

Summary
Background
Type 3 von Willebrand disease (VWD) is the most severe form of the disease and is classically inherited in an autosomal recessive fashion.
Objectives
The aim of the current study was to investigate the molecular pathogenesis of a Canadian cohort of type 3 VWD patients.
Patients/Methods
34 families comprised of 100 individuals were investigated. Phenotypic data, including bleeding scores (BS), von Willebrand factor (VWF) laboratory values, and anti-VWF inhibitor status were included as well as sequence analysis.
Results
We identified 31 different mutations (20 novel): 8 frameshift, 5 splice site, 9 nonsense, 1 gene conversion, 6 missense, and 2 partial gene deletion mutations. The majority of mutations identified were in the propeptide (42%); index cases (IC) with these mutations exhibited more severe bleeding (BS=22) than those with mutations elsewhere in VWF (BS=13). 62 of 68 (91%) mutant alleles were identified. Twenty-nine IC (85%) had a VWF null genotype identified; 17 homozygous, 12 compound heterozygous. In five IC (15%), two mutant VWF alleles were not identified to explain the type 3 VWD phenotype. In four ICs only one mutant VWF allele was identified and in one IC no mutant VWF alleles were identified.
Conclusions
We have investigated the molecular pathogenesis of a Canadian cohort of type 3 VWD patients. Obligate carriers are not phenotypically silent in the Canadian population; 48% have been diagnosed with type 1 VWD. In ~50% of families in this study the inheritance pattern for type 3 VWD is co-dominant and not recessive.
Keywords: obligate carrier, type 3, VWD, VWF
Introduction
Von Willebrand disease (VWD) results from quantitative or qualitative deficiencies of von Willebrand factor (VWF), a large, multimeric glycoprotein that plays a critical role in primary and secondary hemostasis. Von Willebrand disease (VWD) is recognized as the most common inherited bleeding disorder in humans, with symptomatic prevalence estimates of approximately 1 in 1,000 (0.1%) [1]. Type 3 VWD is the most severe form of the disease resulting from markedly decreased or undetectable VWF and reduced factor VIII (FVIII) activity [2].
The prevalence of type 3 VWD varies between countries, ranging from 0.1–5.3 per million, with increased prevalence in areas with more frequent consanguineous marriages [2]. Individuals with type 3 VWD present clinically with moderate to severe mucocutaneous bleeding symptoms as well as muscle hematomas and hemarthroses. Classically, inheritance of type 3 VWD follows an autosomal recessive pattern with affected individuals being homozygous or compound heterozygous for null alleles and with obligate carriers (OC) of the type 3 VWD mutations being phenotypically normal. More recently, however, it is becoming clear that this is not always the case and co-dominance is observed, with OC exhibiting low VWF levels and a bleeding diathesis. Castaman et al. (2006) showed that OC of type 3 VWD report more bleeding symptoms than healthy controls, but are less symptomatic than carriers of type 1 VWD [3]. A commentary written in the same issue of the journal titled “When it comes to von Willebrand disease does 1+1=3?” further highlights the need for additional studies looking at the differences between OC of type 3 VWD and individuals with type 1 VWD in order for that question to be answered [4].
The majority of type 3 VWD mutations are small deletions and insertions, nonsense mutations or other mutations, located throughout the VWF gene that interfere with VWF synthesis and secretion [2]. This is in contrast to type 1 VWD in which the majority of mutations are missense changes (70–75%), with only about 10–15% of mutations identified which lead to null alleles [5–7]. There are presently 108 different reported type 3 VWD mutations according to the International Society on Thrombosis and Haemostasis (ISTH) SSC VWF database (http://www.vwf.group.shef.ac.uk/, accessed May 13th, 2012).
Partial and total VWF deletions have been reported previously including deletions of single exons [8], multiple exons [9–13] and the entire VWF gene [14–17]; these deletions however only constitute approximately 10% of all reported type 3 VWD mutations (http://www.group.shef.ac.uk/). Conventional sequencing of PCR-amplified DNA does not provide an adequate strategy for the detection of all mutations, as partial/total deletions and large duplications may not be apparent in the heterozygous state because the alternate normal allele is amplified and masks the deletion or duplication present on the other allele. Therefore, additional strategies are required in cases where no complete pathogenetic explanation has been obtained through conventional methods. Multiplex ligation-dependant probe amplification (MLPA) has been used recently to identify partial and large gene deletions in VWD patients [18–20].
Identification of both mutant VWF alleles resulting in type 3 VWD has been reported in ~80–90% of cases. Importantly, the majority of type 3 VWD populations reported to date have been homogeneous populations [12,21–24]. In contrast, the Canadian type 3 VWD population is distinct, with areas of significant population homogeneity, as well as areas with heterogeneous populations due to immigration in large Canadian urban centres.
In this paper we report the mutational spectrum of a cohort of Canadian type 3 VWD patients. While a number of type 3 VWD studies have been previously reported, our report here of the Canadian type 3 VWD population represents one of the largest and most thoroughly investigated cohorts of type 3 VWD patients and their family members. Our paper also highlights distinct features of the Canadian population of type 3 VWD patients.
Patients, Materials and Methods
Patients
Eligible subjects were enrolled in the Canadian Type 3 VWD Study from the Inherited Bleeding Disorders/Hemophilia Clinics of the Association of Hemophilia Clinic Directors of Canada (AHCDC). Inclusion criteria included an index case (IC) with a documented history of excessive mucocutaneous bleeding and plasma levels of VWF antigen (VWF:Ag) and/or VWF ristocetin cofactor (VWF:RCo) < 0.05 IU/ml on at least two occasions and a factor FVIII coagulant activity (FVIII:C) of <0.10 IU/ml. A positive family history of bleeding was not required for enrollment because of the recessive inheritance pattern of type 3 VWD. When possible, samples from the IC and both parents were collected, as well as any available siblings and/or other family members. Venous blood samples were collected by phlebotomy in both 3.2% sodium citrate (at a ratio of 9:1 vol/vol) and EDTA. All study participants gave informed consent and study approval was obtained from the Research Ethics Board of Queen’s University, Kingston, Canada and from each of the source institutions.
