathways.
Figure 9. A model for SAG cooperation with Kras for driving lung tumorigenesis.

During lung tumorigenesis, triggered by mutant Kras, SAG is induced and recruits other components of SCF/CRL E3s to promote the ubiquitylation and degradation of tumor suppressor substrates. Sag inactivation by genetic deletion or small molecule inhibitor MLN4924 causes the accumulation of these tumor suppressor substrates to antagonize Kras effects, leading to suppression of lung tumor development.
Methods
PCR-based genotyping.
Genomic DNA was isolated from the tips of mouse tails and genotyped using the primer set of Baygen 08: 5′-CATCCGGGCTACCGGCTAAAACTT-3′ and SAGsi09: 5′-CAGAAGGCTCATGGTAAAACATC-3′ for Sag gene-trapped allele (477 bp) and SAGsi09 and SAGsi14: 5′-CATGGTAATCACCCTGCCTG-3′ for Sag WT allele (585 bp). The primer set for Sag floxed allele is PSAG-KO-F: 5′-ttctggccaggtgtggtgatatc-3′ and PSAG-KO-G: 5′-CTTAGCCTTGGTTGTGTAGAC-3′ to detect floxed allele (140 bp) and WT allele (105 bp). The primer set for detecting the removal of the Sag targeting fragment (1.3 kb, Supplemental Figure 2) is PSAG-KO-Seq-B: 5′-GTAACTCCAGACAATGCTCGCT-3′ and PSAG-KO-Seq-R: 5′-TGAGTTCCAGGACAGCCAGGG-3′ with Sag deletion (275 bp) or without Sag deletion (1.6 kb). The primer set for KrasG12D activation is Kras-CreF: 5′-TCCGAATTCAGTGACTACAGA-3′ and Kras-CreR: 5′-CTAGCCACCATGGTCTGAGT-3′. Unrecombined 2 loxP band is approximately 500 bp, whereas WT is 620 bp. Upon recombination, the 500 bp is lost and a 650 bp 1 loxP band is present, which represents the recombined Kras mutant allele.
Ad-Cre infection of mouse lung.
To activate KrasG12D in mouse lung, intranasal administration of Ad-Cre was performed as described (36, 59). Briefly, 6- to 8-week-old mice were anesthetized with isoflurane via a gas chamber. Ad-Cre (purchased from University of Iowa Vector Delivery Core) at the dose of 3 × 107 pfu in a total volume of 125 μl as CaPi coprecipitates were loaded in a gel-loading tip and administered nasally using two 62.5-μl instillations with a 5-minute interval.
Immunohistochemical staining.
Mouse lung organs were isolated after 10% formalin perfusion, fixed in 10% formalin, and embedded in paraffin (26). Sections that were 5-μm thick were cut for H&E staining and examined under a microscope. Immunohistochemistry was performed using the ABC Vectastain Kit (Vector Laboratories) with Abs against SAG (28), pIκBα, pErK, pS6K (Cell Signaling Technology Inc.), p27, p21 (BD Biosciences), Ki67 (Millipore), and p65 (Santa Cruz Biotechnology). Sections were developed with DAB and counterstained with hematoxylin.
Lung tissue scanning and lung lesion counting.
The lung tissue slides were scanned by an Aperio Whole Slide Scanner (ScanScope XT; Aperio Technologies), which digitizes whole microscope slides at ×20 or ×40 magnification and provides high-resolution images (∼0.5 microns/pixel for ×20 and ∼0.25 microns/pixel for ×40 scans). The lesions (hyperplasia and lung tumors) in all 5 lobes of lung tissues were counted.
ATPlite-based cell proliferation assay, IC50 determination, and clonogenic and soft agar assays.
Cells were seeded in 96-well plates in triplicate, and cell proliferation was measured with an ATPlite Kit (PerkinElmer) (60). To determine the IC50 value of MLN4924 against human lung cancer cells, cells were seeded in 96-well plates in triplicate and treated with MLN4924 at various concentrations for 72 hours, followed by ATPlite assay. Clonogenic survival assay and soft agar assays were performed as described previously (24, 61).
siRNA knockdown.
The lentivirus-based siRNA knockdown of SAG (Lt-SAG) along with scrambled siRNA control (Lt-Con) was performed as described (24). For double silencing, cells were infected with Lt-SAG or Lt-Con for 48 to 72 hours in 60-mm dishes. Cells were then split into three 60-mm dishes and transiently transfected with si-Con, si-p21, or si-DEPTOR, respectively, using Lipofectamine 2000. Forty-eight hours later, cells were harvested for proliferation, clonal survival, and soft agar assays as described (24, 61). The sequences for these siRNA oligonucleotides were as follows: Si-Con: 5′-AUUGUAUGCGAUCGCAGAC-3′; Si-SAG: 5′-GAGGACUGUGUUGUGGUCU-3′; Si-p21:5′-GUGGACAGCGAGCAGCUGA-3′; and Si-DEPTOR 5′-GCCAUGACAAUCGGAAAUCUA-3′.
