

Abstract
Leber congenital amaurosis (LCA) encompasses a set of early-onset blinding diseases that are characterized by vision loss, involuntary eye movement, and nonrecordable electroretinogram (ERG). At least 19 genes are associated with LCA, which is typically recessive; however, mutations in homeodomain transcription factor CRX lead to an autosomal dominant form of LCA. The mechanism of CRX-associated LCA is not understood. Here, we identified a spontaneous mouse mutant with a frameshift mutation in Crx (CrxRip). We determined that CrxRip is a dominant mutation that results in congenital blindness with nonrecordable response by ERG and arrested photoreceptor differentiation with no associated degeneration. Expression of LCA-associated dominant CRX frameshift mutations in mouse retina mimicked the CrxRip phenotype, which was rescued by overexpression of WT CRX. Whole-transcriptome profiling using deep RNA sequencing revealed progressive and complete loss of rod differentiation factor NRL in CrxRip retinas. Expression of NRL partially restored rod development in CrxRip/+ mice. We show that the binding of homeobox transcription factor OTX2 at the Nrl promoter was obliterated in CrxRip mice and ectopic expression of OTX2 rescued the rod differentiation defect. Together, our data indicate that OTX2 maintains Nrl expression in developing rods to consolidate rod fate. Our studies provide insights into CRX mutation-associated congenital blindness and should assist in therapeutic design.
Introduction
Inherited retinal degenerative diseases exhibit tremendous clinical and genetic heterogeneity, with almost 200 genes identified so far (Retinal Information Network) (1). In the 19th century, Theodor Leber described the familial nature of a pigmentary retinopathy and congenital blindness (2), now aptly named Leber congenital amaurosis (LCA). LCA encompasses congenital and early-onset retinopathies that account for 5% of inherited blindness and are characterized by vision loss together with nystagmus and nonrecordable rod and cone photoreceptor response by electroretinogram (ERG) (3). At least 19 LCA genes encoding diverse cellular functions, such as intracellular transport, phototransduction, and transcriptional regulation, have been identified so far (4). While LCA is largely recessive, autosomal dominant inheritance is reported for mutations in CRX and IMPDH1 (5–7). Recent success in effective gene-replacement therapy for patients with LCA2, caused by RPE65 mutations that affect retinoid isomerase activity, underscores the importance of elucidating the molecular basis of disease, functional analysis of associated genes, and relevance of preclinical animal models (8).
During development, distinct neuronal subtypes in the vertebrate retina originate from common pools of progenitor cells in a conserved order of birth, primarily under the control of intrinsic genetic programs (9, 10). The rod and cone photoreceptors constitute over 70% of all cells in the mammalian retina (11, 12). The regulatory mechanisms for generating photoreceptors from retinal progenitors and their subsequent differentiation into unique and functional photon-capturing neurons are slowly being unraveled (13). The homeodomain protein OTX2 is implicated as a key regulator of photoreceptor cell fate and induces the expression of cone-rod homeobox (Crx) transcription factor in postmitotic photoreceptor precursors (14–16). While Otx2 expression decreases in the photoreceptors after birth, Crx is suggested to take over as a primary transcriptional regulator and induce the expression of rod differentiation factor, neural retina leucine zipper (Nrl), in differentiating rods (17). CRX exhibits an intimate and synergistic relationship with NRL in controlling rod gene regulatory networks (18–22). CRX is also reported to collaborate with RAR-related orphan nuclear receptor β (RORβ) to initiate the expression of S-opsin and other cone genes (23). Surprisingly, the photoreceptor cell fate is unaltered in the Crx–/– retina; yet the expression of phototransduction genes is greatly reduced and no outer segments are formed, leading eventually to retinal degeneration (24). The enigma is that Crx is expressed early in newly postmitotic photoreceptor precursors, much before functional maturation; however, its loss of function leads to photoreceptor degeneration. Crx is also suggested to be upstream of Nrl in the rod transcriptional hierarchy (17, 25, 26); nonetheless, Nrl is also expressed in newborn photoreceptors during the final mitosis (27, 28), around the same time as Crx (16). In contrast to Crx, Nrl is both essential and sufficient for determining rod cell fate and rod-specific gene expression (21, 29, 30). We wondered whether Otx2, and not Crx, initiates Nrl expression? The questions pertaining to respective contribution(s) of Crx versus Otx2 in initiating and/or maintaining the expression of Nrl and other rod or cone genes have not been directly addressed in vivo.
A range of diverse clinical phenotypes, from cone-rod dystrophy and retinitis pigmentosa to congenital blindness in LCA, associated with CRX mutations in humans (5, 6, 31–33) reveal its more complex role in photoreceptor development and/or function than that reflected by Crx–/– mouse phenotype. Even though a strict genotype-phenotype correlation does not exist, a majority of missense and truncation mutations in the CRX homeodomain are associated with cone-rod dystrophy and alter its DNA binding properties or transcriptional synergy with NRL (34, 35), thereby influencing gene expression and photoreceptor maturation. In contrast, many human CRX frameshift mutations identified downstream of the homeodomain result in dominant and more severe LCA phenotypes. The molecular events underlying congenital blindness in CRX retinopathies are poorly understood, and no treatment is currently available.
Here, we demonstrate the molecular mechanism of LCA associated with dominant CRX frameshift mutations by taking advantage of a new spontaneous mouse mutant (CrxRip mice). By combined genetic mapping and exome sequencing, we have identified a 1-bp frameshift deletion in Crx coding sequence, similar to many LCA-causing dominant CRX mutations (dominant CRX-LCA). We show that the CRXRip protein, carrying additional unrelated residues at the carboxyl terminus, is not functional in vitro but represses both CRX and OTX2 functions in vivo. LCA-causing CRX mutations exhibit phenotypic manifestations similar to those of CrxRip mice. We demonstrate that the rodless phenotype observed in CrxRip mutants is caused by the loss of Nrl expression later in photoreceptor maturation and can be rescued partially by Nrl. ChIP studies reveal that in vivo binding of OTX2 to Nrl promoter is abrogated in the CrxRip mutant retinas but not in the Crx–/– retinas, suggesting that OTX2 is a direct modulator of Nrl expression. Our studies thus reveal a critical role of OTX2 in consolidating cell fate by maintaining Nrl expression in developing rods. In addition, we establish CrxRip mutant mice as a preclinical model for dominant CRX-LCA and suggest opportunities for gene-based therapies.
Results
Identification of a new mouse mutant with congenital blindness and immature photoreceptors.
As part of our systematic screen to identify genetic models of retinal disease, we identified a mouse mutant with white spots on retinal fundus examination. Breeding of this mutant to WT C57BL/6J mice and at least 5 backcrosses revealed an autosomal dominant inheritance of the observed phenotype. Ocular coherence tomography (OCT) Doppler imaging showed smaller retinal blood vessels and poor blood flow in the mutant mice compared with that in the controls (data not shown). Photopic and scotopic ERGs revealed a complete absence of cone and rod visual response, respectively, in 1-month-old heterozygous mutant animals (Figure 1A), suggesting a defect in photoreceptor development. This mutation was hereafter referred to as Rip (retina with immature photoreceptors).
Figure 1. Dominant congenital blindness in the Rip mutant is caused by a 1-bp deletion in Crx.

