

Abstract
Accurate classification is essential for understanding the pathophysiology of a disease and can inform therapeutic choices. For hematopoietic malignancies, a classification scheme based on the phenotypic similarity between tumor cells and normal cells has been successfully used to define tumor subtypes; however, use of normal cell types as a reference by which to classify solid tumors has not been widely emulated, in part due to more limited understanding of epithelial cell differentiation compared with hematopoiesis. To provide a better definition of the subtypes of epithelial cells comprising the breast epithelium, we performed a systematic analysis of a large set of breast epithelial markers in more than 15,000 normal breast cells, which identified 11 differentiation states for normal luminal cells. We then applied information from this analysis to classify human breast tumors based on normal cell types into 4 major subtypes, HR0–HR3, which were differentiated by vitamin D, androgen, and estrogen hormone receptor (HR) expression. Examination of 3,157 human breast tumors revealed that these HR subtypes were distinct from the current classification scheme, which is based on estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2. Patient outcomes were best when tumors expressed all 3 hormone receptors (subtype HR3) and worst when they expressed none of the receptors (subtype HR0). Together, these data provide an ontological classification scheme associated with patient survival differences and provides actionable insights for treating breast tumors.
Introduction
Common classification terminology is necessary for medical progress. Over the past 2 centuries, normal tissue morphology and function has been successfully used as a reference point to define various diseases. Most notably, such an approach has been used to classify hematopoietic tumors, such as lymphomas and leukemias (1). The discovery of the morphologic and molecular resemblance of various subtypes of leukemias and lymphomas to particular normal hematopoietic cell types was critical in this process.
Based on this insight, hematopoietic malignancies have been classified as B cell and T cell neoplasms (e.g., small lymphocytic, large B cell, lymphoblastic, follicular, and mantle cell) that resemble specific normal cell types. Similarly, myeloproliferative diseases are classified as neutrophilic, granulocytic, lymphoblastic, prolymphocytic, myeloid, promyelocytic, monocytic, erythrocytic, basophilic, and megakaryoblastic neoplasms. Some of the most notable and earliest strides against cancers have been made in the treatment of hematopoietic malignancies (2). While many factors have contributed to this success, the accurate classification of hematopoietic malignancies played an important role. The identification of cell-type specific cluster of differentiation (CD) markers on the surface of these cells permitted efficient immunophenotyping (3). These CD markers were later used to identify lymphomas and leukemias with a phenotype nearly identical to a specific normal cell type, allowing the development of the current classification system of these diseases (4). Despite major successes in rationally classifying and treating hematological malignancies, the use of normal cell types to classify solid tumors has not been widely emulated. A major reason for this has been our lack of understanding of the diversity of cell types in most solid tissues.
Characterization of normal cell subtypes in solid tissues has been challenging. Until recently, only 2 cell types have been morphologically described in the human breast: the inner luminal cells and the outer myoepithelial cells (5). This limited understanding of the cell types comprising the breast ducts has precluded the development of a normal cell type–based classification system. While there has been more recent interest in normal breast cell subtypes, this research has been difficult to correlate with existing human breast tumor phenotypes (6). Numerous markers have been used to describe normal human mammary stem/progenitor cells, including CD44hiCD24lo, aldehyde dehydrogenase–high (ALDHhi), CD10+, Ep-CAM+MUC1–, and Ep-CAMhiCD49f+. Whether these stem/progenitor cell markers identify the same cell populations remains unknown. Furthermore, Tlsty and colleagues discovered that human breast cells can exhibit extensive lineage plasticity (7), which may explain why marker profiles have been difficult to associate with distinct tumor subtypes.
Clinically, human breast cancers are grouped into 3 categories based on the presence of estrogen receptor (ER+), progesterone receptor (PR+), and human epidermal growth factor receptor 2 (HER2+), or by their absence in triple-negative breast cancers (TNBCs; i.e., ER–PR–HER2–).
In the research setting, mRNA profiles have been used to define prognostic subtypes of breast cancer: luminal A, luminal B, basal-like, claudin-low, and HER2-like (8). DNA methylation patterns have also been used to identify 5 distinct DNA methylation groups (9), and 10 different breast cancer clusters have been identified in a genome-driven integrated classification system, each associated with distinct clinical outcomes (10, 11). Several additional mRNA expression–based molecular prognostic panels, such as Oncotype Dx, PAM50, and MammaPrint, have also emerged with potential clinical utility (12).
The main evidence supporting the importance of each of these molecular subtypes has been identification of patient groups with different outcomes. Hence, it is important to recognize that these molecular subtypes are prognostic categories, different from disease taxonomy. Therefore, while these molecular prognostic tools have been useful in the research setting, they have not produced a commonly agreed-upon new system of classification that is uniformly used in the clinic. This is partly because each molecular platform appears to produce a different prognostic classification. A breast cancer task force recently concluded that at the moment, molecular tools do not provide sufficiently robust information beyond histological type, grade, and ER/PR/HER2 status (13). Thus, these molecular tests are not routinely performed at most institutions (14).
It is increasingly becoming clear that a more fundamental breast cancer classification system, one that does not conflate prognostic categories with diagnostic categories, is needed. Ideally, such a system should be robust and not change depending on which technological platform is used to classify breast cancer. Inspired by the classification of hematopoietic malignancies, we hypothesized that differentiation states of normal cell populations in normal human breast may provide such a reference classification system for human breast tumors.
Results
The normal human breast is composed of milk-producing lobules and interlobular ducts that transport the milk to the nipple (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI70941DS1). This anatomical distinction is important for understanding breast cancer, because in addition to ER/PR/HER2 status, human breast tumors are classified by pathologists on morphological grounds, either as ductal carcinomas or as lobular carcinomas, for reasons unrelated to their cell of origin. This arcane terminology has resulted in a common misconception that ductal and lobular breast cancers initiate in the normal ducts and lobules, respectively. However, despite their names, almost all of the early progression steps for both tumor types almost exclusively involve the breast lobules. Thus, in the present study, we specifically examined the normal cells in the lobules using immunohistochemical (IHC) staining, which preserves tissue architecture and allows for discrimination of ducts, lobules, and different layers of the epithelium (see below). For a list of the 37 primary antibodies used in these studies, see Supplemental Table 1.
Analysis of CD markers and intermediate filaments in normal human breast.
An ideal cell type–specific immunostain marker should have a bimodal expression pattern (i.e., one subpopulation is clearly negative, and the other strongly positive). While CD markers have been useful in isolating breast cell types using FACS, we found that they had a gradient-type expression pattern in situ that limited their utility to define cell subtypes using semiquantitative methods such as IHC (Supplemental Figure 1).
In an attempt to identify molecules with bimodal expression patterns in normal human breast, we examined the expression of intermediate filaments. These molecules are differentially expressed in distinct cell types, and their expression is both tissue- and cell type–specific. Furthermore, it has been well recognized that cell type–specific expression of intermediate filaments is preserved in tumors and can be used to determine the tissue origin of tumors (15). We found that keratin 5 (K5), K7, K8, K14, K17, K18, and K19 were useful in identifying subpopulations of human breast cells, because they were expressed in a bimodal pattern (Supplemental Figure 2).
Next, we subjected normal breast tissues from 36 breast reduction mammoplasty procedures to IHC with K5, K7, K8, K14, K17, K18, K19, CD10, SMA, and p63. Normal breast lobules and ducts are lined by a bilayer epithelium, consisting of an inner layer of milk-producing luminal cells and an outer layer of supportive myoepithelial cells. As previously shown (16), we found that K7, K18, and claudin-4 (Cld-4) were expressed in all luminal cells, but not in myoepithelial cells (Figure 1A and Supplemental Figure 2A). In contrast, CD10, SMA, and p63 were expressed in all myoepithelial cells, but not in luminal cells (Supplemental Figure 2). Thus, these markers constitute a pan-luminal versus pan-myoepithelial panel. Interestingly, in some lobules, luminal cells were K19– (Figure 1B); thus, K19 was not a pan-luminal marker.