SAG ectopic expression.
A549, A427, and H358 cells were transfected with FLAG-tagged SAG construct, along with pcDNA3 control. Cells were G418 selected for 2 weeks and all G418-resistant stable clones were pooled and subjected to immunoblotting and cell growth assays.
Immunoblotting analysis.
Lung cancer cells with or without MLN4924 treatment were harvested, lysed in a Triton X-100 lysis buffer and subjected to immunoblotting analysis (60). SAG monoclonal Ab was raised against the RING domain (AA44-113) (28). Other Abs were purchased commercially as follows: S6K1, cyclin D1, and c-Jun (Santa Cruz Biotechnology Inc.), human NOXA (Calbiochem), p21 (BD Transduction Labs), β-actin (Sigma-Aldrich), DEPTOR (Millipore and Cell Signaling Technology), phospho-4E-BP1, 4E-BP1, phospho-S6K1 (Thr389), p53, BIM, Notch-1, and phospho-IκBα (Cell Signaling Technology), and p27 (BD Biosciences — Pharmingen) and c-Myc (Epitomics).
Nuclear and cytoplasmic fractionation.
Cells after siRNA knockdown or MLN4924 treatment were washed with cold PBS and collected by scraping after addition of lysis buffer A (20 mM HEPES, pH 8.0, 20% glycerol, 10 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, pH 8.0, 1 mM DTT, 0.1% NP-40 and a proteinase inhibitor cocktail; Roche). Cell lysates were incubated on ice for 10 minutes, followed by centrifugation at 400 g for 5 minutes at 4°C. The supernatants were collected as the cytoplasmic fractions. The pellets were washed 3 times with lysis buffer A and then lysed in buffer B (20 mM HEPES, pH 8.0, 20% glycerol, 500 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, pH 8.0, 1 mM DTT, 0.1% NP-40 and a proteinase inhibitor cocktail) for 30 minutes on ice, followed by centrifugation at 15,000 g at 4°C for 15 minutes. The supernatants were collected as the nuclear fractions.
p65 DNA binding and NF-κB reporter activity assay.
Five micrograms of nuclear extracts was used to determine p65 DNA-binding activity by an ELISA-base assay kit, according to the manufacturer’s protocols (TransAM NF-κB, Active Motif). Briefly, the κB oligonucleotide–coated plates in a 96-well plate were incubated for 1 hour with the nuclear extract. Specificity was achieved by incubation with anti-p65 primary Abs for 1 hour. HRP-conjugated secondary Abs were used for the detection of p65 bound to the κB sequences (48). Luciferase-based reporter assay for NF-κB transcription activity was performed as described previously (16).
MLN4924 in vivo antitumor study.
Mice with the genotype LSL-KrasG12D in combination with WT or heterozygous Sag at the age of 6 to 8 weeks were administered with Ad-Cre as described above. MLN4924 (60 mg/kg, s.c.) was given once a day, 5 days a week, for 4 weeks, beginning at the 13th week, following Ad-Cre instillation. Mice in the drug control group received 10% 2-hydroxypropyl-β-cyclodextrin (HPBCD) as the vehicle control (2, 51). Mice were euthanized at the end of the 16th week, and their lungs were harvested after 10% formalin perfusion, fixed, sectioned, and stained with H&E. The slides were scanned, and lung lesions were counted.
Statistics.
Overall survival was defined from the date of diagnosis to death. The maximum follow-up time for each patient was 5 years, and patients who were alive were censored at the last follow-up time. Survival functions were plotted using the Kaplan-Meier method, and comparison of survival functions was performed by the log-rank test. In patient survival, Cox proportional hazard models were used to compare the overall survival after controlling for important prognostic factors including age, sex, stage, and grade. Two-tailed Student’s t test was used for tumor cell growth and tumor lesion comparison, as well as for comparing marker expression between different stages and grades. Data represent mean ± SEM. P < 0.05 was considered significant.
Study approval.
All procedures were approved by the University of Michigan Committee for the Use and Care of Animals. Animal care was provided in accordance with the principles and procedures outlined in the National Research Council Guide for the Care and Use of Laboratory Animals.