(A) Dark- and light-adapted ERG recording in 1-month-old WT and Rip mutant. (B) Linkage cross analysis. 75 backcross progenies from the (Rip mutant X C3A.BLiAPde6b+/J) F1 X C3A.BLiA-Pde6b+/J were phenotyped by retinal fundus examination and genotyped for the indicated microsatellite markers. The black boxes represent heterozygosity for Rip-derived allele, and white boxes represent homozygosity for C3A.BLiA-Pde6b+/J–derived alleles. The number of chromosomes sharing the corresponding haplotype is indicated. Genetic map of chromosome 7 in the Rip region. (C) Identification of 1-bp deletion in Crx, visualized by Integrative Genome Viewer displaying sequence reads generated by exome capture sequencing. (D) Sanger sequencing of Crx+/+, CrxRip/+, and CrxRip/Rip mice showing deletion of a G nucleotide in exon 4 in Rip mutant but not WT mice. (E) Schematic of the CRX protein, indicating the position of 1-bp deletion upstream of Otx-like domain. Colored boxes show the functional domains. (F) Alignment of mouse CRX and CRXRip mutant protein predicts that the frameshift mutation would lead to the addition of 88 amino acids, starting at residue 299. Conserved amino acids are indicated in red. (G) Immunoblot analysis of retinal extracts from 1-month-old Crx+/+, CrxRip/+, and CrxRip/Rip mice. Anti-CRX antibody identifies 2 CRX bands in CrxRip/+ retinas. The lower band (34 kDa) corresponds to CRXWT, whereas the 44-kDa isoforms correspond to CRXRip protein. Anti-actin antibody was used as a loading control.
To discover the genetic cause of the Rip mutant phenotype, we performed linkage analysis and identified a locus on chromosome 7 between markers D7Mit340 and D7Mit56 (Figure 1B), homologous to human Chr19q13.3. This critical region spanned 17 Mb and contained over 200 genes. We then performed whole-exome sequencing using DNA from 2 homozygous mutant mice, selected by using microsatellite marker haplotypes, and 1 control WT mouse. After mapping the sequence reads, variant calling, and filtering, we identified only 1 homozygous variant that was present in both mutants but was absent in the control. This variant corresponded to a 1-bp deletion in the Crx gene at position 763 (c.763del1) and was confirmed by Sanger sequencing (Figure 1, C and D). The c.763del1 mutation, located in the last Crx exon, would result in a frameshift that skips the C-terminal Otx-like domain and adds 133 unrelated residues (p.Gly255fs or p.Gly255Alafs*133) (Figure 1, E and F). Immunoblot analysis of adult retina protein extracts, using anti-CRX antibody against an epitope corresponding to the residues 166–285, detected both WT and mutant CRX protein in CrxRip/+ mice (of 34 and 44 kDa, respectively) and only a 44-kDa mutant protein in the CrxRip/Rip retinas (Figure 1G).
CrxRip/+ mouse retinas display long-term preservation of immature cone-like photoreceptors.
We performed a detailed phenotypic analysis of the CrxRip/+ and CrxRip/Rip retinas and compared it to that of the Crx–/– mice. Immunohistochemical analysis of the mature P21 CrxRip/+ and CrxRip/Rip retinas showed no rhodopsin (RHO) expression (Figure 2A). In contrast, Crx–/– retinas contained RHO-positive cells, albeit considerably less intensely stained compared with WT retinas. The cone-specific proteins — S-opsin (OPN1SW), M-opsin (OPN1MW), and cone arrestin (ARR3) — were detected in few cells in P21 Crx–/– retinas; however, these markers were completely absent in the CrxRip/+ and CrxRip/Rip retinas. Furthermore, CrxRip/+ retinas revealed very strong and continuous staining for peanut agglutinin (PNA), a cone-specific cell surface marker, similar to that in Nrl–/– retinas (36, 37). Immunostaining of recoverin, an early marker of photoreceptor differentiation, was also dramatically reduced in CrxRip/+ mice compared with that in WT mice, with only a few recoverin-positive cells in CrxRip/Rip mutants. In contrast, the expression of recoverin was high but restricted to fewer photoreceptor cells in the Crx–/– retinas.
Figure 2. CrxRip/+ retinas display immature cone-like photoreceptors and long-term preservation of the outer nuclear layer.

(A) Immunostaining of RHO, OPN1SW, OPN1MW, ARR3, PNA, and Recoverin in P21 Crx+/+, CrxRip/+, CrxRip/Rip, and Crx–/– retinas. onl, outer nuclear layer; inl, inner nuclear layer. Scale bar: 40 μm. (B) Methacrylate sections followed by H&E staining of retinas from 5-week-, 10-week-, and 9-month-old Crx+/+, CrxRip/+, CrxRip/Rip, and Crx–/– mice. rpe, retinal pigment epithelium; os, outer segment; is, inner segment; gcl, ganglion cell layer. Scale bar: 40 μm. (C) Methacrylate sections followed by H&E staining in 5-week-old Crx+/+, CrxRip/+, CrxRip/Rip, and Crx–/– mice. The arrow indicates characteristic rod photoreceptor nuclei with dense chromatin in the center, and arrowheads indicate characteristic cone photoreceptor nuclei with less condensed chromatin. Scale bar: 10 μm. (D) Immunostaining using ribeye and PKCα antibodies to visualize presynaptic photoreceptor region and ON bipolar cells, respectively, in P21 Crx+/+ and CrxRip/+ retinas. opl, outer plexiform layer. Scale bar: 40 μm (left); 10 μm (right).
The histology of 5-week-old retinas in methacrylate sections revealed abnormal photoreceptor segments in the 3 mutants compared with WT mice (Figure 2B). The thickness of outer nuclear layer in CrxRip/+ mice was relatively well preserved at 5 weeks (Figure 2B) and remained largely unchanged, at least up to 18 months, despite the complete loss of visual function (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI72722DS1). In contrast, the photoreceptor layer in CrxRip/Rip and Crx–/– retinas underwent rapid degeneration between 5 and 10 weeks, and only a few nuclei persisted at 9 months of age (Figure 2B). The chromatin density and organization of the photoreceptors in CrxRip/+ retinas revealed cone-like characteristics (ref. 38 and Figure 2C), similar to those observed in cone-only Nrl–/– mice (29, 36). This observation is consistent with the complete absence of RHO expression (Figure 2A). Immunohistochemical analysis of the mature P21 Crx+/+ and CrxRip/+ retinas using ribeye antibody (a presynaptic marker) and PKCα (to visualize the postsynaptic connection with rod bipolar cells) revealed a certain degree of contact between these two types of cells, even in the absence of visual transduction (Figure 2D). However, neuronal sprouting of the immature photoreceptors into the inner nuclear layer was observed with ribeye staining that was not restricted to the top part of the outer plexiform layer, as in the control retinas. EM of CrxRip/+ mouse retinas confirmed the presence of very short inner segments and a complete absence of outer segments (Figure 3, A and B), but ciliary microtubules were detectable (Figure 3C). The outer limiting membrane was present in CrxRip/+ retinas, and, unlike the WT photoreceptors, a majority of mitochondria surrounded nuclei instead of being in the distal ellipsoid region (Figure 3, A and B).
Figure 3. CrxRip/+ retinal photoreceptors lack outer segments and possess very short inner segments.

Transmission EM images in the dorsal-ventral midline plane taken through the central retinas of 1-month-old (A) Crx+/+ and (B) CrxRip/+ mice. mito, mitochondria; nuc, nucleus; olm, outer limiting membrane. Original magnification, ×1,200. Scale bar: 2 μm. (C) Transmission EM images in the dorsal-ventral midline plane taken through the central retinas of 1-month-old Crx+/+ and CrxRip/+ mice, showing the presence of ciliary microtubules in the mutant photoreceptors. CM, ciliary microtubule. Original magnification, ×5,000. Scale bar: 1 μm.
Our data demonstrate that CrxRip/+ mutant retinas exhibit a more severe phenotype compared with that of Crx–/– mice, as the immature cone-like photoreceptors do not express many of the key rod or cone phototransduction genes.
LCA-associated CRX frameshift mutations mimic the mouse CrxRip phenotype, which can be rescued by WT Crx.