Bleeding questionnaire
A standardized bleeding questionnaire was administered to IC and available family members [25]. The occurrence, frequency and severity of various bleeding symptoms including mucocutaneous symptoms as well as muscle hematomas and hemarthroses were assessed using the questionnaires. Bleeding questionnaires were administered by an experienced person and bleeding scores were generated by summing the score of all bleeding symptoms for a given subject.
Coagulation Studies
Laboratory VWF and FVIII tests were conducted at the patients’ source clinics as per local methods. In order to confirm the type 3 VWD diagnosis, all tests were repeated on frozen plasma samples at the Clinical and Molecular Hemostasis Research Laboratory, Queen’s University, Kingston, Canada. The values reported here are from the central laboratory in Kingston.
Alloantibodies to VWF
A rare complication of type 3 VWD is the development of inhibitory antibodies to VWF following replacement therapy (incidence of 7.5–9.5%) which can lead to ineffective treatment and/or severe and life-threatening anaphylaxis [26]. To test for alloantibodies to VWF in the type 3 patients (IC and type 3 VWD family members) an ELISA-based assay was used. Briefly, plates were coated with either Humate-P®, Wilate®, or locally produced recombinant human VWF (~0.7U/ml) and were run in parallel. The recombinant human VWF was produced by transient transfection into HEK293T cells and secretion into serum-free media. Normal plasma pool (NPP) and “positive” anti-VWF antibody plasma, from an individual with acquired VWD, were run as negative and positive controls respectively for the anti-VWF antibody ELISA on each plate. We acknowledge that the use of an acquired VWD patient sample as a positive control is not ideal, and a limitation of this methodology, but we did not have access to an alloantibody positive control. Plasma samples were diluted 1:200. Anti-Human IgG peroxidase conjugate (Sigma A2290) diluted 1:5000 was used as a secondary antibody.
An assay negative cut off value was determined by taking the mean absorbance value plus three standard deviations of the test samples. Any sample with absorbance values above this cut-off were identified as inhibitor-positive and were further analyzed using mixing studies with NPP and analysis by VWF functional assays. VWF:RCo was analyzed using a standard agglutination test with fixed platelets. ELISA-based FVIII binding and collagen binding assays were also conducted as per previously published methods [27,28].
DNA sequencing
Blood samples from IC and available family members were collected and genomic DNA was extracted from leukocytes using a salt-extraction method [29]. DNA analysis was performed by direct sequencing of the VWF gene as previously reported [7], including exons 1–52 (including exon/introns boundaries and flanking intronic sequence) as well as approximately 1.5 kb of the VWF promoter region. Primer sequences are available upon request. If a mutation was identified in an IC, an additional template was PCR amplified and the opposite strand sequenced in order to confirm the sequence variation. All chromatograms were reviewed by at least two experienced technologists and PCR samples were re-run if there were any doubts in interpretation. Once a mutation was identified in an IC then all enrolled family members were sequenced for that mutation in order to confirm familial transmission.
IC for whom no complete pathogenic explanation could be elucidated through direct sequencing were evaluated for partial/total gene deletions. As well, IC found to be homozygous for a missense mutation through direct sequencing were also investigated to rule out the possibility that the missense mutation was in cis with a partial gene deletion. The SALSA-MLPA VWF assay was performed using the P011-B1 and P012-B2 VWF kit (MRC-Holland, Amsterdam, the Netherlands) according to the manufacturer’s directions. The amplification products were identified on the ABI 3130xl sequencer (Applied Biosystems, Foster City, CA, USA) in the DNA Diagnostic Laboratory, Kingston General Hospital, Kingston, ON, Canada. MLPA analysis spreadsheets (National Genetics Reference Laboratory (NGRL), Manchester, UK) were then used to estimate the DNA dosage. Familial transmission of an identified partial gene deletion was confirmed in available family members by MLPA.
Results
Patients
Thirty-five families comprised of 102 individuals were initially submitted to the study. After phenotypic confirmation in the central laboratory, one family (one IC and one FM) was excluded due to a FVIII level >0.10 IU/ml. Therefore, the study population includes 100 individuals: 42 type 3 VWD patients (34 IC and 8 type 3 VWD siblings), 21 OC (defined as either being the offspring of an individual with type 3 VWD or having offspring with type 3 VWD), 30 individuals diagnosed at their enrolling institution with type 1 VWD (19 OC and 11 siblings or other family members), and 7 unaffected family members (UFM). Characteristics of all study participants are presented in Table 1. The majority of families are comprised of two generations; however there are two 3-generation families and four single IC represented in this study. The majority of IC were Caucasian (n=21, 62%), however the remaining IC were of a number of different ethnicities (South Asian 12%; Middle Eastern 6%; and Lebanese, Scottish/Jamaican, South American, Trinidad/Ethiopian, Asian East Indian, West Asian, Hispanic ~ 3% each). Eighteen (53%) of the families have both individuals diagnosed with type 1 VWD and individuals diagnosed with type 3 VWD.
Table 1.