Supplementary Material
Acknowledgments
We thank Millennium Pharmaceuticals Inc. for providing MLN4924. We also thank Ronald Craig at the Department of Pathology, University of Michigan, for scanning the lung tissue slides. This work is supported by NCI grants CA118762, CA170995, and CA171277 (to Y. Sun).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(2):835–846. doi:10.1172/JCI70297.
References
- 1.Zhao Y, Sun Y. Cullin-RING ligases (CRLs) as attractive anti-cancer targets. Curr Pharm Des. 2013;19(18):3215–3225. doi: 10.2174/13816128113199990300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soucy TA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 3.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 4.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6(5):369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 5.Wei D, Sun Y. Small RING finger proteins RBX1 and RBX2 of SCF E3 ubiquitin ligases: the role in cancer and as cancer targets. Genes Cancer. 2010;1(7):700–707. doi: 10.1177/1947601910382776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu K, et al. The SCF(HOS/β-TRCP)-ROC1 E3 ubiquitin ligase utilizes two distinct domains within CUL1 for substrate targeting and ubiquitin ligation. Mol Cell Biol. 2000;20(4):1382–1393. doi: 10.1128/MCB.20.4.1382-1393.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamura T, Conrad MN, Yan Q, Conaway RC, Conaway JW. The Rbx1 subunit of SCF and VHL E3 ubiquitin ligase activates Rub1 modification of cullins Cdc53 and Cul2. Genes Dev. 1999;13(22):2928–2933. doi: 10.1101/gad.13.22.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu K, Chen A, Pan ZQ. Conjugation of Nedd8 to CUL1 enhances the ability of the ROC1-CUL1 complex to promote ubiquitin polymerization. J Biol Chem. 2000;275(41):32317–32324. doi: 10.1074/jbc.M004847200. [DOI] [PubMed] [Google Scholar]
- 9.Watson IR, Irwin MS, Ohh M. NEDD8 pathways in cancer, Sine Quibus Non. Cancer Cell. 2011;19(2):168–176. doi: 10.1016/j.ccr.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 10.Kamura T, et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science. 1999;284(5414):657–661. doi: 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- 11.Ohta T, Michel JJ, Schottelius AJ, Xiong Y. ROC1, a homolog of APC11, represents a family of cullin partners with an associated ubiquitin ligase activity. Mol Cell. 1999;3(4):535–541. doi: 10.1016/S1097-2765(00)80482-7. [DOI] [PubMed] [Google Scholar]
- 12.Seol JH, et al. Cdc53/cullin and the essential Hrt1 RING-H2 subunit of SCF define a ubiquitin ligase module that activates the E2 enzyme Cdc34. Genes Dev. 1999;13(12):1614–1626. doi: 10.1101/gad.13.12.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan P, et al. Recruitment of a ROC1-CUL1 ubiquitin ligase by Skp1 and HOS to catalyze the ubiquitination of IkBa. Mol Cell. 1999;3(4):527–533. doi: 10.1016/S1097-2765(00)80481-5. [DOI] [PubMed] [Google Scholar]
- 14.Duan H, et al. SAG, a novel zinc RING finger protein that protects cells from apoptosis induced by redox agents. Mol Cell Biol. 1999;19(4):3145–3155. doi: 10.1128/mcb.19.4.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swaroop M, et al. Yeast homolog of human SAG/ROC2/Rbx2/Hrt2 is essential for cell growth, but not for germination: Chip profiling implicates its role in cell cycle regulation. Oncogene. 2000;19(24):2855–2866. doi: 10.1038/sj.onc.1203635. [DOI] [PubMed] [Google Scholar]
- 16.Tan M, et al. Disruption of Sag/Rbx2/Roc2 induces radiosensitization by increasing ROS levels and blocking NF-kB activation in mouse embryonic stem cells. Free Radic Biol Med. 2010;49(6):976–983. doi: 10.1016/j.freeradbiomed.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan M, Davis SW, Saunders TL, Zhu Y, Sun Y. RBX1/ROC1 disruption results in early embryonic lethality due to proliferation failure, partially rescued by simultaneous loss of p27. Proc Natl Acad Sci U S A. 2009;106(15):6203–6208. doi: 10.1073/pnas.0812425106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan M, et al. SAG/RBX2/ROC2 E3 ubiquitin ligase is essential for vascular and neural development by targeting NF1 for degradation. Dev Cell. 2011;21(6):1062–1076. doi: 10.1016/j.devcel.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun Y. Induction of glutathione synthetase by 1,10-phenanthroline. FEBS Lett. 1997;408(1):16–20. doi: 10.1016/S0014-5793(97)00380-3. [DOI] [PubMed] [Google Scholar]