Figure 1. Expression of intermediate filaments and ER in normal human breast.
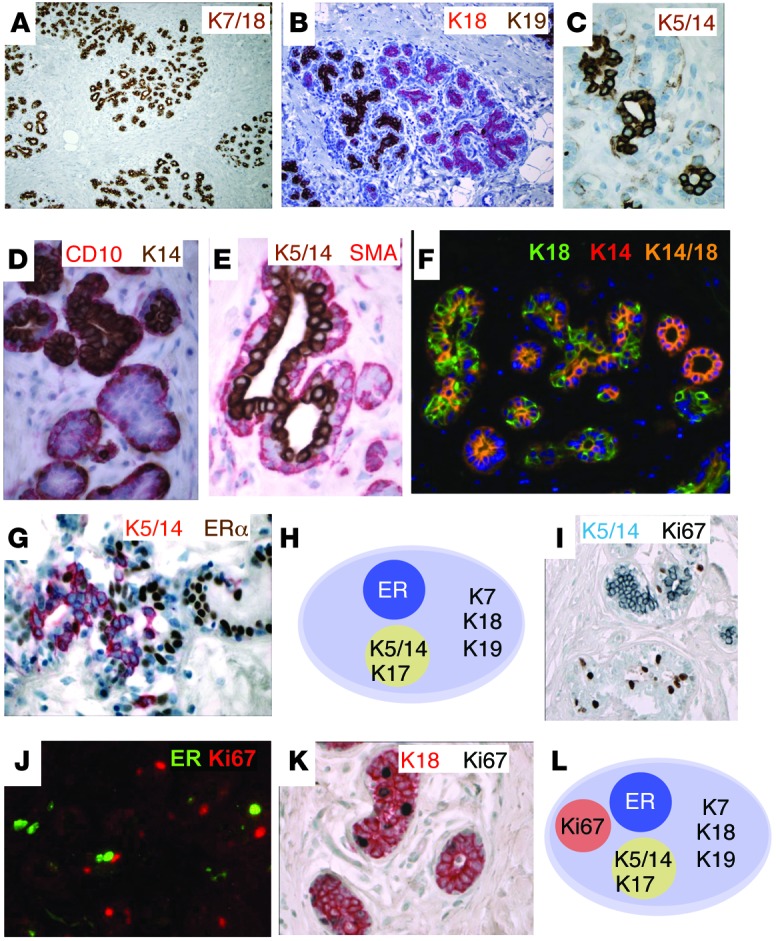
Single and double IHC with immunoperoxidase (A–E, G, I, and K) and merged IHC images (F and J) of normal human FFPE sections are shown. (A) K7/18 (brown). (B) K18 (red) and K19 (brown). (C) K5/14 (brown). (D) CD10 (red) and K14 (brown). (E) K5/14 (brown) and SMA (red). (F) K18 (green) and K14 (red). Merged K14+K18+ appears yellow. (G) K5/14 (red) and ER (brown). We designated this population of cells K5/14/17+ because the tissue sections were not stained simultaneously with these markers. (H) Differentiation states of normal luminal epithelial cells, based on expression of ER and keratins. (I) Ki67 (brown) and K5/14 (blue). (J) ER (green) and Ki67 (red). (K) K18 (red) and Ki67 (brown). (L) Differentiation states of normal luminal epithelial cells, based on ER, keratins, and Ki67. Representative images were selected from multiple patient samples (n = 36). Original magnification, ×20 (A); ×40 (B); ×200 (F); ×400 (C, G, and I–K); ×600 (D and E). See http://sylvester.org/ince for additional high-resolution images.
In human skin, K5/14/17 are exclusively expressed in the basal layers; in mouse mammary tissue, they are expressed in the myoepithelial layer (Supplemental Figure 2C). Hence, these keratins are usually referred to as basal keratins. However, in normal human breast tissue, K5/14/17 were expressed in both luminal and basal layers, depending on location. In the interlobular ducts, K5/14/17 were expressed in the myoepithelial (basal) layer, as expected (Supplemental Figure 1B and Supplemental Figure 2, D–F). However, in the lobules, the site where precursor lesions develop, K5/14/17 were expressed in the luminal layer (Figure 1, C–E, Supplemental Figure 1J, Supplemental Figure 2, G–I, and ref. 6). We confirmed the luminal nature of these cells with double IHC, which demonstrated that the K5+, K14+, or K17+ cells were Ki67/ER– (Figure 1, G–J) and CD10/SMA/K17– (Supplemental Figure 2, J–L) and were located above the CD10/SMA/K17+ myoepithelial cell layer (Supplemental Figure 2, J–L). We did not find luminal K5+ cells in the mouse breast (Supplemental Figure 2C).
We identified luminal K5+, K14+, or K17+ cells in all 36 patients examined; thus, this was a robust and highly reproducible luminal cell subpopulation. Interestingly, while some lobules had a small percent of luminal K5+, K14+, or K17+ cells, adjacent lobules were entirely composed of K5+, K14+, or K17+ luminal cells (Supplemental Figure 2, M–P).
When 2 different cell lineages are defined by mutually exclusive expression of markers, coexpression of these markers in the same cell has been used as evidence of “stemness.” Previously, coexpression of K5/14/17 with K7/8/18 has been interpreted as evidence for bipotential cells. Here, however, some lobules were entirely composed of K14+K18+ or K5+K18+ double-positive cells in nearly every tissue section examined (Figure 1F and Supplemental Figure 2, Q–U). On average, 36% of luminal cells were K14+K18+ (n = 746), and 16% were K5+K18+ (n = 1,339). Importantly, K5/14+ cells also expressed MUC1, a marker of luminal differentiation (Supplemental Figure 2V). It would be extremely unusual to find an epithelial tissue entirely composed of progenitor/stem cells. Thus, our results indicated that the luminal layer cells coexpressing K5/14/17 with K18/19 are more consistent with a differentiated luminal cell variety (6, 17).
Analysis of hormone receptors in normal human breast.
Having identified 2 subtypes of luminal layer cells based on K5/14/17 expression, we next characterized the expression of hormone receptors (HRs) in these cells, because they are involved in differentiation and some have a bimodal expression pattern.
In an initial survey, 3 receptors — ER, androgen receptor (AR), and vitamin D receptor (VDR) — stood out with distinct bimodal expression patterns. Many of the other HRs (i.e., TRHα, TRHβ, PTH1R, OXTR, SSTR1, SSTR2, SSTR3, SSTR5, RARα, RARβ, RXRα, and RXRβ) did not appear to have a bimodal expression pattern. Because PR expression tracks with ER expression, we did not include PR in this study.
Next, we carried out double IHC on normal breast sections and counted cells in 5 different sections for coexpression of various markers (Supplemental Table 2). Double IHC demonstrated that all ER+ cells were luminal and did not overlap with K5/14/17+ luminal cells (<0.3% overlap, n = 3,313) or with Ki67+ proliferating cells (0.1% overlap, n = 1,206) (Figure 1, G and J, and Supplemental Table 2). Nearly all proliferating Ki67+ cells were K18+ luminal cells that were negative for the myoepithelial markers CD10 (0.5% overlap, n = 1,084) and K5/14/17 (0%–1.9% overlap, n = 1,078) (Figure 1K and Supplemental Table 2). These results allowed us to define 4 mutually exclusive subsets of luminal cells in normal human breast that were all positive for the pan-luminal markers K7 and K18 (Figure 1L): (a) ER+ cells, (b) K5/14/17+ cells, (c) ER–K5/14/17– cells, and (d) Ki67+ cells.