Characteristics and phenotypic data for the 100 study subjects.
| Type 3 VWD (n=42) | OC (n=21) | Type 1 VWD (n=30) | UFM (n=7) | p | |
|---|---|---|---|---|---|
| No. of Females (%) | 23 (55) | 11 (52) | 17 (57) | 4 (57) | ns |
| Mean age, years (range) | 29 (1–72) | 51 (32–83) | 39 (2–85) | 49 (22–68) | 0.001 |
| No. Blood group O (%) | 16 (39) | 6 (29) | 10 (33) | 0 (0) | ns |
| Mean VWF:Ag, IU/ml (range) | 0.02 (0.01–0.07) | 0.74 (0.24–1.42) | 0.35 (0.11–0.52) | 1.40 (0.95–2.04) | <0.001 |
| Mean VWF:RCo, IU/ml (range) | 0.02 (0.00–0.05) | 0.66 (0.15–1.15) | 0.30 (0.05–0.57) | 1.04 (0.66–1.96) | <0.001 |
| Mean FVIII:C, IU/ml (range) | 0.03 (0.00–0.09) | 0.88 (0.37–1.56) | 0.53 (0.02–1.32) | 1.23 (0.52–2.12) | <0.001 |
| Median bleeding score (range) | 13 (3–30) | 1 (−1 to 6) | 4 (0–12) | 0 (−1–4) | <0.001 |
OC = obligate carrier; UFM = unaffected family member; ns = no significance. One-way ANOVA followed by Tukey’s post hoc testing for all linear variables. For age: post-hoc analysis p=0.001 between type 3 and OC, all other groups =ns. For bleeding score: Kruskal-Wallis followed by Mann-Whitney U post hoc testing Post-hoc testing BS: p<0.001 between UFM and T1, T1 and OC, and between T3 and all other groups. Chi-squared testing for categorical variables.
Bleeding questionnaire
Bleeding scores (BS) are reported in Table 1. The BS showed an overall inverse correlation with plasma VWF:Ag, VWF:RCo and FVIII levels (p<0.001, Spearman’s rho = −0.746, −0.739, and −0.721, respectively). The BS reported for the type 3 VWD patients in this study (median BS=13: range=3–30) are similar to those reported in other type 3 VWD populations [23,30]. Overall, the median BS were significantly different between the groups (p<0.001, Kruskal-Wallis test), with the BS being higher in the type 3 VWD group compared with each of the other three groups (Mann-Whitney U post hoc testing). OC of type 3 VWD have similar BS compared to UFM (p = 0.394; median BS=1 versus median BS= 0). The OC of type 3 VWD had significantly lower median BS compared to the family members diagnosed with type 1 VWD (p<0.001; median BS=1 compared to BS=4).
IC with mutations identified in the propeptide region of VWF (n=8) had a significantly higher BS (median=22), compared to IC with mutations in other regions of VWF (median=13) (p=0.012, Mann-Whitney U).
Phenotypic analyses
Table 1 summarizes the phenotypic data of all study participants. VWF:Ag, VWF:RCo and FVIII levels were significantly different between all groups (p<0.001, one-way ANOVA followed by Tukey’s post hoc testing). Post-hoc testing showed significant differences between each of the groups for all parameters with the exception of FVIII levels, which were not significantly different between OC and UFM.
Alloantibodies to VWF
The 42 type 3 patients (34 IC+ 8 type 3 VWD family members) were tested for alloantibodies to VWF. One IC (T018) had absorbance levels greater than the assay negative cut-off on all anti-VWF ELISA assays. When the plasma of T018 was mixed 50/50 with NPP, VWF:RCo and FVIII binding function were restored to levels within the normal range. However, collagen binding activity was only restored to 40% of normal upon mixing with NPP.
Genotypic analyses
Mutations were identified in 33 (97%) IC. Sixty-two of 68 (91%) mutant alleles were identified. Twenty-nine IC (85%) had two mutant VWF alleles identified; 17 were homozygous, and 12 were compound heterozygous. Of note, these were not always null mutations; three IC were homozygous for missense mutations and one IC was compound heterozygous for a null mutation and a missense mutation. For four IC (12%) only one VWF mutation was identified, all of which were null mutations. No VWF mutations were identified in one IC.
A total of 31 different mutations (20 novel mutations) were identified, comprised of 8 frameshift, 5 splice site, 9 nonsense, 1 gene conversion, 6 missense, and 2 partial gene deletion mutations. All VWF sequence variations identified, both putative and polymorphic, are listed by IC in Table 2. Figure 1 shows the location of the mutations scattered throughout the VWF gene. While mutations were identified throughout VWF, 42% of the non-splicing mutations were located in the propeptide region. This is in contrast to only 7% of non-splicing mutations being found in the propeptide region in the Canadian Type 1 VWD study [7].