- 20.Sun Y, Li H. Functional characterization of SAG/RBX2/ROC2/RNF7, an antioxidant protein and an E3 ubiquitin ligase. Protein Cell. 2013;4(2):103–116. doi: 10.1007/s13238-012-2105-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He H, Tan M, Pamarthy D, Wang G, Ahmed K, Sun Y. CK2 phosphorylation of SAG at Thr10 regulates SAG stability, but not its E3 ligase activity. Mol Cell Biochem. 2007;295(1–2):179–188. doi: 10.1007/s11010-006-9287-3. [DOI] [PubMed] [Google Scholar]
- 22.Duan H, et al. Promotion of S-phase entry and cell growth under serum starvation by SAG/ROC2/Rbx2/Hrt2, an E3 ubiquitin ligase component: association with inhibition of p27 accumulation. Mol Carcinog. 2001;30(1):37–46. doi: 10.1002/1098-2744(200101)30:1<37::AID-MC1011>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 23.He H, Gu Q, Zheng M, Normolle D, Sun Y. SAG/ROC2/RBX2 E3 ligase promotes UVB-induced skin hyperplasia, but not skin tumors, by simultaneously targeting c-Jun/AP-1 and p27. Carcinogenesis. 2008;29(4):858–865. doi: 10.1093/carcin/bgn021. [DOI] [PubMed] [Google Scholar]
- 24.Gu Q, Tan M, Sun Y. SAG/ROC2/Rbx2 is a novel activator protein-1 target that promotes c-Jun degradation and inhibits 12-O-tetradecanoylphorbol-13-acetate-induced neoplastic transformation. Cancer Res. 2007;67(8):3616–3625. doi: 10.1158/0008-5472.CAN-06-4020. [DOI] [PubMed] [Google Scholar]
- 25.Tan M, et al. SAG/ROC-SCFβ-TrCP E3 ubiquitin ligase promotes pro-caspase-3 degradation as a mechanism of apoptosis protection. Neoplasia. 2006;8(12):1042–1054. doi: 10.1593/neo.06568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu Q, Bowden GT, Normolle D, Sun Y. SAG/ROC2 E3 ligase regulates skin carcinogenesis by stage-dependent targeting of c-Jun/AP1 and IκB-α/NF-κB. J Cell Biol. 2007;178(6):1009–1023. doi: 10.1083/jcb.200612067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan M, Gu Q, He H, Pamarthy D, Semenza GL, Sun Y. SAG/ROC2/RBX2 is a HIF-1 target gene that promotes HIF-1α ubiquitination and degradation. Oncogene. 2008;27(10):1404–1411. doi: 10.1038/sj.onc.1210780. [DOI] [PubMed] [Google Scholar]
- 28.Jia L, et al. Validation of SAG/RBX2/ROC2 E3 ubiquitin ligase as an anticancer and radiosensitizing target. Clin Cancer Res. 2010;16(3):814–824. doi: 10.1158/1078-0432.CCR-09-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang GY, et al. Attenuation of ischemia-induced mouse brain injury by SAG, a redox- inducible antioxidant protein. J Cereb Blood Flow Metab. 2001;21(6):722–733. doi: 10.1097/00004647-200106000-00010. [DOI] [PubMed] [Google Scholar]
- 30.Kim DW, et al. Transduced Tat-SAG fusion protein protects against oxidative stress and brain ischemic insult. Free Radic Biol Med. 2010;48(7):969–977. doi: 10.1016/j.freeradbiomed.2010.01.023. [DOI] [PubMed] [Google Scholar]
- 31.Jia L, Sun Y. SCF E3 ubiquitin ligases as anticancer targets. Curr Cancer Drug Targets. 2011;11(3):347–356. doi: 10.2174/156800911794519734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Y. E3 ubiquitin ligases as cancer targets and biomarkers. Neoplasia. 2006;8(8):645–654. doi: 10.1593/neo.06376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sasaki H, et al. Expression of the sensitive to apoptosis gene, SAG, as a prognostic marker in nonsmall cell lung cancer. Int J Cancer. 2001;95(6):375–377. doi: 10.1002/1097-0215(20011120)95:6<375::AID-IJC1066>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 34.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 35.Riely GJ, Marks J, Pao W. KRAS mutations in non-small cell lung cancer. Proc Am Thorac Soc. 2009;6(2):201–205. doi: 10.1513/pats.200809-107LC. [DOI] [PubMed] [Google Scholar]
- 36.Jackson EL, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15(24):3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia L, Soengas MS, Sun Y. ROC1/RBX1 E3 ubiquitin ligase silencing suppresses tumor cell growth via sequential induction of G2-M arrest, apoptosis, and senescence. Cancer Res. 2009;69(12):4974–4982. doi: 10.1158/0008-5472.CAN-08-4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shedden K, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008;14(8):822–827. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Regala RP, Davis RK, Kunz A, Khoor A, Leitges M, Fields AP. Atypical protein kinase C{iota} is required for bronchioalveolar stem cell expansion and lung tumorigenesis. Cancer Res. 2009;69(19):7603–7611. doi: 10.1158/0008-5472.CAN-09-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kissil JL, et al. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res. 2007;67(17):8089–8094. doi: 10.1158/0008-5472.CAN-07-2300. [DOI] [PubMed] [Google Scholar]