Double IHC demonstrated that all AR+ cells were luminal, and they were also mutually exclusive with K5/14+ cells (0.0% overlap, n = 789) and Ki67+ cells (0.0% overlap, n = 698) (Figure 2, A and B, and Supplemental Table 2). AR+ cells partially overlapped with ER+ cells (44% overlap, n = 429) (Figure 2C and Supplemental Table 2). These results allowed us to describe 3 subsets of HR+ cells: ER+, AR+, and ER+AR+ (Figure 2D). Double IHC demonstrated that VDR+ cells were exclusively in the luminal layer as well, with no overlap with CD10+ myoepithelial cells or proliferating Ki67+ cells (0.0% overlap, n = 179), but they did partially overlap with K5/14+ cells (15%–23% overlap, n = 266), AR+ cells (16%–35% overlap, n = 835), and ER+ cells (22%–74% overlap, n = 749) (Figure 2, E–I, and Supplemental Table 2).
Figure 2. Expression of intermediate filaments, ER, AR, and VDR in normal human breast.

Double IHC (A and J) and merged images (B, C, E–I, and K–M) of normal human breast FFPE sections, as well as differentiation states of luminal (D and N) and myoepithelial (O) cell types, are shown. (A) K5/14 (red) and AR (brown). (B) AR (green) and Ki67 (red). (C) ER (green) and AR (red). Merged ER+AR+ appears yellow. (D) Differentiation states of normal luminal epithelial cells based on presence of ER, keratins, Ki67, and AR. (E) CD10 (green) and VDR (red). (F) VDR (red) and Ki67 (green). (G) K5 (green) and VDR (red). (H) AR (green) and VDR (red). Merged AR+VDR+ appears yellow. (I) ER (green) and VDR (red). Merged ER+VDR+ appears yellow. (J) CD10 (red) and Ki67 (brown). (K) ER (green), AR (red), and VDR (blue). Merged ER+AR+ appears yellow; merged ER+VDR+ appears purple. (L) ER (green), AR (green), and VDR (red) shown individually. In the merged image, ER+AR+VDR+ (i.e., HR3) appears white. (M) HR3 (green), Ki67 (red), and DAPI (blue; nuclear marker). (N and O) Differentiation states of normal luminal (N) and myoepithelial (O) breast cells based on the full marker panel. Representative images were selected from multiple patient samples (n = 36). Original magnification, ×200 (A–C, E–K, and M); ×400 (L). See http://sylvester.org/ince for additional high-resolution images.
Triple IHC also demonstrated the presence of triple-HR+ cells (i.e., ER+AR+VDR+; Figure 2, K and L). These results allowed us to describe 7 subsets of HR+ cells in the luminal layer of human breast lobules: ER+, AR+, VDR+, ER+AR+, ER+VDR+, AR+VDR+, and ER+AR+VDR+ (Figure 2N). Interestingly, only VDR+ cells substantially overlapped with K5/14/17+ luminal cells, and the proliferating K18+Ki67+ luminal cells were ER–AR–VDR–K5/14– (Figure 2M).
Cumulatively, in the luminal layer of normal human breast, we were able to define 11 differentiation states (Table 1), including 3 HR– states (collectively designated group HR0, states L1–L3; ER–AR–VDR–), which were either K5/14/18– (L2; 52%–83%) or K5/14/18+ (L3; 17%–48%, n = 2,085), and 8 HR+ states, grouped as single-HR+ (HR1, states L4–L7; ER+, AR+, or VDR+), double-HR+ (HR2, states L8–L10; ER+AR+, ER+VDR+, or AR+VDR+), or triple-HR+ (HR3, state L11; ER+AR+VDR+).
Table 1.
Cellular differentiation states in normal human breast lobules
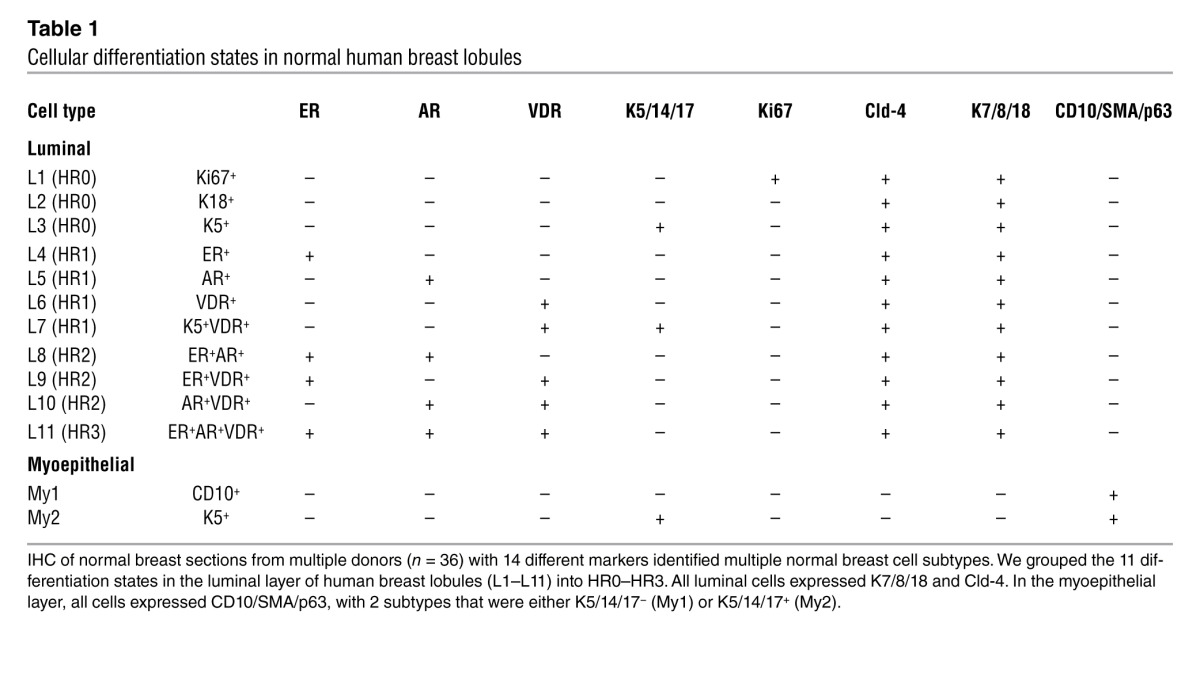
In the myoepithelial layer, all cells expressed CD10, SMA, and p63, with 2 subtypes, K5/14/17– and K5/14/17+ (designated My1 and My2, respectively; Table 1). Proliferating cells were very uncommon in the myoepithelial layer; CD10 and Ki67 overlapped in only 0.5% of the cells (Figure 2J, Table 1, and Supplemental Table 2).
Simultaneous examination of 12 markers in normal human breast with a novel multiplex immunofluorescence method.
In the above experiments, we were able to stain the same formalin-fixed, paraffin-embedded (FFPE) section with up to 3 different antibodies simultaneously. A greater number of antibodies is difficult to multiplex by conventional methods, for multiple reasons (see Supplemental Methods).
To confirm simultaneous coexpression patterns predicted by double and triple IHC for all 12 different markers (ER, AR, VDR, K5, K7, K8/18, Cld-4, CD10, SMA, Ki67, NaKATPase, and DAPI), we wanted to examine their expression in the same cells. Recently, a new technology has been developed by GE Healthcare that allows for immunofluorescence (IF) of the same tissue section with more than 10 different antibodies serially (known as multiplex IF; ref. 18), which was used to confirm all of our results (Figure 3, Supplemental Figure 3, and Supplemental Methods).
Figure 3. Multiplex IF of 12 markers in normal human breast.

(A–I) 1 FFPE section of normal breast epithelium was stained serially with each antibody for the markers (A) pan-keratin (Pan-K, green), (B) K18 (red), (C) K5 (red), (D) DAPI (blue), (E) ER (green), (F) AR (green), (G) VDR (red), (H) Ki67 (red), and (I) SMA (green). (J–O) The individual IF staining images were merged to reveal the coexpression pattern of all markers in each cell. (J) K5 (red) and SMA (green). (K) K5 (red) and K18 (green). (L) ER (red), AR (green), and K5 (blue). (M) VDR (red) and ER (green). (N) VDR (red) and AR (green). (O) AR (red), ER (green), and VDR (blue). (P) Differentiation states of normal luminal breast cells based on the full marker panel. Representative images were acquired using multiplex IF technology (GE Healthcare). Original magnification, ×200 (A–O). See http://sylvester.org/ince for additional high-resolution images, including K7, Cld-4, NaKATPase, and CD10 stains.