The CrxRip mutation (CRXG255fs refers to the corresponding change in humans) is in a conserved domain of CRX, in close proximity of several similar 1-bp deletion mutations associated with dominant LCA in humans (35). We therefore selected 2 human mutations — CRXL237fs and CRXP263fs (Figure 4A) — to test their effect on photoreceptor development in mouse retina. Neonatal WT mouse retinas were cotransfected in vivo with each CRX expression construct, along with Ub-GFP and Rho-DsRed plasmids (Figure 4B). As predicted, a vast majority of WT CRX–transfected (CRXWT-transfected) cells (GFP-positive) in the outer nuclear layer coexpressed DsRed (Rho promoter active) and exhibited elongated outer segments, indicative of rod photoreceptors. However, the transfection of CRXG255fs, CRXL237fs, or CRXP263fs (indicated by GFP) resulted in DsRed-negative cells that lacked outer segments. Our data suggest that LCA-associated CRX frameshift mutations (caused by 1-bp deletion in the last exon) arrest the committed photoreceptors in an early differentiation state and that the CrxRip/+ mutant can be used as a model of dominant LCA.
Figure 4. LCA-associated CRX frameshift mutations mimic CrxRip phenotype that can be rescued by CrxWT.

(A) Alignment of mouse CrxG255fs and the corresponding human mutation CRXG255fs with 2 other human frameshift mutants: CRXL237fs and CRXP263fs. The top line corresponds to human CRX, indicating the degree of conservation between mouse and human. Conserved amino acids are indicated in red. Arrowheads indicate the position of the amino acid changed in CRXL237fs, CrxG255fs, CRXG255fs, and CRXP263fs. (B) Representative images of P21 WT mouse retinas electroporated at P0 with Ub-GFP (green), Rho-DsRed (red), and with one of the following CRX frameshift mutants: CRXL237fs, CRXG255fs, or CRXP263fs. CRX was used as control. Arrows indicate GFP- and RHO-positive cells, and arrowheads indicate GFP-positive and RHO-negative cells. Scale bar: 40 μm. (C) Representative images of P21 CrxRip/+ mouse retinas electroporated at P0 with Ub-GFP (green), RHO-DsRed (red), and either CRXWT or empty vector (Mock). Arrows indicate GFP- and RHO-positive cells. Scale bar: 40 μm.
The dominant inheritance of the mutation would imply that the CrxRip allele is functionally null, but then why does the CrxRip mutant exhibit a dominant retinal phenotype, which is more severe compared with that exhibited by the Crx–/– retina, even though WT protein is still present in the CrxRip/+ retina (see Figure 1G)? We hypothesized that CRXRip mutant protein interferes with the binding of CRXWT protein to CRX target genes in vivo. If this were the case, the expression of additional CRXWT protein would restore photoreceptor differentiation by blocking the function of the available mutant CRXRip protein. Indeed, the transfection of CRXWT construct but not of the empty vector in the newborn CrxRip/+ mouse retina by electroporation in vivo completely ameliorated the photoreceptor morphology (including outer segments) and restored RHO expression in rod cells (Figure 4C).
The CRXRip mutant protein does not transactivate opsin promoters or bind DNA.
To investigate the mechanism of lack of opsin expression in mutants, we examined the function of CRXRip protein in HEK293 cells using 2 established CRX target promoters — Rho and Opn1sw. To test Rho promoter transactivation, the CRXWT or CRXRip construct was cotransfected with two known CRX interactors, NRL and NR2E3, whereas Opn1sw promoter activity was evaluated in presence of RORβ, as described previously (22, 23, 39, 40). In contrast to CRXRip, the CRXRip mutant protein did not exhibit any transactivation of Rho or Opn1sw promoter, either alone or with respective coactivators (Figure 5A), even though the protein was produced and localized in the nucleus (Supplemental Figure 2). To test whether the loss of function of CRXRip protein was due to the lack of Otx-like domain or unrelated residues at C terminus, we generated a CRX1–254 expression construct by introducing a nonsense mutation in Gly255 codon. The truncated CRX1–254 protein was able to transactivate both Rho and Opn1sw promoters, albeit less efficiently compared with CRXWT. Additional cotransfection experiments revealed that increasing amounts of CRXRip construct (0.15–0.55 μg) did not alter the activity of CRXWT (kept constant at 0.15 μg), whereas transcriptional activation of opsin promoters was further augmented by CRX1–254 (Figure 5A). Our results show that Otx2-like C-terminal domain of CRX is not critical for transcriptional activation and that the CRXRip mutant does not act as a dominant-negative suppressor of the CRXWT protein or other coactivators (NRL, NR2E3, or RORβ) in cultured cells.
Figure 5. The CRXRip mutant protein is functionally null in vitro.

(A) Lack of transactivation by CRXRip mutant protein. HEK293 cells were cotransfected with constructs containing bovine Rho or mouse Opn1sw promoters driving firefly Luciferase reporter gene simultaneously with NRL, NR2E3, or RORβ, respectively. In both sets of experiments, different amounts of CRXWT, CRXRip, and/or CRX1–254 (0.15–0.55 μg) were cotransfected. Fold change is relative to the luciferase activity in presence of NRL and NR2E3 only. *P < 0.05. (B) CRXRip protein does not bind DNA. Autoradiograms of EMSA using nuclear extracts from HEK293T cells transfected with pcDNA4c, Xpress-tagged CRXWT, CRXRip, and CRX1–254 were performed using oligonucleotides encompassing CRX binding sites in Rho and Opn1sw promoters. 100 times more unlabeled specific probes were used for competition. Oligonucleotide supershift assays were performed with anti-Xpress antibody. Arrows indicate the CRX-shifted probe. (C) ChIP-qPCR with anti-CRX antibody from P21 WT, CrxRip/Rip, and Crx–/– retinas. Normal IgG was used control. Fold enrichment represents the fold change of qPCR amplification signals for the different genes tested between CRX ChIP DNA and IgG control ChIP DNA.
To examine why CRXRip protein does not transactivate in reporter assays, we performed electrophoretic mobility shift experiments with Rho and Opn1sw promoter elements (both encompassing CRX binding sites) using nuclear extracts from transfected cells (Figure 5B). As predicted, CRXWT and CRX1–254 proteins shifted the mobility of the 2 promoter elements, but the CRXRip protein did not bind cognate CRX binding sites. In concordance, ChIP-qPCR analysis of P21 retinas from CrxRip/Rip and Crx–/– (as control) mice using anti-CRX antibody showed no CRX binding to photoreceptor-specific target genes (Figure 5C). We conclude that the lack of transcriptional activity by CRXRip is a consequence of the loss of DNA binding.
RNA-Seq analysis of CrxRip mutant retinas, unlike that of Crx–/– retinas, reveals a complete lack of phototransduction gene expression.
To gain additional molecular insights, we performed whole-transcriptome sequencing (using RNA sequencing [RNA-Seq]; ref. 41) of P2 (corresponding to the peak of rod birth) and mature (P21) CrxRip/+ and CrxRip/Rip retinas (Figure 6). In addition, we included WT (Crx+/+), Crx–/–, and Nrl–/– retinas for comparative analysis. We used a cutoff of fold change of 2, FPKM (fragments per kilobase of exon per million fragments mapped) value of more than or equal to 2, and an adjusted P value of less than or equal to 0.05 to identify differentially expressed genes (DEGs) at P2 and P21 among different pairwise combinations. As shown in Supplemental Figure 3, volcano plots show the distinctions in gene expression patterns of retinas from various mutants. Only 24 genes were differentially expressed between CrxRip/+ and WT retinas at P2, but many more genes showed differential expression at P21 (Figure 6A and Table 1). Two-way hierarchical clustering of DEGs using a higher normalized FPKM cutoff of 5 revealed divergent gene expression patterns in P2 Rip mutant retinas when compared with corresponding Crx+/+, Crx–/–, and Nrl–/– retinas (Figure 6B); however, the expression profiles of CrxRip mutants at P21 were more similar to Crx–/– retinas then Nrl–/– retinas, relative to WT controls (Figure 6C).