Table 2.
Phenotypic and genotypic data for the 34 index cases.
| Patient ID | Gender/age (M/F year) | Bleeding Score | VWF:Ag (IU/ml) | VWF:RCo (IU/ml) | FVIII:C (IU/ml) | Nucleotide Change, HGVS | Amino Acid Change, HGVS | Exon | Genotype |
|---|---|---|---|---|---|---|---|---|---|
| T001 | M/42 | 19 | 0.01 | 0.02 | 0.02 | c.221-977_532+7059del | p.Asp75_Gly178 del | 4–5 | Homozygous |
| T006B | F/11 | 11 | 0.01 | 0.01 | 0.02 | c.4006C>T | p.Arg1336* | 28 | Homozygous |
| T015 | M/72 | 8 | 0.02 | 0.02 | 0.07 | c.8418_8419insTCCC | p.Ser2807fs | 52 | Homozygous |
| T018 | F/18 | 12 | 0.04 | 0.00 | 0.0.3 | c.7399C>T, c.8418_8419insTCCC | p.Gln2467*, p.Ser2807fs | 43/52 | Compound heterozygous |
| T028 | M/34 | 16 | 0.01 | 0.01 | 0.03 | c.7730-1 G>T, c.7906A>T |
- p.Asn7906Tyr |
46/48 | Homozygous Heterozygous |
| T030 | M/66 | 7 | 0.03 | 0.01 | 0.09 | c.-2328T>G, c.2220G>A, c.3379+7A>C, c.3539-33G>A, c.3539-49C>T, c.4414dupC, c.5620+33-40delA, c.6554G>A, c.6902-5T>A, c.8418_8419insTCCC | p.Met740Ile, p.His1472fs, p.Arg2185Gln, p.Ser2807fs | 28/52 | Compound heterozygous |
| T032 | F/19 | 13 | 0.01 | 0.00 | 0.01 | c.3379+1 G>A | - | 25 | Heterozygous |
| T036 | F/41 | 25 | 0.01 | 0.01 | 0.01 | c.5180_5181insTT | p.Leu1727fs | 30 | Homozygous |
| T040 | F/4 | 11 | 0.01 | 0.03 | 0.01 | c.874+5 G>A | - | 7 | Homozygous |
| T046 | F/16 | 21 | 0.01 | 0.04 | 0.02 | c.385C>A, c.813C>A | p.Leu129Met, p.Tyr271* | 5/7 | Compound heterozygous |
| T050 | F/21 | 24 | 0.01 | 0.00 | 0.01 | c.876delC, c.1255C>T | p.Ser293fs, p.Gln419* | 8/11 | Compound heterozygous |
| T058 | F/1 | n/a | 0.01 | 0.03 | 0.02 | c.2443-?_2685+?del, c.5180_5181insTT |
p.Val815_Gln895del p.Leu1727fs |
19–20/30 | Compound heterozygous |
| T065 | M/15 | 22 | 0.01 | 0.02 | 0.01 | c.3303C>A | p.Cys1101* | 25 | Homozygous |
| T076 | M/12 | 23 | 0.01 | 0.03 | 0.01 | c.1897T>C | p.Cys633Arg | 15 | Homozygous |
| T078 | F/5 | 9 | 0.01 | 0.03 | 0.01 | c.5455+2 T>C | - | 31 | Homozygous |
| T085 | M/17 | 23 | 0.04 | 0.03 | 0.01 | c.1926G>A, c.2438dupG | p.Trp642*, p.Gly813fs | 15/18 | Compound heterozygous |
| T086 | M/1 | 3 | 0.01 | 0.02 | 0.01 | c.1A>G, c.2377C>T | p.Met1Val, p.Gln793* | 2/18 | Compound heterozygous |
| T090 | M/2 | 8 | 0.01 | 0.04 | 0.01 | c.1117C>T, gene conversion (c.3931C>T, c.4027A>G, c.4079T>C, c.4105T>A) | p.Arg373*, p.Gln1311*, p.Ile1343Val, p.Val1360Ala, p.Phe1369Ile | 10/28 | Compound heterozygous |
| T093 | M/8 | 10 | 0.02 | 0.02 | 0.04 | c.6709T>C, c.220+20G>T | p.Cys2237Arg, - | 38 | Homozygous |
| T099 | F/3 | 18 | 0.04 | 0.02 | 0.06 | c.1750_1765delinsCG | p.Cys584_Ser58 9delinsArg | 15 | Heterozygous |
| T103 | M/65 | 6 | 0.02 | 0.04 | 0.02 | c.8418_8419insTCCC | p.Ser2807fs | 52 | Homozygous |
| T108 | F/60 | 13 | 0.07 | 0.01 | 0.06 | c.4146G>T | p.(=) | n/a | n/a |
| T110 | F/41 | 23 | 0.01 | 0.01 | 0.02 | c.8418_8419insTCCC | p.Ser2807fs | 52 | Homozygous |
| T112 | M/48 | 4 | 0.04 | 0.04 | 0.08 | c.8418_8419insTCCC | p.Ser2807fs | 52 | Homozygous |
| T114 | M/47 | 8 | 0.03 | 0.02 | 0.05 | c.7399C>T, c.8418_8419insTCCC | p.Gln2467*, p.Ser2807fs | 43/52 | Compound heterozygous |
| T116 | F/29 | 6 | 0.02 | 0.04 | 0.02 | c.8418_8419insTCCC | p.Ser2807fs | 52 | Homozygous |
| T118 | F/30 | 8 | 0.02 | 0.00 | 0.05 | c.8418_8419insTCCC | p.Ser2807fs | 52 | Homozygous |
| T121 | M/62 | 7 | 0.02 | 0.05 | 0.06 | c.7399C>T, c.8418_8419insTCCC | p.Gln2467*, p.Ser2807fs | 43/52 | Compound heterozygous |
| T136 | M/24 | 24 | 0.01 | 0 | 0 | c.2438dupG | p.Gly813fs | 18 | Heterozygous |
| T141 | F/16 | 13 | 0.04 | 0.04 | 0.06 | c.817T>C | p.Arg273Trp | 7 | Homozygous |
| T143 | F/64 | 24 | 0.04 | 0.04 | 0.03 | c.8043_8044delAG | p.Arg2681fs | 49 | Heterozygous |
| T151 | M/27 | 29 | 0.02 | 0.04 | 0.01 | c.3939G>A, c.5842+1 G>C | p.Arg1313*, - | 28/34 | Compound heterozygous |
| T154 | M/16 | 24 | 0.01 | 0.03 | 0.01 | c.1656_1657insT, c.8418_8419insTCCC | p.Trp552fs, p.Ser2807fs | 14/52 | Compound heterozygous |
| T166 | M/20 | 10 | <0.10 | <0.10 | 0.06 | c.8418_8419insTCCC | p.Ser2807fs | 52 | Homozygous |
Figure 1.
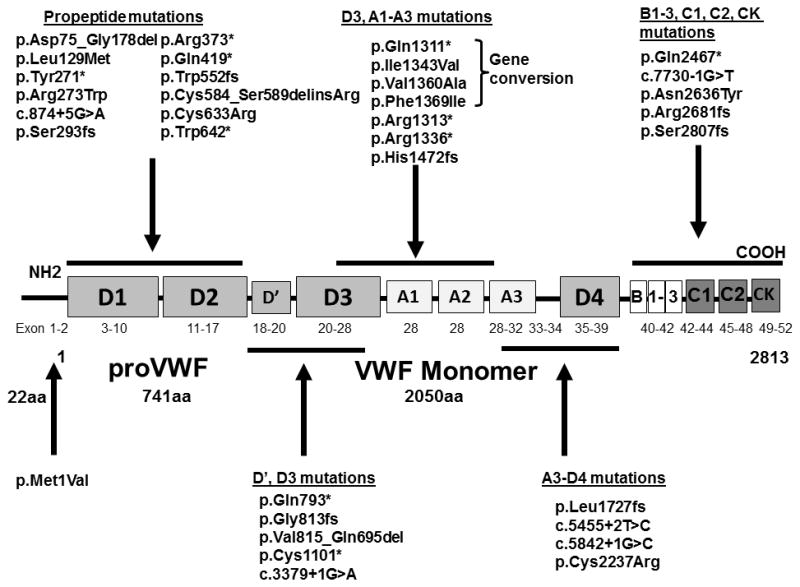
Figure 1 shows the location of the 31 mutations identified in this study (HGVS nomenclature; * represents a nonsense mutation).