We used image analysis software for quantitative analysis of our multiplex IF for ER, AR, VDR, K5, and Ki67 in individual cells. Each cell was numbered by the image analysis software, and the fluorescent signal specifically from the luminal epithelium was measured for each marker. We plotted the results for each marker as a percentage of total fluorescence for each cell. This analysis allowed us to correlate the expression of these markers in >300–500 individual cells in lobules from 8 different patients (Figure 4 and Supplemental Figure 3C). Based on the double IHC analysis (Figures 1 and 2), we had deduced that there were inverse correlations between Ki67 and K5, between Ki67 and ER/AR/VDR, and between K5 and ER/AR (Supplemental Table 2). Multiplex IF allowed us to demonstrate all of these complex trends for the first time in individual cells (Figure 4 and Supplemental Figure 3).
Figure 4. Multiplex analysis of 12 markers in normal human breast.

Histograms of relative ER, AR, VDR, K5, and Ki67 expression in each luminal cell in normal human breast lobules 1–4. Cell number is plotted against percent contribution of each marker to total fluorescence of each cell. See Supplemental Figure 3 for additional lobules.
These observations highlighted 4 predominant, mutually exclusive differentiation patterns in the luminal layer: a HR+ state, a proliferative state (Ki67+), and 2 HR– states, one K5+ and the other K5– (Figure 4 and Supplemental Table 2). Consistent with this, we observed that Ki67+ and K5+ cells were rare in lobules that were enriched in HR+ cells (Supplemental Figure 3D). When K5+ cells expanded, HR+ and Ki67+ cells decreased, and in highly proliferative areas, there were very few HR+ or K5+ cells (Supplemental Figure 3D). Hence, it appears that a given cell can exist in only one of these differentiation states at one time.
Analysis of HR+ and HR– cell types in ER+ breast tumors.
The remarkable heterogeneity observed at the single-cell level in normal breast epithelium was reminiscent of the distinct features of normal hematopoietic cell populations. Because hematological malignancies maintain normal cell type– and differentiation-specific patterns, we next asked whether breast tumors do as well.
We evaluated the staining pattern of 12 protein markers in 20 full FFPE sections using IHC, then confirmed the results using tissue microarrays (TMAs) that contained 216 tumors (51 ER+, 46 HER2+, and 119 TNBC) (Figure 5). Staining was scored by combining percent expression and staining intensity on a 0–25 expression scale (Supplemental Figure 4).
Figure 5. Identification of normal cellular phenotypes in human breast tumors.

Heat maps of Cld-4, K7, K18, VDR, AR, K5, K14, CD10, SMA, p63, PR, ER, and HER2 protein levels in 216 human breast cancer tumors, separated into (A) ER+ (n = 51), (B) HER2+ (n = 46), and (C) TNBC (n = 119). Luminal markers (Cld-4, K7, K18, VDR, and AR) and basal markers (CD10, SMA, and p63) are indicated. TNBCs are separated into luminal 1 (LM1; K5/14–), luminal 2 (LM2; K5/14+), and mixed (M; expressing both luminal and myoepithelial markers) subtypes. TMA sections were subjected to IHC and scored using light microscopy on a scale of 0 (blue, low expression) to 25 (yellow, high expression), with white denoting intermediate expression. Corresponding normal cell counterparts are illustrated next to each heat map.
In both tissue sets, we observed that all ER+ human breast cancers strongly expressed multiple pan-luminal markers (Cld-4, K7, and K18), whereas none were positive for pan-myoepithelial markers (CD10, SMA, and p63) (Figure 5A). All ER+ breast cancers were K5/14–. Interestingly, the majority of ER+ tumors were VDR+ (93%), and two-thirds were AR+ (59%) (Figure 5A). This pattern was identical to that of normal breast ER+ cells, which could coexpress AR or VDR, but were very rarely K5/14/17+ or CD10/SMA+. These results indicate that all ER+ tumors have a luminal phenotype identical to HR+ normal luminal cell type L4, L8, L9, or L11 (Figure 5A and Supplemental Table 3). Intriguingly, we also observed that, similar to normal tissues, most proliferating tumor cells (Ki67+) were ER–AR–VDR–, with focal VDR+ proliferating tumor cells (Supplemental Figure 5).
Analysis of HR+ and HR– cell types in HER2+ breast tumors.
In HER2+ tumors, we observed strong expression of multiple pan-luminal markers (Cld-4, K7, and K18) and none of the pan-myoepithelial markers (CD10, SMA, and p63) (Figure 5B). Nearly all HER2+ tumors (44 of 46) had a luminal phenotype identical to that of HR+ normal breast cells (i.e., L4–L11). A minority of HER2+ tumors (2 of 46) were similar to HR– cells (Figure 5B and Supplemental Table 5).
Analysis of HR+ and HR– cell types in TNBCs.
TNBCs are defined as ER–PR–HER2–. We examined 119 TNBCs for the expression of HRs and keratin markers, which revealed 3 major subgroups (Figure 5C and ref. 19). Nearly 66% of TNBCs (78 of 119) had a pure luminal phenotype, positive for pan-luminal markers and negative for pan-myoepithelial markers; of these, 37 were identical to K5/14– HR0 luminal cells (designated luminal 1), and 41 were identical to K5/14+ HR0 luminal cells (luminal 2) (Figure 5C and Supplemental Table 5). All remaining TNBCs (33%, n = 41) strongly expressed luminal markers (Cld-4, K7, AR, VDR, and K18), but 38 of these tumors also expressed myoepithelial markers (CD10, SMA, and p63) (Figure 5C), consistent with a mixed phenotype.
In summary, 95% of human breast tumors were phenotypically identical to one of the normal luminal breast cell subtypes (Supplemental Table 3), similar to lymphomas and leukemias. For the remaining 5% (HR0 tumors with a mixed phenotype), it is possible that their normal counterparts are rare progenitor cells with a mixed luminal/myoepithelial phenotype (20), or that these tumors exhibit an altered phenotype due to mutations that result in inappropriate expression of these markers.
Expression of normal basal versus luminal-specific mRNAs in TNBC.
The cell of origin of TNBC has been of great interest recently (21). As mentioned above, K5/14/17 are expressed in the basal layers of human skin and rodent mammary glands (Supplemental Figure 2C). Thus, these keratins have been commonly referred to as basal keratins in the literature (17), and TNBCs that express them have been called basal-like carcinoma (BLC) (22). Consequently, some have suggested that these tumors are similar to myoepithelial (basal) cells of the normal breast. However, as we demonstrated here, K5/14/17 were predominantly expressed in the luminal layer of normal human breast lobules, and K5/6+ BLCs expressed markers identical to those of L3 and L7 luminal cells (Table 1 and Supplemental Table 3).
Because the TNBC/BLC category was based on mRNA expression in microarray analysis (23–25), we also carried out an analysis of mRNA in normal human breast cells, by combining results from 3 different studies that profiled highly purified luminal versus myoepithelial cells (26–28). We found that 131 mRNAs were identified as luminal-specific and 90 as myoepithelial-specific in at least 2 of the 3 datasets (Supplemental Table 4), providing a strong consensus signature distinguishing normal luminal versus myoepithelial cells. Next, we examined the expression of these genes in basal-like and non-basal-like human breast tumors (29–32).