Figure 6. RNA-Seq analysis of P2 and P21 CrxRip retinas reveals a progressive loss of expression of rod-specific genes.

(A) Venn diagrams comparing DEGs identified as significant (fold change ≥ 2, adjusted P value ≤ 0.05, and a mean normalized expression of 5 in at least 1 of the groups compared) between WT and CrxRip/+, Crx–/–, and Nrl–/– retinas. (B and C) Heat maps and hierarchical clustering dendrogram from 24 and 460 DEGs at (B) P2 and (C) P21, respectively. Genes showing a minimum fold change of 2 between Crx+/+ and CrxRip/+ retinas, with adjusted P values of less than or equal to 0.05 and mean of normalized expression of more than or equal to 5 were selected. The expression value of each sample is represented with blue indicating lowest expression, with increasing expression from gray to red for the highest expression. Expression value is in log2 scale.
Table 1.
Total number of DEGs at P2 and P21 identified as significant between each mouse mutant
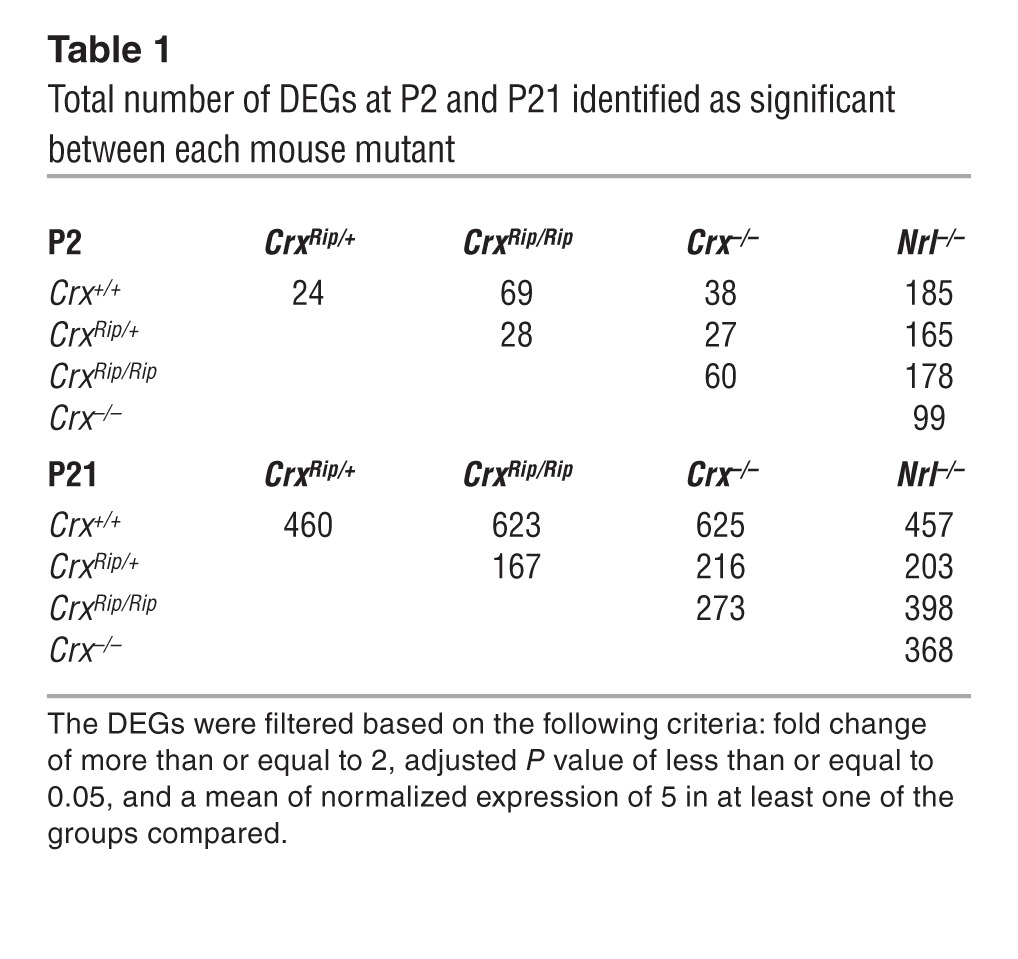
In general, unlike that in Crx–/– and Nrl–/– retinas, the expression of a majority of photoreceptor genes needed for visual transduction was completely undetectable in the CrxRip/+ and CrxRip/Rip mutants. Notable exceptions were a subset of cone-specific genes (such as Gnb3, Gnat2, Pde6c, and Cngb3) that were downregulated at P2 but upregulated in P21 CrxRip/+ retinas (Figure 6C and Supplemental Table 1). In contrast, the expression of these genes was upregulated or unchanged at P2 in Crx–/– retinas.
The pathway analysis of 69 genes showing differential expression between P21 CrxRip/+ and Crx+/+ retinas, but unaltered expression in Crx–/– or Nrl–/– retinas, highlights several cell death and survival genes, including Stat3, that are associated with neuroprotection as well as suppression of rod differentiation (Supplemental Figure 4 and refs. 42, 43). These data are consistent with the preservation of outer nuclear layer in CrxRip/+ mutants, with a concurrent lack of photoreceptor maturation.
Nrl can partially rescue the rod phenotype in CrxRip/+ retinas.
A key finding from RNA-seq analysis was that, unlike Crx–/– retinas, the expression of rod differentiation factor Nrl was detectable in P2 but not P21 CrxRip/+ retinas (Figure 6, B and C, and Supplemental Table 1). A complete absence of rod gene expression (including Nr2e3, Mef2c, Rho, and other phototransduction genes) in mature retina can thus be explained simply by the loss of NRL (21). Immunoblot analysis and qPCR analysis showed that the expression of NRL was dramatically reduced after P6 in CrxRip/+ mutant retinas and was absent at and after P14, whereas NRL expression in Crx–/– retinas decreased at a much slower rate (Figure 7A and Supplemental Figure 5). We hypothesized that the loss of NRL at or immediately after P6 would not permit immature committed precursors to complete the rod differentiation program. To test this hypothesis, we maintained Nrl expression independent of its own promoter by mating CrxRip mutants with Crxp::Nrl mice, in which Nrl expression is driven by the Crx promoter (active in the CrxRip mutant) in all photoreceptor precursors leading to rod-only retina (30). Scotopic ERG recordings revealed a partial rescue of the rod function in Crxp::Nrl;CrxRip/+ retinas, with shorter outer segments expressing RHO and peripherin and with nuclei displaying a rod-like denser chromatin (Figure 7, B–D). As predicted, no cone-mediated photopic ERG response was detectable (data not shown). The expression of Nrl and Nr2e3 (completely absent in Rip mutants) was restored by Crxp::Nrl transgene, with no effect on Crx expression (Figure 7E). The rod-specific transcriptional targets of NRL, including Rho, Gnat1, Cnga1, and Esrrb (21), were expressed in Crxp::Nrl;CrxRip/+ retinas, but cone genes (such as Arr3) remained undetectable. Notably, ectopic Nrl expression did not prevent photoreceptor degeneration in the Crxp::Nrl;CrxRip/Rip retinas, presumably because of the strong inhibitory effect of the mutant protein or complete absence of the CRXWT (data not shown). Our data showing the partial rescue of the rod phenotype in CrxRip/+ retinas by ectopic Nrl expression suggested that CrxRip allele did not block NRL function.
Figure 7. Expression of NRL partially rescues the Rip mutant phenotype.