Four mutations (exon 18 c.2438dupG (p.Gly813fs), exon 30 c.5180_5181insTT (p.Leu1727fs), exon 43 c.7399C>T (p.Gln2467*), and exon 52 c.8418_8419insTCCC (p.Ser2807fs)) were identified in more than one family. The most frequent mutation identified in this cohort was the frameshift mutation in exon 52, p.Ser2807fs, found in 12 IC (35%), six of whom were homozygous and six heterozygous. Additionally, 14 family members were found to be either homozygous (n=3) or heterozygous (n=11) for this mutation. This frameshift mutation was previously identified and reported in two IC from the Canadian type 1 VWD population [7]. The individuals with this mutation are from Eastern Canada and can all be linked to a larger pedigree. The insertion of the four nucleotides (TCCC) results in a frameshift, which abolishes the original stop codon and adds 16 amino acids prior to a new stop codon being created. The molecular pathogenesis of this mutation is currently under further investigation. The Baltic founder mutation in exon 18, c.2435delC (p.812Argfs*31), identified in the original VWD family from the Åland Islands [31], was not found in this population.
Missense mutations
A total of six missense mutations (19% of all mutations) were identified in this study; c.1A>G (p.Met1Val), c.385C>A (p.Leu129Met), c.817T>C (p.Arg273Trp), c.1897T>C (p.Cys633Arg), c.6709T>C (p.Cys2237Arg), and c.7906A>T (p.Asn2636Tyr). Three IC were found to be homozygous for missense mutations and had no other mutations identified throughout VWF. The predicted impact of these mutations was examined using PolyPhen-2 and SIFT in silico programs (Table S1). These individuals were negative for partial gene deletions.
The missense mutation p.Met1Val found in the heterozygous state in T086 replaces the invariant initiator methionine with a valine. This substitution will inevitably result in a null allele due to the marked suppression of VWF translation from the mutant transcript.
The missense mutation p.Arg273Trp, identified in the homozygous state in IC T141, has been previously reported. Expression studies showed that this mutation was responsible for the VWF deficiencies noted in their patients and the aberrant multimer patterns observed [32].
In IC T076 the only mutation identified was a single homozygous nucleotide change in exon 15 resulting in an arginine residue replacing a cysteine at amino acid 633 (p.Cys633Arg). Due to the location of this mutation in the propeptide region and the known importance of cysteine molecules in VWF biosynthesis and the proper folding of VWF, the molecular pathogenesis of this mutation is currently under investigation.
A third IC (T093), was found to be homozygous for a different missense mutation which again results in a cysteine residue being replaced by an arginine, this time at position 2237 (p.Cys2237Arg). This mutation is located in exon 38 which encodes part of the D4 domain; the function of which is not well understood.
Partial/total gene deletions
We identified two different partial VWF deletions in two IC. One IC (T001) was identified as being homozygous for the previously reported exon 4–5 deletion mutation (c.221-977_532+7059del; p. Asp75_Gly178del) [13]. The deletions of exons 4 and 5 were confirmed using the deletion-specific PCR described by Sutherland et al. (2009) and MLPA. Two enrolled family members, a sibling and mother, diagnosed with type 1 VWD (T002 and T156), were screened and determined to be heterozygous for this deletion. The haplotype of these individuals was analyzed and found to be different than the common haplotype reported in the UK population [13]. Similarly, no common haplotype was found between individuals with the same mutations in the Canadian and European type 1 VWD studies [6,7].
T058, for whom only a heterozygous frameshift mutation (p.Leu1727fs) had been identified after direct sequencing was also found to be heterozygous for a deletion of exons 19 and 20 (c.2443-?_2685+?del; p.Val815_Gln895). The deletion of exons 19 and 20 removes 81 amino acids and remains in-frame. The location of the breakpoints have yet to be determined. Both mother and father were subsequently analyzed for this partial gene deletion. The insertion mutation was inherited from the father (T060) and the partial gene deletion from the mother (T059).
Gene Conversion
Gene conversions between VWF and its pseudogene have been previously reported [33]. One IC (T090) in this study was found to have a heterozygous gene conversion, a minimum 175 bp and maximum 395 bp in length, which included the nonsense mutation c.3931C>T (p.Gln1311*), and the missense mutations c.4027A>G (p.Ile1343Val), c.4079T>C (p.Val1360Ala), and c.4105T>A (p.Phe1369Ile). Upon sequencing the family members, it was determined that T090 inherited the gene conversion from the father.
Phenotype-Genotype Correlations
The five individuals for whom incomplete pathogenic explanations could be elucidated had similar VWF:Ag, VWF:RCo and FVIII levels compared to the 29 individuals for whom two mutant VWF alleles were identified (p=ns, Mann-Whitney U testing). As well, these five IC did not have significantly lower bleeding scores when compared to the other IC (p=0.119, Mann-Whitney U test) (BS of the five ICs=13, 13, 18, 24, and 24).