Interestingly, no significant correlation was observed between basal-like tumors in these cohorts and the expression signature of normal basal/myoepithelial cells (P = 0.22, Fisher exact test; Supplemental Figure 6A). Thus, the differentiation state of BLC is most similar to K5/14/17/18+ normal luminal cells of the breast (L3; Supplemental Table 5), and the name basal-like is probably not an accurate description of neither their differentiation state nor their cell of origin (6, 17, 21, 33, 34). In some cohorts, patients with basal-like tumors have a worse outcome than those with TNBC tumors (22); here, we did not observe a significant difference between K5/6+ versus K5/6– TNBC patients (Supplemental Figure 6B).
Distribution of HR0–HR3 breast tumor phenotypes in the NHS cohort.
Based on the above results, we hypothesized that human breast tumors can be classified according to normal breast differentiation states and tested this hypothesis using a breast cancer cohort from the Nurses’ Health Study (NHS), with >25 years of follow-up from a large number of patients (n = 1,731) (35–37). We conducted IHC of NHS TMAs with ER, PR, HER2, VDR, AR, K8/18/Cld-4, K5/6, and CD10/SMA/p63 antibodies and scored them semi-quantitatively into 4 categories based on normal tissue differentiation: HR3 (ER+AR+VDR+), HR2 (ER+AR+, AR+VDR+, or ER+VDR+), HR1 (ER+, VDR+, or AR+), and HR0 (ER–AR–VDR–).
Importantly, the 4 HR categories are different from the current ER+, HER2+, and TNBC classification. For example, based on standard classification, 75% of NHS study patient tumors were ER+ (n = 1,356), 10% were HER2+ (n = 177), and 15% were TNBC (n = 253) (Figure 6A and Supplemental Table 5). These were reclassified as 58.1% HR3 (n = 1,006), 24.8% HR2 (n = 429), 10.7% HR1 (n = 185), and 6.4% HR0 (n = 111) (Figure 6B), because each standard breast cancer subtype was composed of multiple HR groups: of ER+ tumors, 75.1% were HR3, 23.4% were HR2, and 1.5% were HR1; of HER2+ tumors, 29.4% were HR3, 43.5% were HR2, 22.0% were HR1, and 5.1% were HR0; of TNBC tumors, 36.8% were HR0, 44.6% were HR1 (i.e., AR+ or VDR+), and 18.6% were HR2 (i.e., AR+VDR+) (Supplemental Figure 7A and Supplemental Table 5). Thus, our HR-based classification approach does not merely rename existing groups, but organizes tumors in a new way.
Figure 6. Normal cell subtype-based classification identifies breast cancers with different outcomes.

(A) Distribution of ER+, HER2+ and TNBC cases from the full panel of NHS cases analyzed in this study. (B) Reclassification of ER+, HER2+, and TNBC human breast tumors from the full panel of NHS cases analyzed in this study as HR3 (ER+AR+VDR+), HR2 (ER+AR+, AR+VDR+, or ER+VDR+), HR1 (ER+, VDR+, or AR+), and HR0 (ER–AR–VDR–). Breast tumors were divided into the 4 HR0–HR3 categories based on normal tissue differentiation (see Supplemental Table 3). (C) Kaplan-Meier analysis for overall survival of all individuals with invasive breast cancer from the NHS, scored by IHC. (D) Kaplan-Meier analysis of relapse-free survival for all invasive breast cancers from an 855-patient breast tumor dataset (38). Tumors were ranked according to gene expression values for ER, AR, and VDR, scored as high or low based on a 50% cutoff point, and assembled based on HR status (HR0, n = 141; HR1, n = 287; HR2, n = 284; HR3, n = 143).
Analysis of breast cancer outcomes based on normal cell lineage phenotypes.
We next investigated whether the HR0–HR3 categories correlated with breast cancer survival and found a strong association between the total number of positive receptors and outcome. Kaplan-Meier analyses of the NHS cohort showed that patients with HR3 tumors had the best survival, those with HR1 tumors had the worst survival, and those with HR2 tumors had intermediate survival (P < 0.0001; Figure 6C). In multivariate analysis, these differences remained significant: compared with HR3 tumors, the relative hazard ratio (RHR) for HR2 tumors was 2.9 (95% CI, 1.60–5.21); for HR1 tumors, the RHR was 5.3 (95% CI, 2.77–9.97), and for HR0, the RHR was 6.9 (95% CI, 3.37–14.39) (Supplemental Table 6).
Interestingly, the HR0 group had a biphasic outcome curve similar to that of HR1 tumors, with the worst outcome during the first 5 years, followed by a flat curve thereafter (Figure 6C), consistent with an excellent outcome. Thus, we reevaluated the association stratified by time, before and after a 5-year cutoff. During the first 5 years, HR3 tumors had the best outcome; compared with HR3, HR2 tumors had a worse outcome (RHR, 1.69; 95% CI, 1.14–2.50), and HR1 tumors (RHR, 2.44; 95% CI, 1.55–3.84) and HR0 tumors (RHR, 2.7; 95% CI, 1.56–4.70) had the worst outcome (P < 0.0001; Supplemental Figure 7, B and C, and Supplemental Table 6). After 5 years, there was no significant difference among HR3, HR2, and HR1 (P > 0.5), but HR0 had a better outcome (RHR, 0.34; P = 0.02; Supplemental Figure 7D). Analyzing the HER2 groups separately did not change these results (Supplemental Figure 7E). In a multivariate analysis, these differences remained significant even after accounting for other factors, such as age, stage, grade, HER2 status, treatment, and radiation (Supplemental Tables 5 and 6).
We evaluated our survival results at the mRNA expression level by examining a meta-dataset of gene expression TMAs from 855 human breast tumors (38). Kaplan-Meier analyses for relapse-free survival showed that women with HR3 tumors had the best outcome, HR1 and HR0 tumors were the most aggressive, and HR2 tumors were intermediate between these groups. Unlike the IHC-based HR categories, which had significant overall survival differences (P < 0.0001; Figure 6C), there was a more modest overall relapse-free survival difference among the mRNA-based HR groups (P = 0.13; Figure 6D). However, lung metastasis relapse-free survival differences among mRNA-based HR groups were significantly different (P = 0.0014; Supplemental Figure 7F). Taken together, these data support a correlation of ER/AR/VDR with tumor differentiation state: more differentiation correlates with less aggressive behavior. Importantly, these results suggest that measurement of ER, AR, and VDR protein levels may be more relevant than mRNA levels.
Analysis of HR+ and HR– cell types in breast cancer cell lines.
We next examined whether the HR0–HR3 phenotypes are preserved in breast cancer cell lines. Publicly available mRNA expression data from more than 50 breast cancer cell lines were analyzed (39), which revealed that most tumor cell lines fell into one of the normal cell-of-origin categories.
We found that the HR+ pure luminal breast cancer cell lines (ER/AR/VDR+; n = 16) rarely expressed K5/14/CD10/SMA, as expected (Supplemental Figure 8A). This was also true for the HER2+ tumor cells lines (n = 13), which rarely expressed K5/14/CD10/SMA, but were occasionally AR/VDR+, as expected. 7 cell lines had a TNBC luminal 2 phenotype (BT-20, SUM149, HCC38, HCC-1187, BPLER, HCC-1143, HCC-1500), and 6 cell lines had a TNBC mixed phenotype (MDA-MB-468, HCC1937, HCC-70, HMLER, HCC-3153, HCC2157) (Supplemental Figure 8A). We also confirmed these phenotypes at the protein level in breast cancer cell lines, in order to select a subset of lines that closely conformed to in vivo HR phenotypes (Supplemental Figure 8, B and C). This set of breast cancer cell lines was then examined for in vitro drug response studies.
Interestingly, 9 cell lines that are frequently used as models of human TNBC (MDA-MB-231, SUM-159PT, MDA-MB-157, MDA-MB-436, HBL100, BT549, SUM1315M02, MDA-MB-435, and HS578T) had expression profiles that were not present either in normal breast cells or in human breast cancers (i.e., negative for most HRs and epithelial markers; Supplemental Figure 8A). Since this profile is almost never seen in vivo, either these cells have lost their original phenotype, or they were derived from very rare tumor types, cautioning against their frequent use.