(A) Immunoblot analysis using retinal extracts from P2, P6, P10, P21 Crx+/+ and CrxRip/+ mouse retinas that were probed with anti-NRL antibody. Anti-actin antibody was used as a loading control. (B) Dark-adapted ERG recording in 1-month-old Crxp::Nrl;Crx+/+ and Crxp::Nrl;CrxRip/+ mice. (C) Immunolabeling analysis of RHO (green) and peripherin (red) in 1-month-old Crxp::Nrl;Crx+/+ and Crxp::Nrl;CrxRip/+ mice. Scale bar: 40 μm. (D) Methacrylate sections followed by H&E staining on 1-month-old Crxp::Nrl;Crx+/+ and Crxp::Nrl;CrxRip/+ mice. Scale bar: 40 μm. (E) Differential expression analysis by qPCR of photoreceptor transcription factors (Crx, Nrl, Nr2e3, Err) and genes involved in rod (Gnat1, Rho, Cnga1) or cone (Arr3) phototransduction. For each gene, all values were expressed as mean ± SEM from 3 biological replicates and compared to mRNA expression of WT mice after normalizing by the average expression of 2 housekeeping genes: Act and Hprt.
Otx2 binds to promoters of rod genes, including Nrl, in WT and Crx–/– retinas, but not in CrxRip/Rip retinas, and it can restore Rho expression.
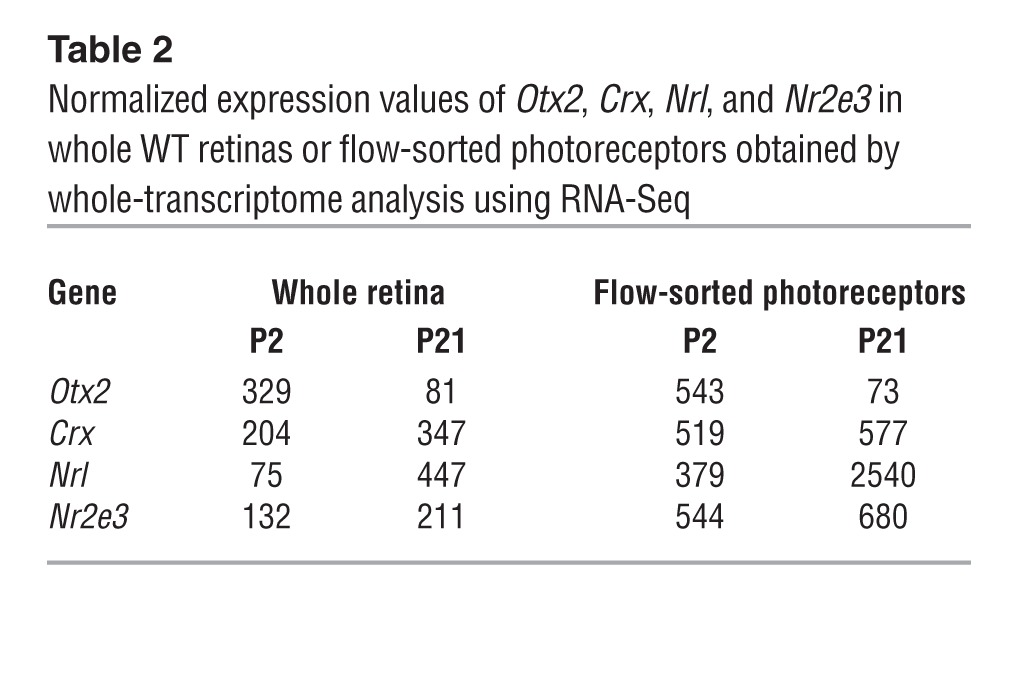
The presence of homeodomain binding sites in the Nrl promoter (17, 44) would argue in favor of CRX and/or OTX2 being the major regulators of Nrl. However, as Nrl expression is rapidly turned off in CrxRip/+ mutants compared with that in Crx–/– mice, we propose that OTX2, and not CRX, is a major contributor for the initiation or maintenance of Nrl transcription in developing retinas. Indeed, RNA-Seq analysis (Supplemental Table 1) showed a small but statistically significant (P < 0.001) increase of Otx2 expression in CrxRip/+, CrxRip/Rip, and Crx–/– retinas (1.6-, 2.9-, and 2.1-fold, respectively), indicating that OTX2 might compensate for the lack of CRX, as demonstrated in Drosophila (45). In addition, transcriptome analysis of flow-sorted WT mouse rod photoreceptors at P2 and P21 revealed continued (though reduced) expression of Otx2 (Table 2).
Table 2.
Normalized expression values of Otx2, Crx, Nrl, and Nr2e3 in whole WT retinas or flow-sorted photoreceptors obtained by whole-transcriptome analysis using RNA-Seq
To further test the potential role of OTX2 in developing and mature retina, we performed OTX2 ChIP-qPCR analysis (Figure 8A). Indeed, the promoters of Nrl and several rod-specific genes exhibited OTX2 binding in P21 WT and Crx–/– retinas; however, the binding of OTX2 to its target genes was completely abrogated in the CrxRip/Rip retinas (Figure 8A). Additionally, OTX2 binding to the cone-specific promoters was observed in WT and Crx–/– retinas but not in the CrxRip/Rip mutant retinas. Our results argue strongly in support of the lack of OTX2 binding being the primary mechanism for the loss of both rod and cone gene expression in the Rip mutants.
Figure 8. OTX2 binds to promoters of rod genes, including Nrl, in WT and Crx–/– retinas but not in CrxRip/Rip retinas.

(A) ChIP-qPCR with anti-OTX2 antibody from P21 WT, CrxRip/Rip, and Crx–/– retinas. Normal IgG was used as control. Fold enrichment represents the fold change of qPCR amplification signals for the different genes tested between OTX2 ChIP DNA and IgG control ChIP DNA. (B) Representative images of P21 CrxRip/+ mouse retinas electroporated at P0 with RHO-DsRed (red) and either OTX2-GFP or Ub-GFP (green; mock). Scale bar: 40 μm.
Based on the dominant phenotype and lack of OTX2 binding to rod and cone gene promoters in the CrxRip/Rip retinas, we wondered whether overexpression of OTX2 could rescue the CrxRip/+ mutant phenotype. In vivo transfection of Ub-GFP (GFP expression identifies the electroporated cells) with Ub-OTX2 construct in retinas of newborn CrxRip/+ mice demonstrated that by P21 a vast majority of Otx2-positive cells (and none of the mock-transfected cells) expressed RHO (Figure 8B). We conclude that ectopic expression of OTX2 in CrxRip/+ mouse retinas could activate the Rho promoter and should be able to rescue the mutant photoreceptor phenotype.
Discussion
Developmental and/or degenerative diseases affecting retinal photoreceptors are a major cause of inherited visual dysfunction and are largely incurable. While cones dominate visual transduction in humans because of their role in mediating day light and color vision, rods represent 95% of the photoreceptors in the human retina and are affected first in the majority of retinal and macular diseases. A better understanding of regulatory networks that establish rod and cone photoreceptor mosaic in developing retina would be valuable for designing knowledge-based strategies for treatment of congenital blinding diseases, such as LCA. OTX2, CRX, and NRL are key transcriptional regulators guiding rod and cone differentiation. Here, we determine a direct role of OTX2 in initiating and maintaining the transcription of Nrl in developing rod photoreceptors by taking advantage of a newly discovered mouse mutant carrying a Crx frameshift mutation. The CRXRip protein acts in a dominant-negative manner in vivo and blocks both CRX and OTX2 in postmitotic precursors, consequently producing a more severe phenotype than the loss of CRX alone (as in Crx–/– retina). Our studies also provide the mechanism of congenital blindness in patients with LCA carrying dominant CRX mutations.