Discussion
This report comprises one of the largest type 3 VWD studies to date, with information on 42 type 3 VWD patients from 34 different families in Canada. Type 3 VWD is classically inherited in an autosomal recessive fashion with individuals homozygous or compound heterozygous for null alleles. However, this is not always the case and interestingly, the original type 3 VWD family from the Åland Islands did not exhibit recessive inheritance and showed evidence of co-dominant inheritance. We have shown that in the Canadian type 3 VWD population as well this is not always the case, with 53% of the families reported in this study having both type 1 VWD and type 3 VWD individuals. Many OC in this cohort are not phenotypically silent, manifesting low VWF levels and mucocutaneous bleeding symptoms. The Canadian type 3 VWD population is comprised of a few homogeneous populations with evidence of founder alleles, yet is also very heterogeneous in nature with approximately 40% of IC from nine different ethnicities. This heterogeneity may account for the distinct nature of this cohort.
Alloantibodies to VWF are a rare complication of type 3 VWD and are frequently associated with homozygous large deletions [8,9,11,14–17], however a few cases of alloantibodies to VWF in patients with homozygous gene conversions [34] and homozygous nonsense mutations [10,35] have also been reported. The one individual (T018) in this study that is positive on the anti-VWF ELISA is compound heterozygous for our commonly reported mutation, p.Ser2807fs, and the nonsense mutation p.Gln2467*. The inhibition of collagen binding activity after mixing suggests that the alloantibody in this patient is affecting this specific function of VWF, however further investigation is required to better define the nature of this inhibitory influence. T018 is not on prophylactic treatment but has been treated with Humate-P® (5000 IU) at times of nosebleeds, surgery or trauma. The patient responds clinically to treatment and has not developed any anaphylactic reactions and/or become refractory to replacement therapy.
As with previous type 3 VWD studies we have identified a number of novel mutations scattered throughout the VWF gene that result in null alleles. An important finding within this study is the high prevalence of mutations located within the propeptide region of VWF (42%). As well, we have shown that IC with mutations in the propeptide have higher BS than IC with mutations in other areas of VWF. Futher Investigation of the contribution of propeptide mutations to type 3 VWD is warranted and is under investigation by our group.
While the number of missense mutations (~19%) identified in this study is consistent with other reported type 3 VWD populations [10] we believe the contribution of these missense mutations warrants further investigation. Recently, other groups have begun to investigate the contribution of missense mutations to type 3 VWD [36,37]. Investigations of the two missense mutations that were found in the homozygous state are ongoing.
In the majority of type 3 VWD studies, pathogenic mutations are identified in 80–90% of patients [12,21–24]. In this study we were able to identify both pathogenic mutations in 85% of IC (62 of 68 (91%) mutant alleles identified). In the remaining five IC we were unable to find two mutant VWF alleles that could explain their VWD disease state. A number of possibilities may exist for this and warrant investigation. Pathogenic possibilities include apparently silent sequence variations in the VWF gene located outside of consensus splice sites that disrupt the normal VWF mRNA splicing, deep intronic mutations that may only be identified through whole gene sequencing, and distant regulatory elements outside of VWF.
In conclusion, our study represents one of the largest and most comprehensive reports of type 3 VWD patients and their family members. We have made important observations regarding the contribution of mutations in the propeptide region to the type 3 VWD phenotype including the increased severity of bleeding in these cases. As well, the phenotype-genotype correlations made in this paper further highlight the differences between OC of type 3 VWD and those diagnosed with type 1 VWD. We have also shown that in the Canadian type 3 VWD population, 48% of OC of type 3 VWD are not phenotypically silent and have been diagnosed with type 1 VWD. This Canadian type 3 VWD population further emphasizes that in a significant proportion of cases the genetic transmission of the VWD phenotype is co-dominant in nature and not recessive.
Supplementary Material
Acknowledgments
The authors acknowledge the contributions of the members of the Association of Hemophilia Clinic Directors of Canada (AHCDC) and the Canadian Association of Nurses in Hemophilia Care. This project was funded by a Canadian Hemophilia Society Research Grant and through the Zimmerman Program for Molecular and Cellular Biology of von Willebrand Disease by The National Institutes of Health Program Project Grant HL081588. MB is the recipient of an Ontario Graduate Scholarship and a Queen’s University Graduate Scholarship. DL holds a Canada Research Chair in Molecular Hemostasis. PJ held a Bayer Hemophilia Awards Program Early Career Investigator Award at the time of this study and currently holds a Clinician Scientist Award from the Southeastern Ontario Academic Medical Assocation (SEAMO).
Footnotes
Addendum
M.B. performed research, analyzed data and wrote the paper. A.T., C.N., C.B., S.T., M.D., J.L., and V.S.B. performed research. D.L. supervised research. P.J. designed and supervised research, analyzed data and wrote the paper.