Response of breast cancer cell lines to HR inhibition.
The HR0–HR3 classification of breast cancers not only correlated with clinically significant outcome groups, but may also provide insights about how the treatment of these patients might be personalized. For example, we envisioned that HR3 tumors might be treated using triple-hormone therapy (ER antagonists plus AR and VDR agonists), and tested some of these concepts in breast cancer cell lines.
There are currently very few effective treatments against TNBCs, because they are ER– and HER2–. However, because 63% of TNBCs were AR+, VDR+, or AR+VDR+, hormone treatment might be possible in a majority of TNBCs, in combination with chemotherapy. Cell lines BT-20, MDA-MB-468, and SUM159 only expressed VDR, corresponding to the HR1/TNBC phenotype. We found that combining the VDR agonist calcitriol with taxol additively inhibited proliferation of these HR1 breast cancer cells more effectively than either drug alone (Supplemental Figure 9A).
A similar combination treatment strategy can be also employed in ER+ tumor cells; for example, the HR2 ZR75B cell line was ER+VDR+, and combining calcitriol with low doses of the ER antagonist ICI182,780 (0.5 nM) additively inhibited proliferation of these cells (Supplemental Figure 9B). In another example, the combination of the AR agonist R1881 (50 nM) with calcitriol (50 nM) additively inhibited proliferation of the HR3 breast cancer cell line T47D (Supplemental Figure 9C).
In HER2+ breast cancer cells, we observed that the combination of the AR antagonist flutamide (45 μM) and the HER2 inhibitor lapatinib (0.5 μM) additively inhibited proliferation of the HR2/HER2+ cell line MDA-MB-453 (Supplemental Figure 9D). Similarly, the combination of ICI182,780 (0.5 nM) and lapatinib (10 nM) additively inhibited proliferation of the HR3/HER2+ cell line BT474 (Supplemental Figure 9E). In control experiments, no inhibition was observed with the ER antagonist ICI182,780 in HR2 AR+VDR+ (i.e., ER–) MDA-MB-453 cells or with the VDR agonist calcitriol in HR2 AR+ER+ (i.e., VDR–) BT549 cells (Supplemental Figure 9, F and G).
Because nearly 95% of HER2 tumors expressed at least 1 HR, and 29% expressed all 3 HRs, these results indicate that hormone treatment might also be possible in a majority of HER2 tumors in combination with anti-HER2 therapy.
Discussion
In the present study, we analyzed more than 15,000 normal breast cells and described 11 previously undefined cell subtypes in the luminal layer of human breast, L1–L11. These 11 normal breast cell types conformed to 4 novel hormonal differentiation groups, HR0–HR3. Analysis of 3,157 human breast tumors revealed that each tumor was similar to 1 of the 11 normal cell types and HR groups. Almost none of the breast cancers exhibited a pure basal-like phenotype, as defined by the expression of true myoepithelial markers and the absence of any luminal markers. Nearly all human breast tumors replicated one of the normal hormonal differentiation groups (HR0–HR3), and we found that these subgroups exhibited significant survival differences. Our ontological classification scheme provides actionable hormonal treatment strategies for all subtypes of human breast cancer.
Taxonomy dilemma: lumpers versus splitters.
Historically, the challenges of taxonomy have led to 2 opposing taxonomic approaches: lumping and splitting. Lumpers prefer a few categories marked by large similarities that have clear practical utility; splitters tend to describe as many entities as possible with small differences, regardless of practical utility. Charles Darwin has been credited with using the terms first, when he wrote in a letter, “Those who make many species are the ‘splitters,’ and those who make few are the ‘lumpers’” (40). In medicine, this divide is exacerbated when a clear mechanistic understanding of a disease entity is incomplete.
High-throughput technologies — such as mRNA, miRNA, SNP, and epigenetic profiling as well as next-generation sequencing — have unveiled a complex heterogeneity of breast tumors in individual patients (9, 10, 22, 41, 42). This has led some to advocate designation of categories based on mutations and genetic alterations (which, at its logical extreme, would place each patient into his own unique category), even as the difficulties of such an approach are increasingly becoming evident (43). As we currently stand, without a clear intellectual underpinning of the origins of breast tumors, a consensus has yet to emerge regarding how many types of breast cancers there are, or should be, and how they should be appropriately lumped or split.
While -omics approaches have tremendous molecular resolution, at the anatomic level, they are hindered by several limitations (21, 22). To begin, -omics approaches have low morphologic resolution. In most cases, RNA/DNA is isolated from mm3–cm3 tissue fragments containing tumor cells with heterogeneous molecular characteristics, admixed with various normal epithelial, stromal, vascular, and inflammatory cells. An additional concern is the self-referential nature of many such tumor datasets, as they are predominantly focused on comparing tumors among each other, not against normal cell counterparts. Finally, although cataloging mutations remains an excellent method by which to identify genomic differences between tumors, it is ill-suited for finding similarities between tumors and normal tissues (10, 22, 41, 42). These shortcomings result in a loss of spatial, architectural, and tumor lineage information.
An alternative method of classifying tumors is an ontological approach, which focuses on defining tumor subtypes based on their similarities with specific normal cell origin subtype, akin to evolutionary biology, in which subspecies are identified based on the degree of similarities to common ancestors. Immunostains are currently the most powerful means by which to define similarities between tumors and specific normal cell types, and they resolve many of the issues raised regarding the -omics approaches. As in situ methods preserve tissue architecture, they provide high morphologic resolution at the subcellular level. Additionally, the normal cells provide an unchanging gold-standard internal control and provide a means to describe similarities between normal and tumor cells that is simply not possible using mutational analysis. Lastly, one of the most important differences between the unsupervised clustering (-omics) and the normal cell type–based (ontological) tumor classification methods is that the latter is hypothesis based, which can be tested and refuted or confirmed.
All of the markers used in this study have been examined previously by other investigators individually. However, to our knowledge, this is the first time they have been examined all together within the framework of a comprehensive taxonomic assessment and, moreover, evaluated within the same tissue sections. The latter was made possible by use of the novel matrix IF technology that was recently developed.
It is important to note that this is just the beginning of efforts to identify markers that define the cells comprising the luminal layer of the breast epithelium. It took several decades to accurately describe all the relevant cell types in the hematopoietic system, with the aid of an ever-increasing number of cell type–specific markers. Likewise, in the case of human breast, additional markers will undoubtedly refine and expand the classification system we propose here.
Our results indicate that the composition of normal breast epithelium is much more complex than previously appreciated. While one might ponder the need for 11 cellular subtypes within the normal breast luminal epithelium, it is well worth considering the evolutionary importance of mammary gland development. This seminal event required emergence of a diverse range of cell types that together produce milk, an extremely complex substance essential for the survival of all mammals.
Potential diagnostic, prognostic, and treatment implications.
Admittedly, the HR0–HR3 categories describe only one aspect of a disease as complex as breast cancer, and we do not propose using this classification alone or in place of existing approaches. Rather, we imagine that the HR0–HR3 categories can be used to refine the ER+, HER2+, and TNBC classification presently in use. In the future, this approach can be improved by more detailed descriptions of normal cell types in human breast and by incorporating this information into clinical evaluation along with integrated molecular genetic approaches.
Nevertheless, even at this early stage, we believe this cell-based classification approach has produced some actionable insights. One of the most intriguing implications of our work is the possibility of expanding the patient population being treated with hormone therapy, by targeting AR and VDR in conjunction with ER (44).
Genotype, cell of origin, and tumor phenotype.
The outcome differences of tumors that arise from the same organ can be associated with genetic differences, presenting an attractive paradigm by which to guide the design of personalized cancer therapeutics (45).