The dominant CrxRip/+ mutant presents a surprising retinal phenotype, with long-term preservation of functionally inactive and immature photoreceptors that display a decondensed chromatin in the nuclei. The cone-like appearance, but without the expression of either cone or rod visual pigment in CrxRip/+ photoreceptors, suggests an early block in the developmental pathway before the decision to become a rod or a cone is finalized, consistent with the proposed model of S-cone being the default photoreceptor cell fate (13). We can now extend the model and propose a novel role of OTX2 in rod differentiation (Figure 9). OTX2, and probably CRX, induces the expression of Nrl (likely in collaboration with RORβ; refs. 44, 46), which competes with Trβ2 to drive the postmitotic precursors to rod fate (28). We propose that OTX2 is needed in developing rods to maintain Nrl levels and that the CRXRip protein inhibits OTX2 binding to Nrl promoter thereby blocking rod differentiation. Crx–/– data (this report, refs. 16, 20, 26) show that CRX does not affect Nrl expression appreciably in early developing rods but rather enhances the expression of phototransduction genes synergistically with NRL to produce mature and functional rods. In the absence of CRX, OTX2 can maintain NRL levels, allowing rod development to proceed, though dramatically reduced expression of RHO and other phototransduction genes does not permit outer segment formation. In concordance, the Crxp::Nrl transgene was able to only partially rescue the rod phenotype in CrxRip/+ mutant retinas, as reflected by ERG and low expression of phototransduction genes (see Figure 7). In CrxRip/+ retinas, the mutant protein acts as a dominant negative and blocks OTX2, leading to the loss of of both CRX and NRL function and arrest of photoreceptor differentiation. Our P2 RNA-Seq data clearly establish the role of OTX2 in inducing and maintaining NRL expression in developing rods.
Figure 9. The molecular mechanism of congenital blindness caused by dominant CRX frameshift mutations.

After cell cycle exit, and under the control of OTX2, postmitotic precursors get restricted to the photoreceptor lineage and are fated to produce both rods and cones. These precursors will differentiate by default into S-cones, unless their fate is directed into rods by the expression of NRL, or into M-cones by the expression of TRβ2. In developing photoreceptors, NRL expression is initiated by OTX2 and RORβ and increases during development to restrict the lineage to rods. OTX2 plays a crucial role in maintaining NRL expression to consolidate rod cell fate. Sustained expression of NRL is needed to induce downstream targets, including NR2E3, that are critical for suppressing cone genes and for rod maturation in collaboration with CRX. In CrxRip/+ mutant retinas, CRXRip protein blocks both OTX2 and CRX, arresting NRL expression and, consequently, rod differentiation pathway. In addition, CRXRip protein prevents the CRXWT protein from forming requisite transcriptional complexes for rod gene expression. Ultimately, the arrest in photoreceptor development does not permit phototransduction, causing congenital blindness.
In humans, almost 50 CRX mutations have been identified in patients with retinopathy (35); a majority of these are fully penetrant and act in a dominant manner (47, 48). The p.G255fs mutation in CrxRip mice (as in several dominant LCA patients) removes the C-terminal Otx-like domain of CRX and instead adds 133 unrelated residues. As the truncated CRX1–254 protein retains DNA binding and transactivation properties consistent with a previous study of CRX functional domains (49), the addition of unrelated residues in CRXRip protein must interfere with the function of CRXWT and OTX2 proteins (as discussed earlier). As two dominant LCA-causing mutations examined here mimic the CrxRip/+ phenotype, our studies provide a plausible molecular mechanism for congenital blindness in dominant CRX-LCA.
The long-term preservation of immature and dysfunctional cones in CrxRip/+ mutant retinas in the absence of rod photoreceptors is consistent with our recent studies (37, 50) and provides opportunities for elucidating cone survival pathways and designing treatment paradigms for degenerating retinal diseases. At this stage, it is unclear whether dominant CRX frameshift mutations arrest photoreceptor differentiation in humans, as disease onset is very early and OCT studies are difficult to perform. Where OCT was possible, significant retinal thinning is observed with some preservation of photoreceptors (34). We believe that CrxRip/+ mutant mice would serve as reasonable model for assessing gene- or cell-based intervention approaches for dominant CRX-LCA. Though disease rescue using AAV-based vectors that express CRX, OTX2, or NRL in target cells may be achievable, the selected gene would have to be introduced very early in infancy. Preliminary investigations of injecting AAV constructs expressing Nrl in 2-month-old CrxRip/+ retinas revealed promising findings, as some RHO expression was restored in photoreceptors (see Supplemental Figure 6). However, more detailed clinical analysis of patients would be clearly desirable to design therapeutic strategies.
We also note that Crx–/– mice have been used previously for evaluating cell replacement therapy by photoreceptor transplantation (51, 52); however, photoreceptor cell death occurring in the Crx–/– host retinas may impair long-term survival of the transplanted cells. The unique phenotype of the CrxRip/+ mutant should provide a better host retinal environment for exploring photoreceptor integration and functional assessment in cell replacement therapy.
Methods
Animals and tissue collection.
The Rip mutant was identified in C57BL/6J background from our mouse colony by fundus examination. The Nrl–/–, Crx–/–, and Crxp::Nrl mice in C57BL/6J background have been described previously (24, 29, 30). Neonatal CD1 mice (Charles River Laboratories) were used for in vivo electroporation. P0 is considered the day of birth. Mice of either sex were used for the study and euthanized by CO2 inhalation. The procedures for tissue preparation for cryopreservation, methacrylate sections, and RNA/protein extraction have been described earlier (37, 53), and additional details are provided in the Supplemental Methods.
Antibodies.
The antibodies used in this study are listed in Supplemental Table 2.
Plasmid constructions and site-directed mutagenesis.
The details of cDNA constructs used in this study are described in the Supplemental Methods.
Retinal phenotyping.
Mice were anesthetized with ketamine and xylazine. Pupils were dilated using topical 0.5% tropicamide and 1% cyclopentolate hydrochloride. The methods for fundus examination, OCT imaging, and ERG have been described previously (37).
Linkage analysis of Rip mutant mice.
We mated Rip mutant mice in the C57BL/6J background with C3A.BLiA-Pde6b+/J mice. For linkage analysis, 75 backcross progenies from the (Rip mutant X C3A.BLiAPde6b+/J)F1 X C3A.BLiA-Pde6b+/J were phenotyped by retinal fundus examination and genotyped using microsatellite markers. Genetic markers defining the critical domain were D7Mit340, D7Mit56, and D7Mit191, spanning a 19.5-Mb genomic region.
Exome sequencing and variant calling.
Exome capture and sequencing were performed as described previously (54). Genomic DNA (3 μg) from 2 homozygous Rip mutants, identified by using the linked markers, and 1 WT mouse was sheared using a Covaris ultrasonicator and subjected to library preparation and whole-exome capture using the SureSelect Human All Exon 50 Mb Kit (Agilent Technologies), following the manufacturer’s instructions. The captured libraries were amplified and converted to clusters using Cluster Station. Single-end sequencing was performed on Illumina GAIIx. The sequence reads were mapped to the mouse genome (NCBI37/mm9) using the Burrows-Wheeler Alignment tool (55). Variants were subsequently called using SAMtools (56), and annotations were obtained using ANNOVAR (57).
Immunoblotting and immunohistochemistry.
Frozen retinas were lysed by sonication in radioimmunoprecipitation assay buffer supplemented with protease inhibitors (Roche Applied Science). The supernatant proteins were resolved by SDS-PAGE under reducing conditions and transferred to polyvinyl difluoride membrane, as previously described (53). Cryosections were probed with selected antibodies (58). Fluorescent staining signals were captured using an Olympus FluoView FV1000 confocal laser-scanning unit (Olympus America Inc.).
In vivo electroporation in mouse retinas.