- 41.Schramek D, et al. The stress kinase MKK7 couples oncogenic stress to p53 stability and tumor suppression. Nat Genet. 2011;43(3):212–219. doi: 10.1038/ng.767. [DOI] [PubMed] [Google Scholar]
- 42.Lehman TA, et al. p53 mutations, ras mutations, and p53-heat shock 70 protein complexes in human lung carcinoma cell lines. Cancer Res. 1991;51(15):4090–4096. [PubMed] [Google Scholar]
- 43.Sasaki AT, et al. Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci Signal. 2011;4(163):ra13. doi: 10.1126/scisignal.2001518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeong WJ, et al. Ras stabilization through aberrant activation of Wnt/beta-catenin signaling promotes intestinal tumorigenesis. Sci Signal. 2012;5(219):ra30. doi: 10.1126/scisignal.2002242. [DOI] [PubMed] [Google Scholar]
- 45.Peterson TR, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137(5):873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(βTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell. 2011;44(2):304–316. doi: 10.1016/j.molcel.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barbie DA, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meylan E, et al. Requirement for NF-κB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462(7269):104–107. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Deshaies RJ, Emberley ED, Saha A. Control of cullin-RING ubiquitin ligase activity by Nedd8. In: Groettrup M, ed. Conjugation and Deconjugation of Ubiquitin Family Modifiers: Subcellular Biochemistry. Vol. 54. New York, New York, USA: Springer Science; 2010. [DOI] [PubMed] [Google Scholar]
- 50.Brownell JE, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. 2010;37(1):102–111. doi: 10.1016/j.molcel.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 51.Wei D, et al. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 2012;72(1):282–293. doi: 10.1158/0008-5472.CAN-11-2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao Y, Xiong X, Jia L, Sun Y. Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis. 2012;3:e386. doi: 10.1038/cddis.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Skaar JR, D’Angiolella V, Pagan JK, Pagano M. SnapShot: F Box Proteins II. Cell. 2009;137(7):1358. doi: 10.1016/j.cell.2009.05.040. [DOI] [PubMed] [Google Scholar]
- 54.Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE. The NEDD8 conjugation pathway and its relevance in cancer biology and therapy. Genes Cancer. 2010;1(7):708–716. doi: 10.1177/1947601910382898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soucy TA, Smith PG, Rolfe M. Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin Cancer Res. 2009;15(12):3912–3916. doi: 10.1158/1078-0432.CCR-09-0343. [DOI] [PubMed] [Google Scholar]
- 56.Nawrocki ST, Griffin P, Kelly KR, Carew JS. MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin Investig Drugs. 2012;21(10):1563–1573. doi: 10.1517/13543784.2012.707192. [DOI] [PubMed] [Google Scholar]
- 57.Xirodimas DP. Novel substrates and functions for the ubiquitin-like molecule NEDD8. Biochem Soc Trans. 2008;36(pt 5):802–806. doi: 10.1042/BST0360802. [DOI] [PubMed] [Google Scholar]
- 58.Rabut G, Peter M. Function and regulation of protein neddylation. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9(10):969–976. doi: 10.1038/embor.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009;4(7):1064–1072. doi: 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bockbrader KM, Tan M, Sun Y. A small molecule Smac-mimic compound induces apoptosis and sensitizes TRAIL- and etoposide-induced apoptosis in breast cancer cells. Oncogene. 2005;24(49):7381–7388. doi: 10.1038/sj.onc.1208888. [DOI] [PubMed] [Google Scholar]
- 61.Zheng M, et al. Growth inhibition and radiosensitization of glioblastoma and lung cancer cells by small interfering RNA silencing of tumor necrosis factor receptor-associated factor 2. Cancer Res. 2008;68(18):7570–7578. doi: 10.1158/0008-5472.CAN-08-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.