An emerging and complementary hypothesis is that phenotypic differences among distinct subtypes of tumors arising in a single tissue may also be imposed by cell-autonomous factors unique to the cell of origin (16, 17, 46). For example, we previously demonstrated that while some normal cell types gave rise to highly tumorigenic and metastatic adenocarcinomas, other breast cell types that were isolated from the same patient and transformed with identical oncogenes gave rise to cells that were morphologically distinct, weakly tumorigenic, and nonmetastatic (16, 47–49). Others have also suggested that the same oncogenes can have vastly different phenotypic consequences depending on the cell of origin (50, 51).
In light of these observations, we contemplate whether the better outcome of HR+ breast tumors may be due to their cell of origin and differentiation lineage. As mutations or amplifications of ER, AR, or VDR genes are very rare in breast cancer, it is likely that HR+ tumors have high HR expression because they either arose in an already HR+ normal cell, or they arose in a HR– precursor that was preordained to differentiate into a HR+ phenotype.
Ontological taxonomy of tumors.
In summary, the use of in situ stains and normal cells as a reference point for classifying tumors solves several of the issues that were raised for -omics approaches: (a) tissue architecture is preserved, (b) morphologic resolution is high, (c) normal cells provide an unchanging gold-standard reference, and (d) phenotypic similarity between normal and tumor cells is maintained. In addition, using normal cell subtypes as a reference point in tumor classification addresses the question of lumping versus splitting. Each normal tissue is designed for a specific function, and each cell subtype is designed to perform different components of this function. Since these functions are finite, the maximum number of biologically important normal cell types is limited, unchanging, and able to be precisely defined. Thus, this method constrains the arbitrary splitting of tumors into endless subclasses. This ontological approach provides a durable infrastructure and context within which molecular data may be appropriately placed in the right cellular context and accurately interpreted.
At first, the molecular heterogeneity of breast cancer appears difficult to reconcile with the robust phenotypic subtypes we observed. However, it is the signaling pathways, not the individual genes, that are responsible for the phenotypes of tumors (43). Thus, a deeper pathway-based understanding will be necessary to correlate the cell type–based subtypes and molecular heterogeneity of breast tumors.
We perceive the cell type–based ontological and high-throughput molecular -omics approaches as complimentary methods. Current in situ examination methods, such as IF and IHC, have several shortcomings: they are low-throughput and semiquantitative and do not allow examination of thousands of proteins simultaneously. As mentioned above, the high-throughput molecular approaches have low morphologic resolution. An ideal approach in the future should combine the subcellular resolution of immunostains with the power of high-content, multigene molecular approaches for studying tumors. We believe that such molecular imaging technologies represent the next frontier in cancer research, and the multiplex IF technique used herein is just one example of how such technologies may advance our understanding of cancer.
Methods
Cell culture.
For cell propagation and studying effects of ER and VDR modulators on cell proliferation, see Supplemental Methods.
IHC, IF, and NHS analysis.
The NHS is a prospective cohort study initiated in 1976 (52). For antibodies and conditions of FFPE section staining, see Supplemental Table 1. For image acquisition and processing; sample scoring and data display in heat maps; and study design, population, and analysis, see Supplemental Methods.
Multiplex IF.
For antibodies and conditions of FFPE section staining, see Supplemental Table 1. For sequential quenching of fluorescence signal, data acquisition, and multiplex IF image analysis, see Supplemental Methods.
Gene expression analysis and cell lines.
For analysis of mRNA expression profiling data of cancer cell lines (39), see Supplemental Methods.
Cell proliferation assays.
For analysis of cell line proliferation responses to calcitriol, taxol, ICI182,780, and R1881, see Supplemental Methods.
Statistics.
Event outcomes were compared using Kaplan-Meier analysis, and P values were determined with the log-rank test. Tumor expression of myoepithelial and luminal genes (26–28) were explored in frozen robust multiarray analysis–normalized (53) gene expression data (GEO accession nos. GSE3744, GSE4922, GSE6532, and GSE7390) using hierarchical cluster analysis (Pearson r, average linkage) using the Bioconductor package MADE4 (54). Global test, available from the Bioconductor package globaltest (55), was used to assess the association between gene expression and luminal or myoepithelial classification. Tumors were classified as basal-like or non-basal-like as previously described (56), except where basal-like subtype classification was provided by the authors (29). Global test (55) was used to determine association between individual genes and basal-like/non-basal-like division. A P value less than 0.05 was considered significant. For statistical methods for NHS protein expression and outcome data analysis, UNC mRNA expression and outcome data analysis, and additional details, see Supplemental Methods.
Study approval.
FFPE blocks from surgical resection specimens of normal breast tissue and of breast tumors were obtained from the archives of Brigham and Women’s Hospital (BWH) in accordance with the regulations for excess tissue use stipulated by the BWH IRB. For TMAs used in this study, see Supplemental Methods.
High-resolution images.
High-resolution files corresponding to all image panels in Figures 1–3, Supplemental Figures 1–3 and 5, and Supplemental Table 2 are available at http://sylvester.org/ince.
Supplementary Material
Acknowledgments
The authors acknowledge funding support from Breast Cancer Research Foundation and Play for P.I.N.K. (to T.A. Ince), Kristin Jordahl and friends (to T.A. Ince); NCI grant R01-CA146445-01, NIH Roadmap Epigenomics Project (to T.A. Ince); Department of Defense CDMRP BCRP grant W8 1XWH-08-1-0282,BC-07456 (to T.A. Ince); DHHS and GlaxoSmithKline grants WE234 and EPI40307 (to R.M. Tamimi); NIH grant K08NS064168 (to S. Santagata); V Foundation for Cancer Research (to S. Santagata); DFCI Women’s Cancers Program (to A.C. Culhane and M. Schwede); and Claudia Adams Barr Program in Innovative Basic Cancer Research (to A.C. Culhane). We thank Q. Li, S. Dinn, A. Santamaria-Pang, and F. Ginty (GE Global Research Center) as well as T.D. Tlsty, J.S. Brugge, J.M. Slingerland, R.D. Cardiff, C. Gomez, M.D. Pegram, S.A. Borowsky, M. Nadji, and M. Jorda for helpful suggestions.
Footnotes
Conflict of interest: Sandro Santagata is a cofounder of and scientific advisor to Bayesian Diagnostics. Tan A. Ince was a scientific advisor to 3DM Inc. (2007–2012) and a consultant to Stemgent (2008–2010).
Citation for this article: J Clin Invest. 2014;124(2):859–870. doi:10.1172/JCI70941.
See the related Commentary beginning on page 478.