The retinas of newborn CD1 and Rip mutant mice were electroporated as previously described (53, 59).
EMSAs.
EMSAs were performed as previously described (60). The method used for this study is described in the Supplemental Methods.
ChIP-qPCR.
ChIP using CRX or OTX2 antibody and normal IgG control was performed as described previously (21) using retinas from P21 WT C57BL/6J, Crx–/–, and CrxRip/Rip mice. Duplicate ChIP was performed, and the ChIP DNA was quantified by real-time qPCR using SYBR Green Super mixture (Bio-Rad) with the primers listed in Supplemental Table 3.
Whole-transcriptome sequencing (RNA-Seq) and data analysis.
RNA extraction, library preparation, and sequencing are detailed in the Supplemental Methods. RNA-seq data are available at GEO (accession no. GSE52006).
Statistics.
Two-way comparisons in Figure 5A used 2-tailed Student’s t tests, and P values of less than 0.05 were considered significant. For RNA-seq data, differential expression analysis was performed using DEseq, and an adjusted P value of less than or equal to 0.05 was considered significant. Data in Figure 5, A and C; Figure 7E; and Figure 8A are represented using mean ± SEM.
Study approval.
All experiments with mice followed the animal protocol approved by the Animal Care and Use Committee of the National Eye Institute, conforming to the Association for Research in Vision and Ophthalmology guidelines.
Supplementary Material
Acknowledgments
We are grateful to Jung-Woong Kim for expression profiles from flow-sorted photoreceptors and Rivka Rachel, Matthew Brooks, and Harsha Rajasimha for advice and assistance. We also thank Peter Colosi, Zhijian Wu, and Suja Hiriyanna for help with the AAV experiment. This research was supported by Intramural Research Program (EY000450, EY000473, EY000474) and EY019943 of the National Eye Institute.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(2):631–643. doi:10.1172/JCI72722.
References
- 1.Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet. 2010;11(4):273–284. doi: 10.1038/nrg2717. [DOI] [PubMed] [Google Scholar]
- 2.Leber T. Über Retinitis pigmentosa und angeborene Amaurose. Archiv für Ophthalmologie. 1869;15:1–25. [Google Scholar]
- 3.Traboulsi EI. The Marshall M. Parks memorial lecture: making sense of early-onset childhood retinal dystrophies — the clinical phenotype of Leber congenital amaurosis. Br J Ophthalmol. 2010;94(10):1281–1287. doi: 10.1136/bjo.2009.165654. [DOI] [PubMed] [Google Scholar]
- 4.den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27(4):391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Freund CL, et al. De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat Genet. 1998;18(4):311–312. doi: 10.1038/ng0498-311. [DOI] [PubMed] [Google Scholar]
- 6.Sohocki MM, et al. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am J Hum Genet. 1998;63(5):1307–1315. doi: 10.1086/302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowne SJ, et al. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2006;47(1):34–42. doi: 10.1167/iovs.05-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cideciyan AV. Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog Retin Eye Res. 2010;29(5):398–427. doi: 10.1016/j.preteyeres.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cayouette M, Barres BA, Raff M. Importance of intrinsic mechanisms in cell fate decisions in the developing rat retina. Neuron. 2003;40(5):897–904. doi: 10.1016/S0896-6273(03)00756-6. [DOI] [PubMed] [Google Scholar]
- 10.Agathocleous M, Harris WA. From progenitors to differentiated cells in the vertebrate retina. Annu Rev Cell Dev Biol. 2009;25:45–69. doi: 10.1146/annurev.cellbio.042308.113259. [DOI] [PubMed] [Google Scholar]
- 11.Jeon CJ, Strettoi E, Masland RH. The major cell populations of the mouse retina. J Neurosci. 1998;18(21):8936–8946. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wikler KC, Rakic P. Distribution of photoreceptor subtypes in the retina of diurnal and nocturnal primates. J Neurosci. 1990;10(10):3390–3401. doi: 10.1523/JNEUROSCI.10-10-03390.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swaroop A, Kim D, Forrest D. Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat Rev Neurosci. 2010;11(8):563–576. doi: 10.1038/nrn2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishida A, et al. Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nat Neurosci. 2003;6(12):1255–1263. doi: 10.1038/nn1155. [DOI] [PubMed] [Google Scholar]
- 15.Omori Y, et al. Analysis of transcriptional regulatory pathways of photoreceptor genes by expression profiling of the Otx2-deficient retina. PLoS One. 2011;6(5):e19685. doi: 10.1371/journal.pone.0019685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muranishi Y, et al. Gene expression analysis of embryonic photoreceptor precursor cells using BAC-Crx-EGFP transgenic mouse. Biochem Biophys Res Commun. 2010;392(3):317–322. doi: 10.1016/j.bbrc.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 17.Montana CL, et al. Transcriptional regulation of neural retina leucine zipper (Nrl), a photoreceptor cell fate determinant. J Biol Chem. 2011;286(42):36921–36931. doi: 10.1074/jbc.M111.279026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen S, et al. Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron. 1997;19(5):1017–1030. doi: 10.1016/S0896-6273(00)80394-3. [DOI] [PubMed] [Google Scholar]
- 19.Furukawa T, Morrow EM, Cepko CL. Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell. 1997;91(4):531–541. doi: 10.1016/s0092-8674(00)80439-0. [DOI] [PubMed] [Google Scholar]
- 20.Corbo JC, et al. CRX ChIP-seq reveals the cis-regulatory architecture of mouse photoreceptors. Genome Res. 2010;20(11):1512–1525. doi: 10.1101/gr.109405.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hao H, et al. Transcriptional regulation of rod photoreceptor homeostasis revealed by in vivo NRL targetome analysis. PLoS Genet. 2012;8(4):e1002649. doi: 10.1371/journal.pgen.1002649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitton KP, Swain PK, Chen S, Xu S, Zack DJ, Swaroop A. The leucine zipper of NRL interacts with the CRX homeodomain. A possible mechanism of transcriptional synergy in rhodopsin regulation. J Biol Chem. 2000;275(38):29794–29799. doi: 10.1074/jbc.M003658200. [DOI] [PubMed] [Google Scholar]
- 23.Srinivas M, Ng L, Liu H, Jia L, Forrest D. Activation of the blue opsin gene in cone photoreceptor development by retinoid-related orphan receptor beta. Mol Endocrinol. 2006;20(8):1728–1741. doi: 10.1210/me.2005-0505. [DOI] [PubMed] [Google Scholar]
- 24.Furukawa T, Morrow EM, Li T, Davis FC, Cepko CL. Retinopathy and attenuated circadian entrainment in Crx-deficient mice. Nat Genet. 1999;23(4):466–470. doi: 10.1038/70591. [DOI] [PubMed] [Google Scholar]
- 25.Hsiau TH, Diaconu C, Myers CA, Lee J, Cepko CL, Corbo JC. The cis-regulatory logic of the mammalian photoreceptor transcriptional network. PLoS One. 2007;2(7):e643. doi: 10.1371/journal.pone.0000643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hennig AK, Peng GH, Chen S. Regulation of photoreceptor gene expression by Crx-associated transcription factor network. Brain Res. 2008;1192:114–133. doi: 10.1016/j.brainres.2007.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akimoto M, et al. Targeting of GFP to newborn rods by Nrl promoter and temporal expression profiling of flow-sorted photoreceptors. Proc Natl Acad Sci U S A. 2006;103(10):3890–3895. doi: 10.1073/pnas.0508214103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng L, Lu A, Swaroop A, Sharlin DS, Swaroop A, Forrest D. Two transcription factors can direct three photoreceptor outcomes from rod precursor cells in mouse retinal development. J Neurosci. 2011;31(31):11118–11125. doi: 10.1523/JNEUROSCI.1709-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mears AJ, et al. Nrl is required for rod photoreceptor development. Nat Genet. 2001;29(4):447–452. doi: 10.1038/ng774. [DOI] [PubMed] [Google Scholar]