References
- 1.Bowman M, Hopman WM, Rapson D, Lillicrap D, James P. The prevalence of symptomatic von Willebrand disease in primary care practice. J Thromb Haemost. 2010;8:213–6. doi: 10.1111/j.1538-7836.2009.03661.x. [DOI] [PubMed] [Google Scholar]
- 2.Eikenboom JC. Congenital von Willebrand disease type 3: Clinical manifestations, pathophysiology and molecular biology. Best Pract Res Clin Haematol. 2001;14:365–79. doi: 10.1053/beha.2001.0139. [DOI] [PubMed] [Google Scholar]
- 3.Castaman G, Rodeghiero F, Tosetto A, Cappelletti A, Baudo F, Eikenboom JC, Federici AB, Lethagen S, Linari S, Lusher J, Nishino M, Petrini P, Srivastava A, Ungerstedt JS. Hemorrhagic symptoms and bleeding risk in obligatory carriers of type 3 von Willebrand disease: An international, multicenter study. J Thromb Haemost. 2006;4:2164–9. doi: 10.1111/j.1538-7836.2006.02070.x. [DOI] [PubMed] [Google Scholar]
- 4.Montgomery RR. When it comes to von Willebrand disease, does 1 + 1 = 3? J Thromb Haemost. 2006;4:2162–3. doi: 10.1111/j.1538-7836.2006.02158.x. [DOI] [PubMed] [Google Scholar]
- 5.Cumming A, Grundy P, Keeney S, Lester W, Enayat S, Guilliatt A, Bowen D, Pasi J, Keeling D, Hill F, Bolton-Maggs PH, Hay C, Collins P the UK Haemophilia Centre Doctors’ Organisation. An investigation of the von Willebrand factor genotype in UK patients diagnosed to have type 1 von Willebrand disease. Thromb Haemost. 2006;96:630–41. [PubMed] [Google Scholar]
- 6.Goodeve A, Eikenboom J, Castaman G, Rodeghiero F, Federici AB, Batlle J, Meyer D, Mazurier C, Goudemand J, Schneppenheim R, Budde U, Ingerslev J, Habart D, Vorlova Z, Holmberg L, Lethagen S, Pasi J, Hill F, Hashemi Soteh M, Baronciani L, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the european study, molecular and clinical markers for the diagnosis and management of type 1 von Willebrand disease (MCMDM-1VWD) Blood. 2007;109:112–21. doi: 10.1182/blood-2006-05-020784. [DOI] [PubMed] [Google Scholar]
- 7.James PD, Notley C, Hegadorn C, Leggo J, Tuttle A, Tinlin S, Brown C, Andrews C, Labelle A, Chirinian Y, O’Brien L, Othman M, Rivard G, Rapson D, Hough C, Lillicrap D. The mutational spectrum of type 1 von Willebrand disease: Results from a Canadian cohort study. Blood. 2007;109:145–54. doi: 10.1182/blood-2006-05-021105.. [DOI] [PubMed] [Google Scholar]
- 8.Peake IR, Liddell MB, Moodie P, Standen G, Mancuso DJ, Tuley EA, Westfield LA, Sorace JM, Sadler JE, Verweij CL. Severe type III von Willebrand’s disease caused by deletion of exon 42 of the von willebrand factor gene: Family studies that identify carriers of the condition and a compound heterozygous individual. Blood. 1990;75:654–61. [PubMed] [Google Scholar]
- 9.Mancuso DJ, Tuley EA, Castillo R, de Bosch N, Mannucci PM, Sadler JE. Characterization of partial gene deletions in type III von Willebrand disease with alloantibody inhibitors. Thromb Haemost. 1994;72:180–5. [PubMed] [Google Scholar]
- 10.Baronciani L, Cozzi G, Canciani MT, Peyvandi F, Srivastava A, Federici AB, Mannucci PM. Molecular defects in type 3 von Willebrand disease: Updated results from 40 multiethnic patients. Blood Cells Mol Dis. 2003;30:264–70. doi: 10.1016/s1079-9796(03)00033-0. [DOI] [PubMed] [Google Scholar]
- 11.Xie F, Wang X, Cooper DN, Chuzhanova N, Fang Y, Cai X, Wang Z, Wang H. A novel Alu-mediated 61-kb deletion of the von Willebrand factor (VWF) gene whose breakpoints co-locate with putative matrix attachment regions. Blood Cells Mol Dis. 2006;36:385–91. doi: 10.1016/j.bcmd.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 12.Mohl A, Marschalek R, Masszi T, Nagy E, Obser T, Oyen F, Sallai K, Bodo I, Schneppenheim R. An Alu-mediated novel large deletion is the most frequent cause of type 3 von Willebrand disease in Hungary. J Thromb Haemost. 2008;6:1729–35. doi: 10.1111/j.1538-7836.2008.03107.x. [DOI] [PubMed] [Google Scholar]
- 13.Sutherland MS, Cumming AM, Bowman M, Bolton-Maggs PHB, Bowen DJ, Collins PW, Hay CRM, Will AM, Keeney S. A novel deletion mutation is recurrent in von Willebrand disease types 1 and 3. Blood. 2009;114:1091–8. doi: 10.1182/blood-2008-08-173278. [DOI] [PubMed] [Google Scholar]
- 14.Shelton-Inloes BB, Chehab FF, Mannucci PM, Federici AB, Sadler JE. Gene deletions correlate with the development of alloantibodies in von Willebrand disease. J Clin Invest. 1987;79:1459–65. doi: 10.1172/JCI112974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ngo KY, Glotz VT, Koziol JA, Lynch DC, Gitschier J, Ranieri P, Ciavarella N, Ruggeri ZM, Zimmerman TS. Homozygous and heterozygous deletions of the von Willebrand factor gene in patients and carriers of severe von Willebrand disease. Proc Natl Acad Sci U S A. 1988;85:2753–7. doi: 10.1073/pnas.85.8.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneppenheim R, Krey S, Bergmann F, Bock D, Budde U, Lange M, Linde R, Mittler U, Meili E, Mertes G. Genetic heterogeneity of severe von Willebrand disease type III in the German population. Hum Genet. 1994;94:640–52. doi: 10.1007/BF00206958. [DOI] [PubMed] [Google Scholar]
- 17.Eikenboom JC, Castaman G, Vos HL, Bertina RM, Rodeghiero F. Characterization of the genetic defects in recessive type 1 and type 3 von Willebrand disease patients of Italian origin. Thromb Haemost. 1998;79:709–17. [PubMed] [Google Scholar]
- 18.Acquila M, Bottini F, DI Duca M, Vijzelaar R, Molinari AC, Bicocchi MP. Multiplex ligation-dependent probe amplification to detect a large deletion within the von Willebrand gene. Haemophilia. 2009;15:1346–8. doi: 10.1111/j.1365-2516.2009.02078.x. [DOI] [PubMed] [Google Scholar]