References
- 1.Swerdlow SH, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue. 4th ed. Lyon, France: World Health Organization; 2008. [Google Scholar]
- 2.Hamilton A, Gallipoli P, Nicholson E, Holyoake TL. Targeted therapy in haematological malignancies. J Pathol. 2010;220(4):404–418. doi: 10.1002/path.2669. [DOI] [PubMed] [Google Scholar]
- 3.Wood GS, Warnke RA. The immunologic phenotyping of bone marrow biopsies and aspirates: frozen section techniques. Blood. 1982;59(5):913–922. [PubMed] [Google Scholar]
- 4.Drexler HG. Classification of acute myeloid leukemias--a comparison of FAB and immunophenotyping. Leukemia. 1987;1(10):697–705. [PubMed] [Google Scholar]
- 5.Jones C, et al. Expression profiling of purified normal human luminal and myoepithelial breast cells: identification of novel prognostic markers for breast cancer. Cancer Res. 2004;64(9):3037–3045. doi: 10.1158/0008-5472.CAN-03-2028. [DOI] [PubMed] [Google Scholar]
- 6.Gusterson BA, Ross DT, Heath VJ, Stein T. Basal cytokeratins and their relationship to the cellular origin and functional classification of breast cancer. Breast Cancer Res. 2005;7(4):143–148. doi: 10.1186/bcr1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roy S, et al. Rare somatic cells from human breast tissue exhibit extensive lineage plasticity. Proc Natl Acad Sci U S A. 2013;110(12):4598–4603. doi: 10.1073/pnas.1218682110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 2010;5(1):5–23. doi: 10.1016/j.molonc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dawson SJ, Rueda OM, Aparicio S, Caldas C. A new genome-driven integrated classification of breast cancer and its implications. EMBO J. 2013;32(5):617–628. doi: 10.1038/emboj.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prat A, Ellis MJ, Perou CM. Practical implications of gene-expression-based assays for breast oncologists. Nat Rev Clin Oncol. 2012;9(1):48–57. doi: 10.1038/nrclinonc.2011.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guiu S, et al. Molecular subclasses of breast cancer: how do we define them? The IMPAKT 2012 Working Group Statement. Ann Oncol. 2012;23(12):2997–3006. doi: 10.1093/annonc/mds586. [DOI] [PubMed] [Google Scholar]
- 14.Schnitt SJ. Classification and prognosis of invasive breast cancer: from morphology to molecular taxonomy. Mod Pathol. 2010;23(suppl 2):S60–S64. doi: 10.1038/modpathol.2010.33. [DOI] [PubMed] [Google Scholar]
- 15.Chu PG, Weiss LM. Keratin expression in human tissues and neoplasms. Histopathology. 2002;40(5):403–439. doi: 10.1046/j.1365-2559.2002.01387.x. [DOI] [PubMed] [Google Scholar]
- 16.Ince TA, et al. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell. 2007;12(2):160–170. doi: 10.1016/j.ccr.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 17.Molyneux G, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7(3):403–417. doi: 10.1016/j.stem.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 18.Ginty F, et al. The relative distribution of membranous and cytoplasmic met is a prognostic indicator in stage I and II colon cancer. Clin Cancer Res. 2008;14(12):3814–3822. doi: 10.1158/1078-0432.CCR-08-0180. [DOI] [PubMed] [Google Scholar]
- 19.Collins LC, et al. Basal cytokeratin and epidermal growth factor receptor expression are not predictive of BRCA1 mutation status in women with triple-negative breast cancers. Am J Surg Pathol. 2009;33(7):1093–1097. doi: 10.1097/PAS.0b013e31819c1c93. [DOI] [PubMed] [Google Scholar]
- 20.Boecker W, Buerger H. Evidence of progenitor cells of glandular and myoepithelial cell lineages in the human adult female breast epithelium: a new progenitor (adult stem) cell concept. Cell Prolif. 2003;36(suppl 1):73–84. doi: 10.1046/j.1365-2184.36.s.1.7.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molyneux G, Smalley MJ. The cell of origin of BRCA1 mutation-associated breast cancer: a cautionary tale of gene expression profiling. J Mammary Gland Biol Neoplasia. 2011;16(1):51–55. doi: 10.1007/s10911-011-9202-8. [DOI] [PubMed] [Google Scholar]
- 22.Lavasani MA, Moinfar F. Molecular classification of breast carcinomas with particular emphasis on “basal-like” carcinoma: a critical review. J Biophotonics. 2012;5(4):345–366. doi: 10.1002/jbio.201100097. [DOI] [PubMed] [Google Scholar]
- 23.Livasy CA, et al. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod Pathol. 2006;19(2):264–271. doi: 10.1038/modpathol.3800528. [DOI] [PubMed] [Google Scholar]
- 24.Sorlie T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sorlie T, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100(14):8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grigoriadis A, et al. Establishment of the epithelial-specific transcriptome of normal and malignant human breast cells based on MPSS and array expression data. Breast Cancer Res. 2006;8(5):R56. doi: 10.1186/bcr1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lakhani SR, et al. Prediction of BRCA1 status in patients with breast cancer using estrogen receptor and basal phenotype. Clin Cancer Res. 2005;11(14):5175–5180. doi: 10.1158/1078-0432.CCR-04-2424. [DOI] [PubMed] [Google Scholar]
- 28.Raouf A, et al. Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell Stem Cell. 2008;3(1):109–118. doi: 10.1016/j.stem.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 29.Richardson AL, et al. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9(2):121–132. doi: 10.1016/j.ccr.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 30.Loi S, et al. Predicting prognosis using molecular profiling in estrogen receptor-positive breast cancer treated with tamoxifen. BMC Genomics. 2008;9:239. doi: 10.1186/1471-2164-9-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ivshina AV, et al. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006;66(21):10292–10301. doi: 10.1158/0008-5472.CAN-05-4414. [DOI] [PubMed] [Google Scholar]
- 32.Desmedt C, et al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res. 2007;13(11):3207–3214. doi: 10.1158/1078-0432.CCR-06-2765. [DOI] [PubMed] [Google Scholar]
- 33.Gusterson B. Do ‘basal-like’ breast cancers really exist? Nat Rev Cancer. 2009;9(2):128–134. doi: 10.1038/nrc2571. [DOI] [PubMed] [Google Scholar]
- 34.Lim E, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15(8):907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 35.Collins LC, Cole KS, Marotti JD, Hu R, Schnitt SJ, Tamimi RM. Androgen receptor expression in breast cancer in relation to molecular phenotype: results from the Nurses’ Health Study. Mod Pathol. 2011;24(7):924–931. doi: 10.1038/modpathol.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu R, et al. Androgen receptor expression and breast cancer survival in postmenopausal women. Clin Cancer Res. 2011;17(7):1867–1874. doi: 10.1158/1078-0432.CCR-10-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santagata S, et al. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc Natl Acad Sci U S A. 2011;108(45):18378–18383. doi: 10.1073/pnas.1115031108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harrell JC, et al. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res Treat. 2012;132(2):523–535. doi: 10.1007/s10549-011-1619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neve RM, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10(6):515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Darwin C, Darwin F. The life and letters of Charles Darwin: Including an autobiographical chapter. Vol. 1. New York, NY: D. Appleton and Co.; 1911. [Google Scholar]
- 41.Banerji S, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486(7403):405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stephens PJ, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462(7276):1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yaffe MB. The scientific drunk and the lamppost: massive sequencing efforts in cancer discovery and treatment. Sci Signal. 2013;6(269):pe13. doi: 10.1126/scisignal.2003684. [DOI] [PubMed] [Google Scholar]
- 44.Ni M, et al. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell. 2011;20(1):119–131. doi: 10.1016/j.ccr.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.MacConaill LE, et al. Profiling critical cancer gene mutations in clinical tumor samples. PLoS One. 2009;4(11):e7887. doi: 10.1371/journal.pone.0007887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yalcin-Ozuysal O, Brisken C. From normal cell types to malignant phenotypes. Breast Cancer Res. 2009;11(6):306. doi: 10.1186/bcr2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Merritt MA, t al. Gene expression signature of normal cell-of-origin predicts ovarian tumor outcomes. PLoS One. 2013;8(11):e80314. doi: 10.1371/journal.pone.0080314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Godar S, et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134(1):62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McAllister SS, et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell. 2008;133(6):994–1005. doi: 10.1016/j.cell.2008.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 51.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127(4):679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 52.Tamimi RM, et al. Comparison of molecular phenotypes of ductal carcinoma in situ and invasive breast cancer. Breast Cancer Res. 2008;10(4):R67. doi: 10.1186/bcr2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCall MN, Bolstad BM, Irizarry RA. Frozen robust multi-array analysis (fRMA). Biostatistics. 2010;11(2):242–253. doi: 10.1093/biostatistics/kxp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Culhane AC, Thioulouse J, Perriere G, Higgins DG. MADE4: an R package for multivariate analysis of gene expression data. Bioinformatics. 2005;21(11):2789–2790. doi: 10.1093/bioinformatics/bti394. [DOI] [PubMed] [Google Scholar]
- 55.Goeman JJ, van de Geer SA, de Kort F, van Houwelingen HC. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics. 2004;20(1):93–99. doi: 10.1093/bioinformatics/btg382. [DOI] [PubMed] [Google Scholar]
- 56.Culhane AC, Quackenbush J. Confounding effects in “A six-gene signature predicting breast cancer lung metastasis”. Cancer Res. 2009;69(18):7480–7485. doi: 10.1158/0008-5472.CAN-08-3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.