- 30.Oh EC, Khan N, Novelli E, Khanna H, Strettoi E, Swaroop A. Transformation of cone precursors to functional rod photoreceptors by bZIP transcription factor NRL. Proc Natl Acad Sci U S A. 2007;104(5):1679–1684. doi: 10.1073/pnas.0605934104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freund CL, et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell. 1997;91(4):543–553. doi: 10.1016/S0092-8674(00)80440-7. [DOI] [PubMed] [Google Scholar]
- 32.Swaroop A, et al. Leber congenital amaurosis caused by a homozygous mutation (R90W) in the homeodomain of the retinal transcription factor CRX: direct evidence for the involvement of CRX in the development of photoreceptor function. Hum Mol Genet. 1999;8(2):299–305. doi: 10.1093/hmg/8.2.299. [DOI] [PubMed] [Google Scholar]
- 33.Swain PK, et al. Mutations in the cone-rod homeobox gene are associated with the cone-rod dystrophy photoreceptor degeneration. Neuron. 1997;19(6):1329–1336. doi: 10.1016/S0896-6273(00)80423-7. [DOI] [PubMed] [Google Scholar]
- 34.Nichols LL, 2nd, et al. Two novel CRX mutant proteins causing autosomal dominant Leber congenital amaurosis interact differently with NRL. Hum Mutat. 2010;31(6):E1472–E1483. doi: 10.1002/humu.21268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang L, et al. CRX variants in cone-rod dystrophy and mutation overview. Biochem Biophys Res Commun. 2012;426(4):498–503. doi: 10.1016/j.bbrc.2012.08.110. [DOI] [PubMed] [Google Scholar]
- 36.Daniele LL, et al. Cone-like morphological, molecular, and electrophysiological features of the photoreceptors of the Nrl knockout mouse. Invest Ophthalmol Vis Sci. 2005;46(6):2156–2167. doi: 10.1167/iovs.04-1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roger JE, et al. Preservation of cone photoreceptors after a rapid yet transient degeneration and remodeling in cone-only Nrl–/– mouse retina. . J Neurosci. 2012;32(2):528–541. doi: 10.1523/JNEUROSCI.3591-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solovei I, et al. Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell. 2009;137(2):356–368. doi: 10.1016/j.cell.2009.01.052. [DOI] [PubMed] [Google Scholar]
- 39.Cheng H, Khanna H, Oh EC, Hicks D, Mitton KP, Swaroop A. Photoreceptor-specific nuclear receptor NR2E3 functions as a transcriptional activator in rod photoreceptors. Hum Mol Genet. 2004;13(15):1563–1575. doi: 10.1093/hmg/ddh173. [DOI] [PubMed] [Google Scholar]
- 40.Peng GH, Ahmad O, Ahmad F, Liu J, Chen S. The photoreceptor-specific nuclear receptor Nr2e3 interacts with Crx and exerts opposing effects on the transcription of rod versus cone genes. Hum Mol Genet. 2005;14(6):747–764. doi: 10.1093/hmg/ddi070. [DOI] [PubMed] [Google Scholar]
- 41.Brooks MJ, Rajasimha HK, Swaroop A. Retinal transcriptome profiling by directional next-generation sequencing using 100 ng of total RNA. Methods Mol Biol. 2012;884:319–334. doi: 10.1007/978-1-61779-848-1_23. [DOI] [PubMed] [Google Scholar]
- 42.Ozawa Y, et al. Downregulation of STAT3 activation is required for presumptive rod photoreceptor cells to differentiate in the postnatal retina. Mol Cell Neurosci. 2004;26(2):258–270. doi: 10.1016/j.mcn.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 43.Ueki Y, Wang J, Chollangi S, Ash JD. STAT3 activation in photoreceptors by leukemia inhibitory factor is associated with protection from light damage. J Neurochem. 2008;105(3):784–796. doi: 10.1111/j.1471-4159.2007.05180.x. [DOI] [PubMed] [Google Scholar]
- 44.Kautzmann MA, Kim DS, Felder-Schmittbuhl MP, Swaroop A. Combinatorial regulation of photoreceptor differentiation factor, neural retina leucine zipper gene NRL, revealed by in vivo promoter analysis. J Biol Chem. 2011;286(32):28247–28255. doi: 10.1074/jbc.M111.257246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Terrell D, et al. OTX2 and CRX rescue overlapping and photoreceptor-specific functions in the Drosophila eye. Dev Dyn. 2012;241(1):215–228. doi: 10.1002/dvdy.22782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jia L, et al. Retinoid-related orphan nuclear receptor ROR{beta} is an early-acting factor in rod photoreceptor development. Proc Natl Acad Sci U S A. 2009;106(41):17534–17539. doi: 10.1073/pnas.0902425106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rivolta C, Berson EL, Dryja TP. Dominant Leber congenital amaurosis, cone-rod degeneration, and retinitis pigmentosa caused by mutant versions of the transcription factor CRX. Hum Mutat. 2001;18(6):488–498. doi: 10.1002/humu.1226. [DOI] [PubMed] [Google Scholar]
- 48.Silva E, Yang JM, Li Y, Dharmaraj S, Sundin OH, Maumenee IH. A CRX null mutation is associated with both Leber congenital amaurosis and a normal ocular phenotype. Invest Ophthalmol Vis Sci. 2000;41(8):2076–2079. [PubMed] [Google Scholar]
- 49.Chen S, et al. Functional analysis of cone-rod homeobox (CRX) mutations associated with retinal dystrophy. Hum Mol Genet. 2002;11(8):873–884. doi: 10.1093/hmg/11.8.873. [DOI] [PubMed] [Google Scholar]
- 50.Cideciyan AV, et al. Centrosomal-ciliary gene CEP290/NPHP6 mutations result in blindness with unexpected sparing of photoreceptors and visual brain: implications for therapy of Leber congenital amaurosis. Human Mutat. 2007;28(11):1074–1083. doi: 10.1002/humu.20565. [DOI] [PubMed] [Google Scholar]
- 51.Homma K, et al. Developing rods transplanted into the degenerating retina of crx-knockout mice exhibit neural activity similar to native photoreceptors. Stem Cells. 2013;31(6):1149–1159. doi: 10.1002/stem.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lamba DA, Gust J, Reh TA. Transplantation of human embryonic stem cell-derived photoreceptors restores some visual function in Crx-deficient mice. Cell Stem Cell. 2009;4(1):73–79. doi: 10.1016/j.stem.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roger JE, Nellissery J, Kim DS, Swaroop A. Sumoylation of bZIP transcription factor NRL modulates target gene expression during photoreceptor differentiation. J Biol Chem. 2010;285(33):25637–25644. doi: 10.1074/jbc.M110.142810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Priya RR, Rajasimha HK, Brooks MJ, Swaroop A. Exome sequencing: capture and sequencing of all human coding regions for disease gene discovery. Methods Mol Biol. 2012;884:335–351. doi: 10.1007/978-1-61779-848-1_24. [DOI] [PubMed] [Google Scholar]
- 55.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roger J, et al. Involvement of Pleiotrophin in CNTF-mediated differentiation of the late retinal progenitor cells. Dev Biol. 2006;298(2):527–539. doi: 10.1016/j.ydbio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 59.Matsuda T, Cepko CL. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci U S A. 2004;101(1):16–22. doi: 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hao H, et al. The transcription factor neural retina leucine zipper (NRL) controls photoreceptor-specific expression of myocyte enhancer factor Mef2c from an alternative promoter. J Biol Chem. 2011;286(40):34893–34902. doi: 10.1074/jbc.M111.271072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.