- 19.Cabrera N, Casana P, Cid AR, Moret A, Moreno M, Palomo A, Aznar JA. First application of MLPA method in severe von Willebrand disease. confirmation of a new large VWF gene deletion and identification of heterozygous carriers. Br J Haematol. 2011;152:240–2. doi: 10.1111/j.1365-2141.2010.08400.x. [DOI] [PubMed] [Google Scholar]
- 20.Yadegari H, Driesen J, Hass M, Budde U, Pavlova A, Oldenburg J. Large deletions identified in patients with von Willebrand disease using multiple ligation-dependent probe amplification. J Thromb Haemost. 2011;9:1083–6. doi: 10.1111/j.1538-7836.2011.04260.x. [DOI] [PubMed] [Google Scholar]
- 21.Zhang ZP, Blomback M, Egberg N, Falk G, Anvret M. Characterization of the von Willebrand factor gene (VWF) in von Willebrand disease type III patients from 24 families of Swedish and Finnish origin. Genomics. 1994;21:188–93. doi: 10.1006/geno.1994.1241. [DOI] [PubMed] [Google Scholar]
- 22.Sutherland MS, Keeney S, Bolton-Maggs PH, Hay CR, Will A, Cumming AM. The mutation spectrum associated with type 3 von Willebrand disease in a cohort of patients from the north west of England. Haemophilia. 2009;15:1048–57. doi: 10.1111/j.1365-2516.2009.02059.x. [DOI] [PubMed] [Google Scholar]
- 23.Mohl A, Boda Z, Jager R, Losonczy H, Marosi A, Masszi T, Nagy E, Nemes L, Obser T, Oyen F, Radvanyi G, Schlammadinger A, Szelessy Z, Varkonyi A, Vezendy K, Vilimi B, Schneppenheim R, Bodo I. Common large partial VWF gene deletion does not cause alloantibody formation in the Hungarian type 3 von Willebrand disease population. J Thromb Haemost. 2011;9:945–52. doi: 10.1111/j.1538-7836.2011.04250.x. [DOI] [PubMed] [Google Scholar]
- 24.Gupta PK, Saxena R, Adamtziki E, Budde U, Oyen F, Obser T, Schneppenheim R. Genetic defects in von Willebrand disease type 3 in Indian and Greek patients. Blood Cells Mol Dis. 2008;41:219–22. doi: 10.1016/j.bcmd.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Bowman M, Mundell G, Grabell J, Hopman WM, Rapson D, Lillicrap D, James P. Generation and validation of the condensed MCMDM-1VWD bleeding questionnaire for von Willebrand disease. J Thromb Haemost. 2008;6:2062–6. doi: 10.1111/j.1538-7836.2008.03182.x. [DOI] [PubMed] [Google Scholar]
- 26.Mannucci PM, Federici AB. Antibodies to von Willebrand factor in von Willebrand disease. Adv Exp Med Biol. 1995;386:87–92. doi: 10.1007/978-1-4613-0331-2_7. [DOI] [PubMed] [Google Scholar]
- 27.Casonato A, Pontara E, Zerbinati P, Zucchetto A, Girolami A. The evaluation of factor VIII binding activity of von Willebrand factor by means of an ELISA method: Significance and practical implications. Am J Clin Pathol. 1998;109:347–52. doi: 10.1093/ajcp/109.3.347. [DOI] [PubMed] [Google Scholar]
- 28.Favaloro EJ. Collagen binding assay for von Willebrand factor (VWF:CBA): Detection of von Willebrand disease (VWD) and discrimination of VWD subtypes, depends on collagen source. Thromb Haemost. 2000;83:127–35. [PubMed] [Google Scholar]
- 29.Lahiri DK, Nurnberger JI., Jr A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res. 1991;19:5444. doi: 10.1093/nar/19.19.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shahbazi S, Mahdian R, Ala FA, Lavergne JM, Denis CV, Christophe OD. Molecular characterization of Iranian patients with type 3 von Willebrand disease. Haemophilia. 2009;15:1058–64. doi: 10.1111/j.1365-2516.2009.02046.x. [DOI] [PubMed] [Google Scholar]
- 31.Zhang ZP, Blomback M, Nyman D, Anvret M. Mutations of von Willebrand factor gene in families with von Willebrand disease in the Åland Islands. Proc Natl Acad Sci U S A. 1993;90:7937–7940. doi: 10.1073/pnas.90.17.7937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allen S, Abuzenadah AM, Hinks J, Blagg JL, Gursel T, Ingerslev J, Goodeve AC, Peake IR, Daly ME. A novel von Willebrand disease-causing mutation (Arg273Trp) in the von Willebrand factor propeptide that results in defective multimerization and secretion. Blood. 2000;96:560–8. [PubMed] [Google Scholar]
- 33.Corrales I, Ramirez L, Altisent C, Parra R, Vidal F. Rapid Molecular Diagnosis of von Willebrand disease by direct sequencing. Detection of 12 novel putative mutations in VWF gene. Thromb Haemos. 2009;101:570–576. doi: 10.1160/th08-08-0500. [DOI] [PubMed] [Google Scholar]
- 34.Surdhar GK, Enayat MS, Lawson S, Williams MD, Hill FG. Homozygous gene conversion in von Willebrand factor gene as a cause of type 3 von Willebrand disease and predisposition to inhibitor development. Blood. 2001;98:248–50. doi: 10.1182/blood.v98.1.248. [DOI] [PubMed] [Google Scholar]
- 35.Zhang ZP, Lindstedt M, Falk G, Blomback M, Egberg N, Anvret M. Nonsense mutations of the von Willebrand factor gene in patients with von Willebrand disease type III and type I. Am J Hum Genet. 1992;51:850–8. [PMC free article] [PubMed] [Google Scholar]
- 36.Tjernberg P, Castaman G, Vos HL, Bertina RM, Eikenboom JC. Homozygous C2362F von Willebrand factor induces intracellular retention of mutant von Willebrand factor resulting in autosomal recessive severe von Willebrand disease. Br J Haematol. 2006;133:409–18. doi: 10.1111/j.1365-2141.2006.06055.x. [DOI] [PubMed] [Google Scholar]
- 37.Baronciani L, Federici AB, Cozzi G, La Marca S, Punzo M, Rubini V, Canciani MT, Mannucci PM. Expression studies of missense mutations p.D141Y, p. C275S located in the propeptide of von Willebrand factor in patients with type 3 von Willebrand disease. Haemophilia. 2008;14:549–55. doi: 10.1111/j.1365-2516.2008.01682.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.